

Key Points
The CARD11- or MyD88-mutant Eµ-myc– transgenic lymphoma secretome activates macrophages to mediate lymphoma cell senescence via TGF-β.
MyD88- or CARD11-mutant lymphomas counter senescence-preferential T-cell recognition by PD-L1 expression, as seen in human DLBCL profiles.

Abstract
Aberrant B-cell receptor/NF-κB signaling is a hallmark feature of B-cell non-Hodgkin lymphomas, especially in diffuse large B-cell lymphoma (DLBCL). Recurrent mutations in this cascade, for example, in CD79B, CARD11, or NFKBIZ, and also in the Toll-like receptor pathway transducer MyD88, all deregulate NF-κB, but their differential impact on lymphoma development and biology remains to be determined. Here, we functionally investigate primary mouse lymphomas that formed in recipient mice of Eµ-myc transgenic hematopoietic stem cells stably transduced with naturally occurring NF-κB mutants. Although most mutants supported Myc-driven lymphoma formation through repressed apoptosis, CARD11- or MyD88-mutant lymphoma cells selectively presented with a macrophage-activating secretion profile, which, in turn, strongly enforced transforming growth factor β (TGF-β)-mediated senescence in the lymphoma cell compartment. However, MyD88- or CARD11-mutant Eµ-myc lymphomas exhibited high-level expression of the immune-checkpoint mediator programmed cell death ligand 1 (PD-L1), thus preventing their efficient clearance by adaptive host immunity. Conversely, these mutant-specific dependencies were therapeutically exploitable by anti–programmed cell death 1 checkpoint blockade, leading to direct T-cell–mediated lysis of predominantly but not exclusively senescent lymphoma cells. Importantly, mouse-based mutant MyD88- and CARD11-derived signatures marked DLBCL subgroups exhibiting mirroring phenotypes with respect to the triad of senescence induction, macrophage attraction, and evasion of cytotoxic T-cell immunity. Complementing genomic subclassification approaches, our functional, cross-species investigation unveils pathogenic principles and therapeutic vulnerabilities applicable to and testable in human DLBCL subsets that may inform future personalized treatment strategies.
Introduction
Activated B-cell receptor (BCR)/NF-κB signaling is a prominent feature of B-cell non-Hodgkin lymphomas (B-NHLs), especially in diffuse large B-cell lymphoma (DLBCL)and follicular lymphoma.1,2 In addition to antigen-dependent and independent BCR signaling, mutational events downstream of the BCR critically modulate the effector output of the cascade. Specifically, gain- and loss-of-function defects in gene loci such as CD79B, CARD11, A20/TNFAIP3, or NFKBIZ,3-6 and in the Toll-like receptor signaling mediator MyD887 account for constitutive NF-κB activity (collectively referred to here as the “NF-κB–deregulating mutants [NDMs]”), whose individual functional properties in lymphoma development and progression remain elusive. The molecular landscape of DLBCL, the most prevalent aggressive B-NHL entity, is very heterogenous. Gene-expression profiling (GEP) unveiled distinct cell-of-origin (COO) subentities, termed germinal center B-cell (GCB) and activated B-cell (ABC) subtypes.8-10 Further underscoring the highly diverse genetic composition of DLBCL, in-depth genome analyses identified recurrent mutations and a variety of novel molecular subgroups,1,11-17 with NDMs predominantly but not exclusively found in ABC DLBCL.18
Although approximately two-thirds of DLBCL patients can be cured with the rituximab plus cyclophosphamide, hydroxydaunorubicin, oncovin, and prednisone (CHOP; R-CHOP) immune-chemotherapy induction, standard, refractory, or relapsing patients are hard to salvage. Numerous randomized phase 3 trials failed to improve the standard by an additional targeting agent.19-22 Underlying reasons for such a series of discouraging results besides the overwhelming molecular lymphoma heterogeneity are understudied functional aspects of the lymphoma biology linked to lead mutational lesions, specifically the dynamics of important NDM-governed drug-effector– and cell-fate–relevant mechanisms such as cellular senescence.23
Evoked by activated oncogenes or DNA-damaging chemotherapeutic agents, cellular senescence primarily operates as a key tumor-suppressive mechanism in preneoplastic or full-blown malignant lesions.24-26 The senescence hallmark feature, the lasting G1-phase cell-cycle arrest due to trimethylated histone H3-lysine 9 (H3K9)-involving chromatin compaction in the vicinity of S-phase-promoting E2F target genes, is mediated by H3K9 methyltransferases such as Suv39h1.27,28 Moreover, the senescence-associated secretory phenotype (SASP), largely consisting of NF-κB-driven proinflammatory cytokines,23,29 appears as a particularly important component in aggressive lymphoma exhibiting high constitutive BCR/NF-κB activity.23 However, the dynamic interplay between NDMs and cellular senescence during development or later course of DLBCL is poorly understood.30-33
Preclinical platforms based on established multipassage cancer cell lines, primary tumor cells grown in 2-dimensional- or 3-dimensional cultures, or patient-derived xenograft models in immunocompromised mice are instrumental to study functional implications of defined genetic lesions for lymphoma biology but lack the lymphoma/host immune synapse.34-43 Thus, we exploit here cell-autonomous properties and non–cell-autonomous interdependencies of transplantable Eµ-myc transgenic mouse lymphomas in immune-competent syngeneic recipients.23,24 We undertook a systematic investigation of individual patient-derived NDMs stably propagated in primary Eµ-myc transgenic mouse lymphomas as a single-oncogene–driven but secondary hit–dependent model platform that gives rise to a broad spectrum of early to rather mature and quite heterogeneous aggressive B-cell lymphomas.44,45 Remarkably, our functional genomics investigation of NDMs in this model platform focused on biology, senescence capacity, and host immune interference in vivo, and uncovered hitherto unknown immunogenic vulnerabilities in the MyD88- and CARD11-deregulated subsets, for which we retrieved matching genetic patterns in DLBCL where novel lesion-based classifiers may inform alternative, immune-oncology–based therapeutic strategies.
Materials and methods
In vivo treatments
All mouse experiments were approved by the governmental review board (Landesamt für Gesundheit und Soziales, Berlin, Germany) and conform to the regulatory standards. Retroviral infection of fetal liver cells (FLCs) and lymphoma cells, and subsequent transplantation are described in supplemental Methods (available on the Blood Web site). For macrophage depletion, C57BL/6N mice transplanted with MyD88-L265P MSCV–green fluorescent protein (GFP)-infected lymphoma cells were intraperitoneally exposed to liposomal clodronate (Clophosome; FormuMax; 5.5 mg/kg body weight every 48 hours) until lymphoma manifestation.46 Anti–programmed cell death 1 (PD1) treatment of lymphoma-bearing C57/BL6 mice was performed using the InVivoMAb anti-mouse PD1 antibody (BioXcell), which was injected IV (10 mg/kg body weight) every 2 days after first diagnosis of a palpable lymphadenopathy.
T-cell ex vivo killing assay
Lymphoma cells and splenic cytotoxic T cells were obtained as matched isolates from the same hematopoietic stem cell (HSC)-generated MyD88-L265P tumor-bearing mice using the B-cell and CD8a+ isolation kits (Miltenyi). Lymphoma cells were labeled with the fluorescent SA–β-gal substrate C12RG (Thermo Fisher Scientific) before B and cytotoxic T cells were treated with anti–programmed cell death–ligand 1 (PD-L1; Thermo Fisher Scientific) and anti-PD1 (eBioscience) antibodies. After washing, B and CD8+ T cells were cocultured for 24 hours prior to their viability labeling with Ghost-Dye-Red780 (Tonbo Bioscience).
RNA sequencing
Details of library preparation and sequencing can be found in supplemental Methods (available on the Blood Web site). Analysis scripts are available from L.C. and A.D. upon request.
Applying the mouse-informed MyDness and CARDness signatures to human DLBCL GEP
The top 200 genes with the highest fold-changes (upregulation) compared with the respective vector control lymphomas were identified by RNA sequencing (RNA-seq) for all NF-κB mutants tested in the Eµ-myc mouse model. A mutant-specific unique set of genes was identified for MyD88-L265P as well as CARD11-L244P mutant lymphomas, accordingly named MyDness and CARDness. Three large DLBCL cohorts comprising 137 transcriptomes (GSE9858815 ), 522 transcriptomes (National Cancer Institute [NCI] cohort16 ), and 726 transcriptomes,14 respectively, were stratified into 2 groups based on mean expression of genes compiled from these murine MyDness and/or CARDness signatures. Microarray-based expression data were filtered for the genes of interest, row (gene) mean-centered, and scaled to unit variance. Subsequently, probe sets were collapsed to the gene level using a correlation-based approach as described followed by computation of the average gene-expression value for each sample and signature. See Schleich et al regarding the generation and functional validation of the 22-gene-comprising Suvarness signature (see Figure 6C for the complete gene list), which marks senescence-capable aggressive B-cell lymphomas in mice and men.47
Data analysis
Detailed analysis methods gene set enrichment and differential gene expression can be found in supplemental Methods.
Statistical evaluation
Statistical differences between 2 groups were calculated using the unpaired, 2-tailed Student's t test. P values <.05 were considered significant. The log-rank (Mantel-Cox) test was applied to survival analyses. Analysis was performed using GraphPad Prism.
Results
Naturally occurring NDM accelerate Myc-driven lymphomagenesis
GFP-coexpressing murine homologs of the naturally, human lymphoma-occurring mutations in MyD88, CARD11, CD79B, and NFKBIZ were first stably introduced into preneoplastic Eµ-myc transgenic spleen cell preparations, and the dynamics of the mutant/GFP+ population was monitored. Compared with an empty vector control, all mutants tested were positively selected in vitro and presented with enhanced viability (Figure 1A). Next, Eµ-myc transgenic HSCs stably infected with the NDM adjusted to ∼10% GFP+ cells were transplanted, and GFP enrichment, now reflecting lymphoma formation, was monitored in recipient mice. Although MyD88- and NFKBIZ-mutant lymphomas invariably developed as GFP+, that is, mutation-driven malignancies, lymphomas in the CD79B-mutant cohort were GFP+ in approximately half of the cases, and unexpectedly none of the lymphomas arising in recipients of the CARD mutation (Figure 1B). Notably, the immature-to-mature B-cell differentiation spectrum of these aggressive malignancies was quite heterogeneous, with MyD88-L265P– and NFKBIZ–mutant lymphomas exhibiting a more activated B-cell state (supplemental Figure 1).
![Naturally occurring NDMs cooperate with oncogenic Myc to promote B-cell lymphomagenesis in vivo. (A) Mutant enrichment in B-cell–purified preneoplastic Eµ-myc transgenic splenocyte preparations in vitro (n ≥ 3 spleens from 4-week-old transgenic mice with no signs of lymphadenopathy), retrovirally transduced with the indicated NDM (coexpressing GFP) or an empty vector, respectively. The initial percentage of GFP+, GhostRed− (ie, viable) B lymphocytes was adjusted to 8.0% ± 4%. After 3 days in culture, the relative changes in the fraction of GFP+ and viable cells were measured. Error bars denote the standard deviation. (B) Eµ-myc transgenic murine FLCs, a source of HSCs, were stably transduced with mutant moieties as in panel A, and IV transplanted into lethally irradiated strain-matched mice. Recipient mice were monitored for up to 300 days until lymph nodes became well palpable. Shown here is the fraction of GFP+ Eµ-myc lymphomas detected in each mutant cohort, with the number of mice under observation indicated for every cohort. (C) Tumor latencies reflecting the time between HSC transplantation and first-time detectability of a palpable lymphadenopathy for empty vector (ie, GFP−; n = 19) or the indicated mutants (all GFP+; n = 18 [MyD88] and n = 13 [NFKBIZ]); P < .0001 for each mutant compared with empty vector. (D) NDMs interfere with Myc-induced apoptosis in HSC-generated Eµ-myc–driven lymphomas in vivo. Percentages of apoptotic B-lymphoma cells (TUNEL+/B220+) in n ≥ 3 individual cases per genotype (with error bars denoting the standard deviation). TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling.(E) Principal component (PC) analysis of transcriptome profiles in n ≥ 3 individual HSC-derived Eµ-myc lymphoma cases per genotype. Asterisks indicate significance (P < .05).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2020005244/1/m_bloodbld2020005244r2f1.png?Expires=1769803549&Signature=n1oyAmtAp15h3oO3eV4nPYgXVhlXom-~icQSVczmfS4W733vzPklmtJsvqp8QOys-duEs1vXX3ZK~MqPyDn1KyvRu19N-B5jKG-nLDmuC-2NCZmsqhcoDtZcGtFiL0xis9AvpHulPQnvkOg-uBIQzdNnzd9~X6Gff8iH0o9a8WCkaYqdkkPhGXGlMlxUf4L1P30TTJalyv-UyZ8jDWe8rze78H3jE~rYosFM59M0VVhBk1j6OzVvitUPVnJBBvnqeLyTiriDWF6CnLJ7a8I-4T5C9ImF3AmG6ate99Jzi9uJzfGVpaP250rttsqHpJIT-bPddZBny-2mbrviKFfO3Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Naturally occurring NDMs cooperate with oncogenic Myc to promote B-cell lymphomagenesis in vivo. (A) Mutant enrichment in B-cell–purified preneoplastic Eµ-myc transgenic splenocyte preparations in vitro (n ≥ 3 spleens from 4-week-old transgenic mice with no signs of lymphadenopathy), retrovirally transduced with the indicated NDM (coexpressing GFP) or an empty vector, respectively. The initial percentage of GFP+, GhostRed− (ie, viable) B lymphocytes was adjusted to 8.0% ± 4%. After 3 days in culture, the relative changes in the fraction of GFP+ and viable cells were measured. Error bars denote the standard deviation. (B) Eµ-myc transgenic murine FLCs, a source of HSCs, were stably transduced with mutant moieties as in panel A, and IV transplanted into lethally irradiated strain-matched mice. Recipient mice were monitored for up to 300 days until lymph nodes became well palpable. Shown here is the fraction of GFP+ Eµ-myc lymphomas detected in each mutant cohort, with the number of mice under observation indicated for every cohort. (C) Tumor latencies reflecting the time between HSC transplantation and first-time detectability of a palpable lymphadenopathy for empty vector (ie, GFP−; n = 19) or the indicated mutants (all GFP+; n = 18 [MyD88] and n = 13 [NFKBIZ]); P < .0001 for each mutant compared with empty vector. (D) NDMs interfere with Myc-induced apoptosis in HSC-generated Eµ-myc–driven lymphomas in vivo. Percentages of apoptotic B-lymphoma cells (TUNEL+/B220+) in n ≥ 3 individual cases per genotype (with error bars denoting the standard deviation). TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling.(E) Principal component (PC) analysis of transcriptome profiles in n ≥ 3 individual HSC-derived Eµ-myc lymphoma cases per genotype. Asterisks indicate significance (P < .05).
Naturally occurring NDMs cooperate with oncogenic Myc to promote B-cell lymphomagenesis in vivo. (A) Mutant enrichment in B-cell–purified preneoplastic Eµ-myc transgenic splenocyte preparations in vitro (n ≥ 3 spleens from 4-week-old transgenic mice with no signs of lymphadenopathy), retrovirally transduced with the indicated NDM (coexpressing GFP) or an empty vector, respectively. The initial percentage of GFP+, GhostRed− (ie, viable) B lymphocytes was adjusted to 8.0% ± 4%. After 3 days in culture, the relative changes in the fraction of GFP+ and viable cells were measured. Error bars denote the standard deviation. (B) Eµ-myc transgenic murine FLCs, a source of HSCs, were stably transduced with mutant moieties as in panel A, and IV transplanted into lethally irradiated strain-matched mice. Recipient mice were monitored for up to 300 days until lymph nodes became well palpable. Shown here is the fraction of GFP+ Eµ-myc lymphomas detected in each mutant cohort, with the number of mice under observation indicated for every cohort. (C) Tumor latencies reflecting the time between HSC transplantation and first-time detectability of a palpable lymphadenopathy for empty vector (ie, GFP−; n = 19) or the indicated mutants (all GFP+; n = 18 [MyD88] and n = 13 [NFKBIZ]); P < .0001 for each mutant compared with empty vector. (D) NDMs interfere with Myc-induced apoptosis in HSC-generated Eµ-myc–driven lymphomas in vivo. Percentages of apoptotic B-lymphoma cells (TUNEL+/B220+) in n ≥ 3 individual cases per genotype (with error bars denoting the standard deviation). TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling.(E) Principal component (PC) analysis of transcriptome profiles in n ≥ 3 individual HSC-derived Eµ-myc lymphoma cases per genotype. Asterisks indicate significance (P < .05).
MyD88 or NFKBIZ mutations also accelerated Myc-driven lymphoma onset (Figure 1C). We detected reduced apoptosis throughout the manifest mutant-expressing lymphomas (Figure 1D), underscoring the prosurvival capacity of these NDMs. Interestingly, a principal component analysis (PCA) of GEP obtained by RNA-seq analysis indicated the proximity of lymphomas driven by the respective individual mutants (Figure 1E). In essence, most of the patient-derived NDMs contribute to lymphoma development by suppressing apoptosis, but individual mutations present with globally distinct expression profiles.
MyD88-L265P and CARD11-L244P induce a profound senescence response
Given the role of cellular senescence as another oncogene-induced tumor-suppressive mechanism, especially in Ras-driven preneoplastic lesions, and also in Myc-driven lymphomagenesis,28,46,48 we quantified hallmark features of senescent growth control in manifest NF-κB–mutant Eµ-myc lymphomas. Strikingly, MyD88-L265P–mutant lymphomas presented with significantly higher fractions of cells positive for senescence-associated β-galactosidase (SA–β-gal) activity and the repressive H3K9me3 chromatin mark, as well as a lower percentage of cells positive for the cell-cycle indicator Ki67, and lacked enrichment of S-phase–promoting E2F target genes, collectively underscoring a profound senescent phenotype in this genotype. In contrast, NFKBIZ- and CD79B-mutant lymphomas exhibited basal senescence within the range of empty vector lymphomas (Figure 2A-C; supplemental Figure 2A). Accordingly, only GEP of MyD88-L265P lymphomas, but not of NFKBIZ- or CD79B-mutant lymphomas, enriched for transcripts related to gene sets defining “senescence” and the senescence-associated secretory phenotype (“SASP”) by gene-set enrichment analysis (GSEA) (Figure 2D). Importantly, MyD88-L265P lymphomas, when analyzed solely in the proliferating fraction of cells, divided at a much higher pace compared with nonsenescent NFKBIZ lymphoma cells, as demonstrated by a much higher fraction of cells positive for serine-10-phosphorylated H3 (H3S10-P), a well-established proliferation marker at the G2/M transition49 within the viable, nonsenescent (ie, p16INK4a−) lymphoma cell population (supplemental Figure 2B). Moreover, karyotyping analyses comparing vector control to MyD88-L265P–mutant Eµ-myc lymphomas further underscored the relevance of NF-κB–activating lesions for Myc-driven lymphomagenesis in general and the functional contribution of the MyD88 mutant in this regard in particular (supplemental Figure 2C; supplemental Table 2).
![Prosenescent phenotypes of MyD88 and CARD11 mutations in Myc-driven lymphomagenesis. (A) Percentages (left) of SA–β-gal+ cells in cytospin preparations of n ≥ 3 individual cases per indicated genotype (with error bars denoting the standard deviation) of B220+-purified B-lymphoma cell populations at the time of lymphoma manifestation. Representative photomicrographs of SA–β-gal stainings (right). Scale bars, 100 µm. (B) As in panel A, but lymphoma sections were stained for H3K9me3 and hematoxylin and eosin (H&E). Scale bars, 100 µm. (C) As in panel A, but lymphoma sections stained for Ki67 (H&E stain). Scale bars, 100 µm (identical magnification throughout the figure). (D) GSEA for terms “SASP” and “senescence“ (supplemental Table 2) in RNA-seq–based GEP of lymphoma cells as in panel A; P < .05 and false discovery rate (FDR) <0.25 indicates significance. (E) Fractions of SA–β-gal+ cells within B220+- and GFP-sorted lymphocytes isolated 3 months after transplantation (with no signs of manifest lymphoma) from spleens of Eµ-myc transgenic HSCs stably transduced with GFP-coexpressing CARD11-L225LI, CARD11-L244P, or an empty vector construct as control (left; n = 4 for each mutant; error bars denote the standard deviation). Representative photomicrographs of SA–β-gal stainings (right). Scale bars, 100 µm. (F) Tumor latencies in recipients of Eµ-myctransgenic and Suv39h1-deficient HSCs infected with either the CARD11-L244P mutant, MyD88-L265P mutant, or an empty vector for comparison (n ≥ 4 each; CARD11-L244P;suv39h1−/− vs empty vector;suv39h1−/−P = 0.0018, MyD88-L265P;suv39h1+/+ vs MyD88-L265P;suv39h1−/− [median survival 36 vs. 20 days; compare to Figure 1C] P < 0.001). Asterisks indicate significance (P < .05). NES, normalized enrichment score; n.s., not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2020005244/1/m_bloodbld2020005244r2f2.png?Expires=1769803549&Signature=QlQphtKmqFLRIV9tAx4EvtxB6BjiHSxjax1xuoF64gluDOg8JMPwuXMQLpzOXm8w64tArF7b9H3ccE0v17ZZi~hk9zyX-RZV3mJSozkNhKta3M5YXc5gDJl40mzNN9rc4LytkVqIWA7H~WvB24hfgRZOKz-ZyjVo23u4-bEi6bsuVDwbVzq7IAMM4wglHW-eN-lTgMwdznK2vgypD7aKr7A1jQs3-S7Y9-lr3uVbFCaoIhy-I~AOO2tT3ig5719dCBKZSlB5ZhRwNzL3DAcVw0w~aaf6RQrujutz4wR7o0Eb8AWEoBGppr7Sh~TOKQ34YQCBouP~WuRpzr8dXt~QFA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Prosenescent phenotypes of MyD88 and CARD11 mutations in Myc-driven lymphomagenesis. (A) Percentages (left) of SA–β-gal+ cells in cytospin preparations of n ≥ 3 individual cases per indicated genotype (with error bars denoting the standard deviation) of B220+-purified B-lymphoma cell populations at the time of lymphoma manifestation. Representative photomicrographs of SA–β-gal stainings (right). Scale bars, 100 µm. (B) As in panel A, but lymphoma sections were stained for H3K9me3 and hematoxylin and eosin (H&E). Scale bars, 100 µm. (C) As in panel A, but lymphoma sections stained for Ki67 (H&E stain). Scale bars, 100 µm (identical magnification throughout the figure). (D) GSEA for terms “SASP” and “senescence“ (supplemental Table 2) in RNA-seq–based GEP of lymphoma cells as in panel A; P < .05 and false discovery rate (FDR) <0.25 indicates significance. (E) Fractions of SA–β-gal+ cells within B220+- and GFP-sorted lymphocytes isolated 3 months after transplantation (with no signs of manifest lymphoma) from spleens of Eµ-myc transgenic HSCs stably transduced with GFP-coexpressing CARD11-L225LI, CARD11-L244P, or an empty vector construct as control (left; n = 4 for each mutant; error bars denote the standard deviation). Representative photomicrographs of SA–β-gal stainings (right). Scale bars, 100 µm. (F) Tumor latencies in recipients of Eµ-myctransgenic and Suv39h1-deficient HSCs infected with either the CARD11-L244P mutant, MyD88-L265P mutant, or an empty vector for comparison (n ≥ 4 each; CARD11-L244P;suv39h1−/− vs empty vector;suv39h1−/−P = 0.0018, MyD88-L265P;suv39h1+/+ vs MyD88-L265P;suv39h1−/− [median survival 36 vs. 20 days; compare to Figure 1C] P < 0.001). Asterisks indicate significance (P < .05). NES, normalized enrichment score; n.s., not significant.
Prosenescent phenotypes of MyD88 and CARD11 mutations in Myc-driven lymphomagenesis. (A) Percentages (left) of SA–β-gal+ cells in cytospin preparations of n ≥ 3 individual cases per indicated genotype (with error bars denoting the standard deviation) of B220+-purified B-lymphoma cell populations at the time of lymphoma manifestation. Representative photomicrographs of SA–β-gal stainings (right). Scale bars, 100 µm. (B) As in panel A, but lymphoma sections were stained for H3K9me3 and hematoxylin and eosin (H&E). Scale bars, 100 µm. (C) As in panel A, but lymphoma sections stained for Ki67 (H&E stain). Scale bars, 100 µm (identical magnification throughout the figure). (D) GSEA for terms “SASP” and “senescence“ (supplemental Table 2) in RNA-seq–based GEP of lymphoma cells as in panel A; P < .05 and false discovery rate (FDR) <0.25 indicates significance. (E) Fractions of SA–β-gal+ cells within B220+- and GFP-sorted lymphocytes isolated 3 months after transplantation (with no signs of manifest lymphoma) from spleens of Eµ-myc transgenic HSCs stably transduced with GFP-coexpressing CARD11-L225LI, CARD11-L244P, or an empty vector construct as control (left; n = 4 for each mutant; error bars denote the standard deviation). Representative photomicrographs of SA–β-gal stainings (right). Scale bars, 100 µm. (F) Tumor latencies in recipients of Eµ-myctransgenic and Suv39h1-deficient HSCs infected with either the CARD11-L244P mutant, MyD88-L265P mutant, or an empty vector for comparison (n ≥ 4 each; CARD11-L244P;suv39h1−/− vs empty vector;suv39h1−/−P = 0.0018, MyD88-L265P;suv39h1+/+ vs MyD88-L265P;suv39h1−/− [median survival 36 vs. 20 days; compare to Figure 1C] P < 0.001). Asterisks indicate significance (P < .05). NES, normalized enrichment score; n.s., not significant.
Although positively selected for in homotypic culture in vitro, the CARD11-L244P mutant apparently failed to produce GFP+ Eµ-myc lymphomas in vivo. Therefore, we isolated splenic B cells from healthy recipients without palpable lymphadenopathy 3 months after transplantation with Eµ-myc transgenic HSCs that were stably transduced with 2 different CARD11 mutants (ie, CARD11-L244P and CARD11-L225LI50 ) or an empty GFP-coexpressing vector. Although cells from the empty vector control group or GFP− cells all exhibited basal SA–β-gal frequencies of no more than 15%, both CARD11 mutants stained close to 100% SA–β-gal+ (Figure 2E). We previously reported a selective senescence defect in cells lacking the H3K9 methyltransferase Suv39h1.28,46,47,51 When now expressing the CARD11-L244P mutant in Eµ-myc transgenic Suv39h1-deficient HSCs and transplanting them into recipient mice, CARD11-mutant (ie, GFP+) lymphomas formed in all cases tested, presented with a much smaller fraction of senescent cells (supplemental Figure 2D, compared with Figure 2E), and came up even faster than the already accelerated empty vector control lymphomas in this Suv39h1-deprived context, thereby uncovering how effectively CARD11-L244P–evoked senescence masked the strong protumorigenic potential of this mutant (Figure 2F). Importantly, we observed a similarly accelerating effect regarding the MyD88-L265P mutant in a Suv39h1-deficient background (Figure 2F). We also verified this prosenescent phenotype of MyD88-L265P on the transcriptome level in another murine MyD88-L265P–mutant, conditional Bcl2-codriven, and CD19-Cre–activated (“MBC”) lymphoma model (supplemental Figure 2E).
In essence, MyD88-L265P and CARD11-L244P not only promote cell survival by suppressing apoptosis, but also evoke a profound senescence response that counters accelerated tumor onset, and, hence, may render these genotypes particularly susceptible to cooperating senescence-inactivating lesions, for example, at the INK4a/ARF gene locus. Strikingly, this has been found in human DLBCL, where MyD88 mutations strongly correlate with CDKN2A/B loss, and where the majority of CARD11-mutant lymphomas exhibit either inactivating alterations of the CDKN2A/B or TP53 gene locus.52
MyD88-L265P and CARD11-L244P evoke macrophage chemoattraction
Using the Eµ-myc model system with subsequent confirmation in DLBCL, we previously reported a non–cell-autonomous prosenescence mechanism mediated by transforming growth factor-β1 (TGF-β1) secretion emanating from infiltrating macrophages that became activated through engulfment of apoptotic lymphoma remainders.46 To interrogate this principle in an NDM-specific manner, we scanned manifest lymphomas that arose from NDM-infected Eµ-myc transgenic HSCs (Suv39h1-deficient in case of CARD11-L244P), and found macrophage infiltration markedly enhanced selectively in MyD88-L265P– and CARD11-L244P–mutant lymphomas (Figure 3A; supplemental Figure 2F). Among candidate chemokines capable of attracting macrophages are the colony-stimulating factor-1 (CSF-1; also known as macrophage colony-stimulating factor [M-CSF]) and the CC chemokine ligand 2 (CCL2, also known as monocyte chemoattractant protein 1 [MCP1]); hence, we quantified their respective transcript levels in isolated B-lymphoma cells, and specific protein levels in their culture supernatant, with only MyD88-mutant cells exhibiting significantly elevated expression of CCL2 and CARD11-mutant cells of CSF-1 (Figure 3B-C). In vivo administration of macrophage-depleting clodronate led to a profound reduction in MyD88-mutant lymphoma cell senescence (Figure 3D), consistent with much higher, macrophage density-correlated TGF-β1 protein in situ levels in MyD88-L265P– and CARD11-L244P–mutant lymphomas (Figure 3E). Accordingly, TGF-β1 secretion turned out to be the quantitatively leading mechanism of non–cell-autonomous lymphoma cell senescence induction because stable coexpression of a non–membrane-bound, and, thus, dominant-negative TGF receptor mutant (dn-TGFRII-ED) resulted in a strong reduction of the senescent lymphoma cell fraction (Figure 3F). Conversely, in vitro detectable lymphoma cell senescence remained unaffected by cell-autonomous expression of mutant MyD88 or CARD11 moieties (supplemental Figure 2G). Hence, macrophage-derived TGF-β1 operates in a prosenescent fashion downstream of lymphoma-borne chemoattracting factors in a distinct subset of NDM.
![Macrophage-dependent senescence induction in MyD88-L265P– and CARD11-L244P–mutant lymphomas. (A) Fraction of F4/80+/CD11b+/GFP−/B220− macrophages within the overall population of viable cells in freshly isolated single-cell suspensions from n ≥ 3 individual enlarged lymph node samples per indicated genotype (with error bars denoting the standard deviation) analyzed by flow cytometry. Notably, macrophages were GFP− in all cases, that is, did not express the NF-κB–deregulating mutant. (B) Transcript-level expression of the macrophage chemoattractants Ccl2 (green) and Csf-1 (blue) in B220-sorted mutant-driven lymphoma cells relative to the empty vector cohort assessed by RNA-seq in the indicated numbers of samples and with error bars denoting the standard error in log2 scale. Gray bars indicate nonsignificant results with a P ≥ .05. (C) ELISA to detect secreted CCL2 and CSF-1 in the supernatant of manifest Eµ-myc lymphomas (n = 5) stably transduced with either the GFP-encoding vector or the indicated mutants. After 10 days in culture, lymphoma cells were GFP-sorted and supernatants from 2 × 105 GFP+ cells were collected (24 hours incubation time) and analyzed. Notably, mutant-expressing lymphoma cells do not show any signs of senescence induction in vitro (supplemental Figure 2F) (D) Fraction of viable macrophages (left; as in panel A) in MyD88-L265P–driven lymphomas with and without (n = 4 each) Clodronate administration in vivo. Corresponding fractions of senescent lymphoma cells by SA–β-gal staining of cytospin preparations from GFP+ lymphoma cell isolates (right). Scale bars, 100 µm. (E) TGF-β1 expression by immunohistochemistry (ematoxylin and eosin [H&E] stain) in tissue sections of NDM-driven Eµ-myc lymphomas. Representative photomicrographs of n = 3 independent analyses with percentage of positively stained area quantified for each genotype. Scale bars, 100 µm. (F) Detection of the fraction of senescent cells in MyD88-L265P-mutant lymphomas generated with or without stable coexpression of the dominant-negative TGF receptor (dnTGFR) by SA–β-gal staining of cytospin preparations, and Ki67 immunostaining of lymphoma sections in situ (H&E stain). Representative photomicrographs of 3 independent analyses; percentages indicate means ± standard deviation. Scale bars, 100 µm. Asterisks indicate significance (P < .05).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2020005244/1/m_bloodbld2020005244r2f3.png?Expires=1769803549&Signature=qgESIPH-KQxNWoOGwxVHNoSqlRWBmqaf7B1liJjCYxn8M7qB2df8QYQD6cxvkWZMlwIjMO2UiO07qQ66zP-Rex931bqb2KUmdjdpy6Kaap27UMSSSA4nkziZ4ZkzLUZ7gy7k4yPskQH4-R3wpFbHDzWSZNpPI7QkU2Y6ly~9Hnsnvu-evuTnxXJaBw1j5Q09uyZelZM~BvrpBHuAqlXvny~epG2f3jXLWcQWRPLiYZOt~4MiJUHS~MVm63Ap-b2Evbn~lSqTN42ABvO-YspxHAj~n4h9KsqDsbUainUyJpvQ662UAQffKDyNzPrI-DyTiTktycTihhUEgeUnrTsaaQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Macrophage-dependent senescence induction in MyD88-L265P– and CARD11-L244P–mutant lymphomas. (A) Fraction of F4/80+/CD11b+/GFP−/B220− macrophages within the overall population of viable cells in freshly isolated single-cell suspensions from n ≥ 3 individual enlarged lymph node samples per indicated genotype (with error bars denoting the standard deviation) analyzed by flow cytometry. Notably, macrophages were GFP− in all cases, that is, did not express the NF-κB–deregulating mutant. (B) Transcript-level expression of the macrophage chemoattractants Ccl2 (green) and Csf-1 (blue) in B220-sorted mutant-driven lymphoma cells relative to the empty vector cohort assessed by RNA-seq in the indicated numbers of samples and with error bars denoting the standard error in log2 scale. Gray bars indicate nonsignificant results with a P ≥ .05. (C) ELISA to detect secreted CCL2 and CSF-1 in the supernatant of manifest Eµ-myc lymphomas (n = 5) stably transduced with either the GFP-encoding vector or the indicated mutants. After 10 days in culture, lymphoma cells were GFP-sorted and supernatants from 2 × 105 GFP+ cells were collected (24 hours incubation time) and analyzed. Notably, mutant-expressing lymphoma cells do not show any signs of senescence induction in vitro (supplemental Figure 2F) (D) Fraction of viable macrophages (left; as in panel A) in MyD88-L265P–driven lymphomas with and without (n = 4 each) Clodronate administration in vivo. Corresponding fractions of senescent lymphoma cells by SA–β-gal staining of cytospin preparations from GFP+ lymphoma cell isolates (right). Scale bars, 100 µm. (E) TGF-β1 expression by immunohistochemistry (ematoxylin and eosin [H&E] stain) in tissue sections of NDM-driven Eµ-myc lymphomas. Representative photomicrographs of n = 3 independent analyses with percentage of positively stained area quantified for each genotype. Scale bars, 100 µm. (F) Detection of the fraction of senescent cells in MyD88-L265P-mutant lymphomas generated with or without stable coexpression of the dominant-negative TGF receptor (dnTGFR) by SA–β-gal staining of cytospin preparations, and Ki67 immunostaining of lymphoma sections in situ (H&E stain). Representative photomicrographs of 3 independent analyses; percentages indicate means ± standard deviation. Scale bars, 100 µm. Asterisks indicate significance (P < .05).
Macrophage-dependent senescence induction in MyD88-L265P– and CARD11-L244P–mutant lymphomas. (A) Fraction of F4/80+/CD11b+/GFP−/B220− macrophages within the overall population of viable cells in freshly isolated single-cell suspensions from n ≥ 3 individual enlarged lymph node samples per indicated genotype (with error bars denoting the standard deviation) analyzed by flow cytometry. Notably, macrophages were GFP− in all cases, that is, did not express the NF-κB–deregulating mutant. (B) Transcript-level expression of the macrophage chemoattractants Ccl2 (green) and Csf-1 (blue) in B220-sorted mutant-driven lymphoma cells relative to the empty vector cohort assessed by RNA-seq in the indicated numbers of samples and with error bars denoting the standard error in log2 scale. Gray bars indicate nonsignificant results with a P ≥ .05. (C) ELISA to detect secreted CCL2 and CSF-1 in the supernatant of manifest Eµ-myc lymphomas (n = 5) stably transduced with either the GFP-encoding vector or the indicated mutants. After 10 days in culture, lymphoma cells were GFP-sorted and supernatants from 2 × 105 GFP+ cells were collected (24 hours incubation time) and analyzed. Notably, mutant-expressing lymphoma cells do not show any signs of senescence induction in vitro (supplemental Figure 2F) (D) Fraction of viable macrophages (left; as in panel A) in MyD88-L265P–driven lymphomas with and without (n = 4 each) Clodronate administration in vivo. Corresponding fractions of senescent lymphoma cells by SA–β-gal staining of cytospin preparations from GFP+ lymphoma cell isolates (right). Scale bars, 100 µm. (E) TGF-β1 expression by immunohistochemistry (ematoxylin and eosin [H&E] stain) in tissue sections of NDM-driven Eµ-myc lymphomas. Representative photomicrographs of n = 3 independent analyses with percentage of positively stained area quantified for each genotype. Scale bars, 100 µm. (F) Detection of the fraction of senescent cells in MyD88-L265P-mutant lymphomas generated with or without stable coexpression of the dominant-negative TGF receptor (dnTGFR) by SA–β-gal staining of cytospin preparations, and Ki67 immunostaining of lymphoma sections in situ (H&E stain). Representative photomicrographs of 3 independent analyses; percentages indicate means ± standard deviation. Scale bars, 100 µm. Asterisks indicate significance (P < .05).
MyD88-L265P and CARD11-L244P mutants drive PD-L1/2–mediated immune checkpoint control
Because immune-mediated mechanisms may shorten tumor latencies in senescence-prone MyD88-mutant lymphomas, we first interrogated lymphoma RNA-seq data regarding major histocompatibility complex (MHC)-mediated pathways by GSEA (using the gene ontology [GO] terms “Antigen processing and presentation of peptide antigen via MHC class I” [GO:0002474] and “Antigen processing and presentation of exogenous peptide antigen via MHC class II” [GO:0019886]). No significant alterations were revealed, and no mutations of murine MHCII homologs (H2-K1, H2-D1, H2-Q2/6/7/10) were found in MyD88-L265P–driven lymphomas (data not shown). Next, we probed transcript levels of the immune-checkpoint ligands PD-L1 and PD-L2. Strikingly, MyD88-L265P– or CARD11-L244P–mutant B cells selectively presented with significantly upregulated PD-L1/2 transcript expression (Figure 4A), as well as surface PD-L1 and PD-L2 protein expression (Figure 4B; supplemental Figure 3A). Given the inducibility of PD ligands by NF-κB,53 we investigated canonical and noncanonical NF-κB DNA-binding activities, and found the noncanonical p52 component exclusively elevated in the 2 prosenescent mutant scenarios, which also selectively exhibited a significant enrichment for transcripts belonging to the GO term “NF-κB activation” (supplemental Figure 3B-C). Notably, the tumor-promoting phenotype of MyD88-L265P was completely ablated in an NF-κB–blunted context, that is, if the MyD88-L265P mutant was stably coexpressed with the NF-κB superrepressor moiety IκBαΔN on a bicistronic construct in Eµ-myc transgenic HSCs prior to their propagation in myeloablated recipients (supplemental Table 1).
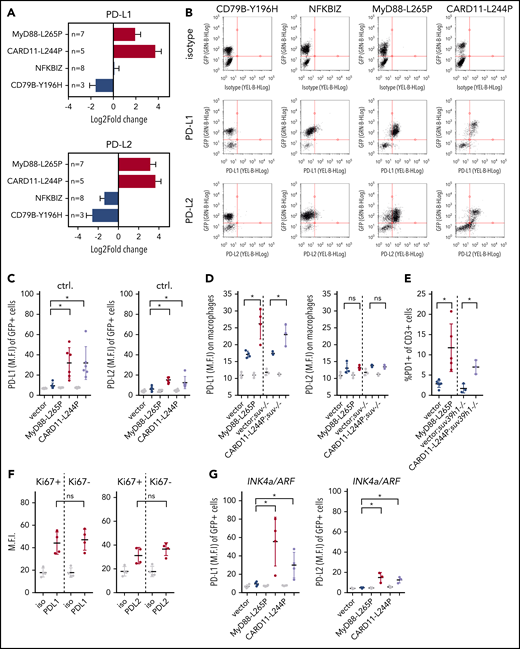
Immune-checkpoint inhibitors PD-L1/2 are upregulated on both MyD88- and CARD11-mutant Eµ-myc lymphomas. (A) PD-L1 (top) and PD-L2 (bottom) RNA-seq transcript levels (relative to vector cohort) in B220-isolated NDM-carrying lymphoma cells. Sample numbers as indicated; error bars denote the standard error, and red color bar indicates significance (P < .05) as compared with the respective vector control cohorts. (B) PD-L1/2 surface expression on B220/GFP double-positive lymphoma cells by flow cytometry. Representative cases of n ≥ 3 independent experiments are shown. (C) Flow cytometric surface PD-L1/2 expression on individual GFP+ Eµ-myc lymphomas (n = 6) that were stably transduced in vitro with the MyD88-L265P or CARD11-L244P moiety vs an empty vector as a control (with error bars denoting the standard deviation). Gray dots represent mean fluorescence intensity (M.F.I.) of isotype controls. (D) PD-L1/2 expression measured as in panel B on F4/80+;B220−;GFP− tumor-infiltrating macrophages isolated from bulk MyD88-L265P– and CARD11-L244P–mutant Eµ-myc lymphomas in n ≥ 3 individual cases per genotype (with error bars denoting the standard deviation). Gray dots represent M.F.I. of isotype controls. (E) Fraction of PD1+ T cells within the CD3+ cell population of bulk lymphomas as in panel D. (F) Costaining for the proliferation marker Ki67 and surface PD-L1 or PD-L2 in viable GFP+;B220+ MyD88-mutant lymphomas as in panel D. Gray dots represent M.F.I. of isotype controls. (G) Flow cytometric surface PD-L1/2 expression on individual GFP+ senescence-incapable INK4a/ARF-deficient (n ≥ 3) Eµ-myc lymphomas that were stably transduced in vitro with the MyD88-L265P or CARD11-L244P moiety vs an empty vector as a control (with error bars denoting the standard deviation). Gray dots represent M.F.I. of isotype controls. Asterisks indicate significance (P < .05); n.s., not significant.
Immune-checkpoint inhibitors PD-L1/2 are upregulated on both MyD88- and CARD11-mutant Eµ-myc lymphomas. (A) PD-L1 (top) and PD-L2 (bottom) RNA-seq transcript levels (relative to vector cohort) in B220-isolated NDM-carrying lymphoma cells. Sample numbers as indicated; error bars denote the standard error, and red color bar indicates significance (P < .05) as compared with the respective vector control cohorts. (B) PD-L1/2 surface expression on B220/GFP double-positive lymphoma cells by flow cytometry. Representative cases of n ≥ 3 independent experiments are shown. (C) Flow cytometric surface PD-L1/2 expression on individual GFP+ Eµ-myc lymphomas (n = 6) that were stably transduced in vitro with the MyD88-L265P or CARD11-L244P moiety vs an empty vector as a control (with error bars denoting the standard deviation). Gray dots represent mean fluorescence intensity (M.F.I.) of isotype controls. (D) PD-L1/2 expression measured as in panel B on F4/80+;B220−;GFP− tumor-infiltrating macrophages isolated from bulk MyD88-L265P– and CARD11-L244P–mutant Eµ-myc lymphomas in n ≥ 3 individual cases per genotype (with error bars denoting the standard deviation). Gray dots represent M.F.I. of isotype controls. (E) Fraction of PD1+ T cells within the CD3+ cell population of bulk lymphomas as in panel D. (F) Costaining for the proliferation marker Ki67 and surface PD-L1 or PD-L2 in viable GFP+;B220+ MyD88-mutant lymphomas as in panel D. Gray dots represent M.F.I. of isotype controls. (G) Flow cytometric surface PD-L1/2 expression on individual GFP+ senescence-incapable INK4a/ARF-deficient (n ≥ 3) Eµ-myc lymphomas that were stably transduced in vitro with the MyD88-L265P or CARD11-L244P moiety vs an empty vector as a control (with error bars denoting the standard deviation). Gray dots represent M.F.I. of isotype controls. Asterisks indicate significance (P < .05); n.s., not significant.
Importantly, we observed elevated PD-L1/2 expression levels in Eµ-myc transgenic lymphomas after infection with MyD88-L265P or CARD11-L244P in vitro, indicating a direct mutant-driven mechanism not primarily requiring the presence of potentially threatening cytotoxic T cells (Figure 4C). Interestingly, the extensive chemoattraction of macrophages by these 2 mutant moieties amplified the putative immune-checkpoint mechanism because these lymphoma-infiltrating macrophages were PD-L1+ as well (Figure 4D). We also detected increased numbers of CD3+ T cells in bulk isolates from MyD88-L265P– or CARD11-L244P–driven lymphomas exhibiting elevated PD1 expression, underscoring the functional relevance of PD-L1/2 expression to keep T-cell activity in check (Figure 4E; supplemental Figure 4A). Notably, the positive PD-L1/2 lymphoma expression status was independent of the senescent condition of the cells: PD-L1/2 surface expression was comparable on Ki67− (senescent) and Ki67+ (proliferating) MyD88-mutant B-cell lymphomas (Figure 4F), and equally detectable on senescence-incapable INK4a/ARF-deficient Eµ-myc lymphomas24 expressing MyD88-L265P or CARD11-L244P moieties in vitro (Figure 4G; supplemental Figure 4B-C).
An adaptive cellular immune response keeps MyD88- or CARD11-mutant lymphomas in check and can be therapeutically exploited
To unleash the tumor-suppressive potential cell-mediated immunosurveillance may exert during lymphomagenesis, we ablated PD-L1 and PD-L2 expression by a small hairpin cotargeting both ligands (sh-PDL) in MyD88-L265P–infected Eµ-myc transgenic HSCs (supplemental Figure 5A). Intriguingly, tumor onset of MyD88-L265P–driven lymphomas was significantly delayed by sh-PDL (Figure 5A). Remarkably, lymphoma-infiltrating macrophages, not expressing sh-PDL, exhibited strongly reduced PD-L1 surface expression, suggesting a lymphoma-instructed effect (supplemental Figure 5B). Conversely, Eµ-myc lymphomas engineered to solely overexpress PD-L1 displayed an accelerated onset but no signs of senescence (supplemental Figure 5C-D). Interestingly, the PD ligand–ablated malignancies presented with increased proliferation markers and a reduced senescence-related transcriptional profile as well as reduced T-cell infiltration (Figure 5B-D). Hypothesizing a preferential ablation of senescent lymphoma cells by unleashed T-cell surveillance, we tested whether MyD88-L265P lymphoma-exposed cytotoxic T cells would exhibit differential cytotoxic activities in vitro when cocultured with either senescent or nonsenescent lymphoma cells that were flow-sorted according to a C12-RG fluorescence-based SA–β-gal assay. Specifically, anti-PD1 checkpoint-derepressed splenic T cells were exposed to C12-RG+ (ie, senescent) vs C12-RG− (ie, nonsenescent) fractions of the very same MyD88-L265P lymphoma, which developed in the individual animal from which the T cells were derived. Strikingly, these lymphoma-educated and PD1-blocked T cells completely eliminated the senescent lymphoma population (Figure 5E). Interestingly, the reactivated T cells also attacked nonsenescent lymphoma cells, albeit less efficiently (Figure 5E). These data demonstrate that the senescence-independent induction of PD ligands in MyD88-L265P–expressing lymphoma cells keeps an adaptive immune response in check, which, if unlocked, is predominantly but not exclusively directed against lymphoma cells in senescence, a state switch evoked by another, PD ligand–independent functionality of the MyD88-L265P mutation.
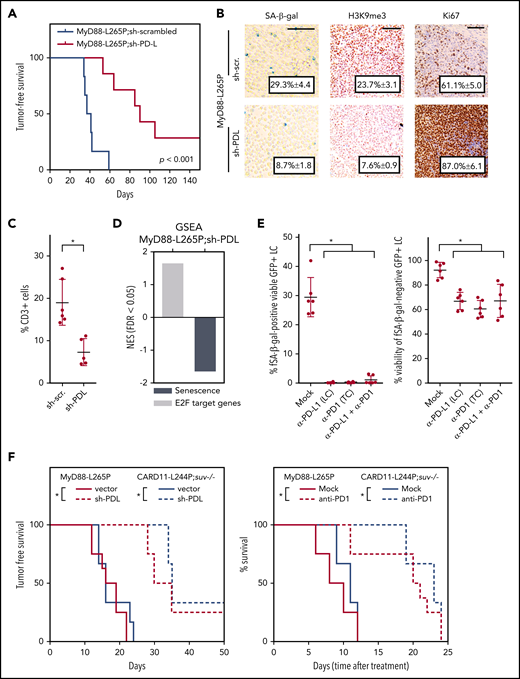
MyD88-L265P- or CARD11-L244P-driven immune evasion phenotypes prevent senescence-preferential immune clearance of lymphoma cells. (A) Lymphoma onset in recipient mice of stably MyD88-L265P;sh-scrambled (n = 6) or MyD88-L265P;sh-PDL (targeting both PD-L1 and PD-L2; n = 7) cotransduced Eµ-myc transgenic HSCs (P = .001). Of note, all lymphomas generated were GFP+ (data not shown). For knockdown efficiency, see supplemental Figure 4A. (B) In situ expression (with representative cases shown) of the indicated senescence-related markers of lymphomas as in panel A. SA–β-gal staining of cytospin preparations and lymphoma sections stained with hematoxylin and eosin and antibodies against H3K9me3 or Ki67. Scale bars, 100 µm. (C) Fraction of CD3+ cells within the overall population of viable cells in freshly isolated single-cell suspensions from n ≥ 3 individual enlarged lymph node samples per indicated genotype (with error bars denoting the standard deviation) analyzed by flow cytometry. (D) GSEA in RNA-seq–based GEP of GFP-sorted B-cell lymphoma cells as in panel A for the indicated gene signatures. (E) Cytotoxic potential of anti-PD1–derepressed splenic cytotoxic CD8a+ T cells coincubated with C12-RG+ vs C12-RG− fractions of the same, but ex vivo anti–PD-L1 antibody-treated MyD88-L265P B-cell lymphoma cells that developed in the same mouse the T cells were isolated from. Left, percentage of senescent viable GFP+ lymphoma cells after T-cell coculture and indicated antibody treatments. Right, viability of nonsenescent (proliferating) GFP+ lymphoma cells after T-cell coculture and indicated antibody treatments, n = 3 different tumor mice (with error bars denoting the standard deviation). (F) Tumor-free/OS, plotted in Kaplan-Meier format, of mice transplanted with manifest MyD88-L265P– or CARD11–L244P-driven lymphomas either engineered to stably coexpress sh-PDL (vs empty vector) at the time of transplantation (left), or exposed to an anti-PD1 antibody (vs mock) at the time of well-palpable lymphoma manifestation (right); n ≥ 3 samples per genotype and treatment arm. Asterisks indicate significance (P < .05).
MyD88-L265P- or CARD11-L244P-driven immune evasion phenotypes prevent senescence-preferential immune clearance of lymphoma cells. (A) Lymphoma onset in recipient mice of stably MyD88-L265P;sh-scrambled (n = 6) or MyD88-L265P;sh-PDL (targeting both PD-L1 and PD-L2; n = 7) cotransduced Eµ-myc transgenic HSCs (P = .001). Of note, all lymphomas generated were GFP+ (data not shown). For knockdown efficiency, see supplemental Figure 4A. (B) In situ expression (with representative cases shown) of the indicated senescence-related markers of lymphomas as in panel A. SA–β-gal staining of cytospin preparations and lymphoma sections stained with hematoxylin and eosin and antibodies against H3K9me3 or Ki67. Scale bars, 100 µm. (C) Fraction of CD3+ cells within the overall population of viable cells in freshly isolated single-cell suspensions from n ≥ 3 individual enlarged lymph node samples per indicated genotype (with error bars denoting the standard deviation) analyzed by flow cytometry. (D) GSEA in RNA-seq–based GEP of GFP-sorted B-cell lymphoma cells as in panel A for the indicated gene signatures. (E) Cytotoxic potential of anti-PD1–derepressed splenic cytotoxic CD8a+ T cells coincubated with C12-RG+ vs C12-RG− fractions of the same, but ex vivo anti–PD-L1 antibody-treated MyD88-L265P B-cell lymphoma cells that developed in the same mouse the T cells were isolated from. Left, percentage of senescent viable GFP+ lymphoma cells after T-cell coculture and indicated antibody treatments. Right, viability of nonsenescent (proliferating) GFP+ lymphoma cells after T-cell coculture and indicated antibody treatments, n = 3 different tumor mice (with error bars denoting the standard deviation). (F) Tumor-free/OS, plotted in Kaplan-Meier format, of mice transplanted with manifest MyD88-L265P– or CARD11–L244P-driven lymphomas either engineered to stably coexpress sh-PDL (vs empty vector) at the time of transplantation (left), or exposed to an anti-PD1 antibody (vs mock) at the time of well-palpable lymphoma manifestation (right); n ≥ 3 samples per genotype and treatment arm. Asterisks indicate significance (P < .05).
Anti-PD1 antibody-based immune-checkpoint blockade reportedly produced rather modest signals of efficacy in unselected relapsed/refractory B-NHL patient populations,54,55 indicating the need of defining susceptible subsets better. Thus, we tested whether genetic or pharmacological PD-L1/2:PD1 interference might produce beneficial long-term effects in recipients of MyD88- or CARD11-mutant lymphomas. Indeed, mice transplanted with manifest MyD88-L265P– or CARD11-L244P–driven lymphomas either engineered to stably coexpress sh-PDL (vs empty vector) or exposed to an anti-PD1 antibody (vs mock) lived significantly longer (Figure 5F). Notably, recent elegant work in an ABC-DLBCL–reminiscent mouse lymphoma model driven by IKK2ca-enforced canonical NF-κB activation and impaired differentiation due to loss of Blimp-1 unveiled anti-PD1–sensitive PD-L1 upregulation in these lymphomas,56 as therapeutic anti-PD1 susceptibility was reported in the MBC model mentioned above in “MyD88-L265P and CARD11-L244P induce a profound senescence response,”57 thus, presumably describing overlapping immune biologies in all 3 model systems investigated, but with specific emphasis on a T-cell–recognized senescence-related immunogenic switch here.
Mouse-informed functional MyD88- or CARD11-mutant states in human DLBCL
Recent analyses conducted in large DLBCL patient cohorts at diagnosis sought to integrate genomics and additional molecular information including GEP data,14-17 allowing us to revisit these publicly available data sets with respect to MyD88 and CARD11 mutations as well as transcriptome-encoded signatures that might indicate senescence and immune-evasion states. Specifically, when scanning 726 DLBCL transcriptomes, we found PD-L1, albeit not PD-L2, transcript levels significantly increased in the MyD88-L265P-mutant subset compared with DLBCL samples without the MyD88-L265P mutation14 (Figure 6A; supplemental Figure 6A). The prosenescent phenotype we observed in Eµ-myc–driven MyD88-mutant lymphomas in vivo suggested that manifest human MyD88-L265P DLBCL might have experienced strong selective pressure for senescence-disabling lesions. Indeed, as reported in the "MyD88-L265P and CARD11-L244P induce a profound senescence response" section from a different set of DLBCL samples,52 we noted a 5 times higher CDKN2A copy-number loss in MyD88-L265P-mutated DLBCL14 (Figure 6B). To probe, on the contrary, whether some MyD88-L265P DLBCL may present at diagnosis, like MyD88-mutant Eµ-myc lymphomas, with remaining signs of cellular senescence, we used a therapy-induced senescence (TIS)-derived gene signature as a classifier termed “Suvarness” in reference to TIS-capable Suv39h1-proficient, as compared with TIS-incapable Suv39h1-deficient, lymphomas,51,58 consisting of 22 largely proliferation-independent senescence-associated genes.47 When tested in a small panel of formalin-fixed paraffin-embedded DLBCL biopsies (n = 9) harboring the MyD88-L265P point mutation, we found a heterogenous expression pattern for p16INK4a as well as 2 of the strongest “Suvarness” markers GAS1 and FKBP9 in situ (supplemental Figure 6B; 2 of 9 cases exhibiting strong staining reactivity). Notably, this was verified in many more cases on the transcript level, when we applied the senescence-associated Suvarness signature to GEP of 66 structurally CDKN2A-unaffected MyD88-L265P DLBCL cases,14 and found a cluster of 15 cases (considered “Suvarnesshigh”) that consistently exhibited a transcriptional senescence profile (Figure 6C). Indeed, subsequent GSEA comparing MyD88-L265P DLBCL based on their Suvarness status showed significant enrichment of senescence, SASP genes, an adult tissue stem cell signature related to senescence-associated stemness (SAS),58 and suppression of E2F target genes in Suvarnesshigh samples (Figure 6D). Notably, SAS-reprogrammed MyD88-L265P DLBCL cells that eventually overcame senescence via loss of CDKN2A, thereby enabling the acquired latent stemness capacity, might represent the dominant population that drives the aggressive clinical behavior of the C5 or a subgroup based on co-occurrence of MyD88-L265P and CD79B mutations (MCD) in DLBCL.15,16 Of note, R-CHOP–exposed DLBCL patients14 diagnosed with MyD88-L265P;Suvarnesshigh lymphomas (n = 15) appeared to have a superior overall survival (OS) when compared with the remaining, Suvarnesslow MyD88-L265P (n = 68) patient population (2-year OS, 83.3% vs 67.4%; P value did not reach significance).
![MyD88- and CARD11-mutant–specific phenotypes recapitulated in DLBCL patients. (A) PD-L1 (CD274) expression in DLBCL with (MyD88-L265P [L265P]; n = 66) or without (wild-type [WT]; n = 660) a MyD88-L265P mutation (CDKN2A copy number loss cases are excluded, and shown are log2-transformed and size factor–normalized counts). (B) Frequency of CDKN2A copy number loss in DLBCL with or without the MyD88-L265P mutation (L265P, n = 83; WT, n = 690). (C) MyD88-L265P;CDKN2A-WT DLBCL (n = 66)14 were grouped based on mean expression of genes compiled from the murine Suvarness signature into 2 cohorts (Suvarnesshigh cohort is labeled with an asterisk, n = 15). Senescence-impaired MyD88-L265P;CDKN2A-loss DLBCL are shown for comparison (n = 17). (D) GSEA comparing Suvarnesshigh vs Suvarnesslow MyD88-L265P;CDKN2A-WT groups from panel C, using senescence- and immune evasion–associated gene signatures (“SASP,” “E2F target genes,” “Senescence,” “Stemness,” “Negative regulation of immune response,” “Negative regulation of T-cell activation,” and “Positive regulation of cellular response to TGF-β”) and the Eµ-myc–derived MyDness signature (see panel E). Notably, the murine MyDness signature is significantly enriched in the human MyD88-L265P;CDKN2A-WT DLBCL cohort that exhibits a senescence-associated GEP (P < .001, FDR = 0.001). Both Suvarness and MyDness signatures comprise a nonoverlapping set of genes. P ≤ .05 and FDR <0.05 indicate significance. (E) Venn diagram showing overlap of the top 200 genes with highest fold-changes (upregulation; compared with the respective vector control lymphomas) as identified by RNA-seq for all NDM tested in the Eµ-myc mouse model. A mutant-specific unique set of genes was identified for MyD88-L265P- as well as CARD11-L244P-mutant lymphomas, accordingly named MyDness (123 genes) and CARDness (182 unique genes), respectively. (F) Transcriptome-analyzed DLBCL14 were stratified into 2 groups based on mean expression of genes compiled from the murine MyDness (left) and CARDness signatures (right), respectively, as shown in panel E. Top, GSEA comparing both cohorts (30% of highest vs 30% of lowest signature expressers; n = 231 cases for each group) was performed using gene sets defining senescence-related phenotypes (senescence, SASP, E2F target genes, stemness), T-cell evasion, as well as positive NF-κB and TGF-β responses. P ≤ .05 indicates significance. Bottom, expression levels (log2-transformed and sf-normalized counts) of PD-L1 (CD274) and PD-L2 (PDCD1LG2), the macrophage chemoattractant Ccl2 and Csf1 as well as the macrophage markers CD68 and Itgam in both cohorts. (G) MyD88-L265P derived MyDness signature identifies a DLBCL subgroup as analyzed in 3 large DLBCL patient cohorts14-16 that display increased expression of immune-checkpoint inhibitors (blue; CD274, PDCD1LG2), macrophage markers and chemoattractants (green; ITGAM, CD68, CSF-1, CCL2) as well as senescence-associated gene-expression profiles (orange) (see panel F, supplemental Figure 6D, and supplemental Figure 6F-G). Indicated n numbers refer to transcriptomes analyzed. Asterisks indicate significance (P < .05)](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/20/10.1182_blood.2020005244/1/m_bloodbld2020005244r2f6.png?Expires=1769803549&Signature=vXS9JW6h7bJXohWSzhY-efKVmDZOtFTJVVPn0AIobtmSCBjVlQBrCuaRq~PKScft2F0UqXqOOjLnH1UhlzP2tAzK~~h12r3LByzrQcOBPL1V8xF2ise8WKr~RtlWAaGKx9s8SMuG2GIRTdHpbj5BonhVXPyOg8MneFjVQZ5-74GprxOzzlFLJJwNMPuxzi6kWUpsZ5Jv~N1DYNRwUzjADBNgS-03czaYyecZ47H6OWknGBsFaTDccEUbcD~2TIuLg1131pWk7T4MlsllzTfkP4bCt6nMxAjeWZRclMnyiMz5lOCVEUKOuSBJWRDW897DUaUJ6WtkPmx2VJiYbmLgcw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MyD88- and CARD11-mutant–specific phenotypes recapitulated in DLBCL patients. (A) PD-L1 (CD274) expression in DLBCL with (MyD88-L265P [L265P]; n = 66) or without (wild-type [WT]; n = 660) a MyD88-L265P mutation (CDKN2A copy number loss cases are excluded, and shown are log2-transformed and size factor–normalized counts). (B) Frequency of CDKN2A copy number loss in DLBCL with or without the MyD88-L265P mutation (L265P, n = 83; WT, n = 690). (C) MyD88-L265P;CDKN2A-WT DLBCL (n = 66)14 were grouped based on mean expression of genes compiled from the murine Suvarness signature into 2 cohorts (Suvarnesshigh cohort is labeled with an asterisk, n = 15). Senescence-impaired MyD88-L265P;CDKN2A-loss DLBCL are shown for comparison (n = 17). (D) GSEA comparing Suvarnesshigh vs Suvarnesslow MyD88-L265P;CDKN2A-WT groups from panel C, using senescence- and immune evasion–associated gene signatures (“SASP,” “E2F target genes,” “Senescence,” “Stemness,” “Negative regulation of immune response,” “Negative regulation of T-cell activation,” and “Positive regulation of cellular response to TGF-β”) and the Eµ-myc–derived MyDness signature (see panel E). Notably, the murine MyDness signature is significantly enriched in the human MyD88-L265P;CDKN2A-WT DLBCL cohort that exhibits a senescence-associated GEP (P < .001, FDR = 0.001). Both Suvarness and MyDness signatures comprise a nonoverlapping set of genes. P ≤ .05 and FDR <0.05 indicate significance. (E) Venn diagram showing overlap of the top 200 genes with highest fold-changes (upregulation; compared with the respective vector control lymphomas) as identified by RNA-seq for all NDM tested in the Eµ-myc mouse model. A mutant-specific unique set of genes was identified for MyD88-L265P- as well as CARD11-L244P-mutant lymphomas, accordingly named MyDness (123 genes) and CARDness (182 unique genes), respectively. (F) Transcriptome-analyzed DLBCL14 were stratified into 2 groups based on mean expression of genes compiled from the murine MyDness (left) and CARDness signatures (right), respectively, as shown in panel E. Top, GSEA comparing both cohorts (30% of highest vs 30% of lowest signature expressers; n = 231 cases for each group) was performed using gene sets defining senescence-related phenotypes (senescence, SASP, E2F target genes, stemness), T-cell evasion, as well as positive NF-κB and TGF-β responses. P ≤ .05 indicates significance. Bottom, expression levels (log2-transformed and sf-normalized counts) of PD-L1 (CD274) and PD-L2 (PDCD1LG2), the macrophage chemoattractant Ccl2 and Csf1 as well as the macrophage markers CD68 and Itgam in both cohorts. (G) MyD88-L265P derived MyDness signature identifies a DLBCL subgroup as analyzed in 3 large DLBCL patient cohorts14-16 that display increased expression of immune-checkpoint inhibitors (blue; CD274, PDCD1LG2), macrophage markers and chemoattractants (green; ITGAM, CD68, CSF-1, CCL2) as well as senescence-associated gene-expression profiles (orange) (see panel F, supplemental Figure 6D, and supplemental Figure 6F-G). Indicated n numbers refer to transcriptomes analyzed. Asterisks indicate significance (P < .05)
MyD88- and CARD11-mutant–specific phenotypes recapitulated in DLBCL patients. (A) PD-L1 (CD274) expression in DLBCL with (MyD88-L265P [L265P]; n = 66) or without (wild-type [WT]; n = 660) a MyD88-L265P mutation (CDKN2A copy number loss cases are excluded, and shown are log2-transformed and size factor–normalized counts). (B) Frequency of CDKN2A copy number loss in DLBCL with or without the MyD88-L265P mutation (L265P, n = 83; WT, n = 690). (C) MyD88-L265P;CDKN2A-WT DLBCL (n = 66)14 were grouped based on mean expression of genes compiled from the murine Suvarness signature into 2 cohorts (Suvarnesshigh cohort is labeled with an asterisk, n = 15). Senescence-impaired MyD88-L265P;CDKN2A-loss DLBCL are shown for comparison (n = 17). (D) GSEA comparing Suvarnesshigh vs Suvarnesslow MyD88-L265P;CDKN2A-WT groups from panel C, using senescence- and immune evasion–associated gene signatures (“SASP,” “E2F target genes,” “Senescence,” “Stemness,” “Negative regulation of immune response,” “Negative regulation of T-cell activation,” and “Positive regulation of cellular response to TGF-β”) and the Eµ-myc–derived MyDness signature (see panel E). Notably, the murine MyDness signature is significantly enriched in the human MyD88-L265P;CDKN2A-WT DLBCL cohort that exhibits a senescence-associated GEP (P < .001, FDR = 0.001). Both Suvarness and MyDness signatures comprise a nonoverlapping set of genes. P ≤ .05 and FDR <0.05 indicate significance. (E) Venn diagram showing overlap of the top 200 genes with highest fold-changes (upregulation; compared with the respective vector control lymphomas) as identified by RNA-seq for all NDM tested in the Eµ-myc mouse model. A mutant-specific unique set of genes was identified for MyD88-L265P- as well as CARD11-L244P-mutant lymphomas, accordingly named MyDness (123 genes) and CARDness (182 unique genes), respectively. (F) Transcriptome-analyzed DLBCL14 were stratified into 2 groups based on mean expression of genes compiled from the murine MyDness (left) and CARDness signatures (right), respectively, as shown in panel E. Top, GSEA comparing both cohorts (30% of highest vs 30% of lowest signature expressers; n = 231 cases for each group) was performed using gene sets defining senescence-related phenotypes (senescence, SASP, E2F target genes, stemness), T-cell evasion, as well as positive NF-κB and TGF-β responses. P ≤ .05 indicates significance. Bottom, expression levels (log2-transformed and sf-normalized counts) of PD-L1 (CD274) and PD-L2 (PDCD1LG2), the macrophage chemoattractant Ccl2 and Csf1 as well as the macrophage markers CD68 and Itgam in both cohorts. (G) MyD88-L265P derived MyDness signature identifies a DLBCL subgroup as analyzed in 3 large DLBCL patient cohorts14-16 that display increased expression of immune-checkpoint inhibitors (blue; CD274, PDCD1LG2), macrophage markers and chemoattractants (green; ITGAM, CD68, CSF-1, CCL2) as well as senescence-associated gene-expression profiles (orange) (see panel F, supplemental Figure 6D, and supplemental Figure 6F-G). Indicated n numbers refer to transcriptomes analyzed. Asterisks indicate significance (P < .05)
Moreover, it was recently reported that the tumor-suppressive TGF-β/SMAD1/ sphingosine-1-phosphate receptor-2 (S1PR2) signaling pathway is recurrently inactivated in DLBCL.59 Interestingly, we found TGF-β response signatures to be significantly enriched in the CDKN2A-unaffected Suvarnesshigh MyD88-L265P DLBCL cohort when compared with the Suvarnesslow samples, suggesting that, beside loss of the CDKN2A gene locus, disruption of the TGF-β axis may represent another mechanism to evade stroma-mediated lymphoma senescence elicited by MyD88-L265P mutations during DLBCL pathogenesis (Figure 6D).
Functional (especially non–cell-autonomous and immune-related mouse model–derived) findings cannot easily be recapitulated in human DLBCL material because no adequate immune-competent in vivo models exist. Therefore, we decided to interrogate data sets from large DLBCL patient cohorts comprising information about individual transcriptome profiles and clinical courses regarding key molecular features unveiled by our functional mouse investigations. We hypothesized that MyD88-L265P– or CARD11-L244P–governed Eµ-myc lymphomagenesis may produce lymphomas with distinct GEP whose human-homolog top-discriminating transcripts, termed MyDness or CARDness, respectively, might be instrumental in identifying human DLBCL subsets that present with a common, MyD88-L265P– or CARD11-L244P–characteristic biology irrespective of their actual MyD88 or CARD11 mutational status (Figure 6E). As important confirmatory evidence, we found MyDness to be significantly enriched for in the CDKN2A-intact MyD88-L265P;Suvarnesshigh DLBCL cohort (Figure 6D; supplemental Figure 6C). When dichotomizing a large transcriptomics-profiled DLBCL cohort14 into subgroups based on mean expression of the genes compiled in the MyDness and CARDness signatures, we unveiled striking similarities to our functional mouse model observations (Figure 6F). GSEA revealed that both MyDnesshigh and CARDnesshigh DLBCL samples were enriched for NF-κB and TGF-β response genes. Importantly, genes promoting T-cell immune evasion/T-cell exhaustion as well as senescence (including SASP and SAS) were also found significantly enriched in the mutanthigh cohorts (P < .05, corrected for multiple testing). Accordingly, E2F target gene expression was reciprocally suppressed. Notably, despite the striking biological similarities of these 2 signature-assigned subgroups, >40% of the MyDnesshigh or CARDnesshigh DLBCL cohorts are nonoverlapping patients. Confirming key MyD88 or CARD11 mutant-governed features detected in the Eµ-myc mouse model, we observed increased expression of PD-L1 (CD274) and PD-L2 (PDCD1LG2), macrophage chemoattractants as well as macrophage markers CD68 and Itgam in patients scoring high for either mutant signature (Figure 6F). Key findings were also confirmed on the transcriptome level in 2 other large DLBCL cohorts (GSE 9858815 [supplemental Figure 6D] and the National Cancer Institute [NCI] cohort16 [supplemental Figure 6E-G]). Notably, both MyDness and CARDness signatures recognized DLBCL patients independent of their COO status (tested in the NCI cohort16 ; data not shown). When probing the aforementioned PD1 blockade-susceptible Blimp1F/FIKK2caGFPstopF/stopFCγ1Cre/+ (BIC)-driven murine ABC-like lymphomas56 for MyDness, we found that not only this classifier but also the Suvarness signature as well as the GO term “macrophage activation” significantly enriched for in BIC lymphomas as compared with control GCB cells (supplemental Figure 7A). Strikingly, BIC lymphomas expressed significantly higher levels of transcripts encoding p16INK4a, PD-L2, macrophage markers Itgam and CD68, and the macrophage chemoattractant Csf1 (supplemental Figure 7B), prompting us to speculate that lymphomas from this genetically distinctly initiated DLBCL model may entertain a similar senescent state switch that, in turn, evokes a comparable cell-based immunological response. In essence, our comprehensive analysis of DLBCL-derived NF-κB–hyperactivating mutations in the Eµ-myc and other DLBCL-reminiscent mouse models of aggressive B-cell lymphomas led to far-reaching functional discoveries regarding the triad of senescence induction, macrophage attraction, and evasion of cytotoxic T-cell immunity, with subsequent confirmation of these cardinal features in corresponding human lymphoma subsets representing hundreds of DLBCL patients (Figure 6G).
Discussion
Aberrant BCR/NF-κB signaling is a hallmark of B-cell malignancies in general and DLBCL in particular.2 Molecular profiling analyses of sizeable DLBCL cohorts linked mutant NF-κB pathway mediators, especially in the ABC subtype, to constitutive activity of this cascade, but provided limited insights into differential functions individual NDMs might possess.1,8-16,60 A refined approach covering mutations, copy-number alterations, and structural variants led to novel classifications whose clusters, designated as C1-C5, or whose 4 distinct subtypes (termed MCD, BN2, N1, and EZB), according to leading genomic lesions, present with distinct clinical behavior.15,16 Interestingly, these analyses unveiled particularly dismal outcomes for the MyD88 mutation-enriched subgroups, the MCD subtype, or cluster C5, respectively, under R-CHOP therapy.15,16 However, these subgroups, statically assigned at diagnosis prior to any treatment encounter, have yet to demonstrate their power to stratify patients for distinct molecularly informed and outcome-improving therapies. Notably, ABC-subtype DLBCL was not only found to exhibit significantly higher levels of PD-L1 expression,14,57 but recent reanalyses of the MCD subtype also reported that almost three-quarters of the cases presented with immune-evading features, and specifically highlighted PD-L1/2 expression in this MyD88-mutant subgroup, albeit without connecting T-cell exhaustion to underlying senescence-prone biology.17
Here, we undertook a primarily mouse model–based approach to investigate the dynamic role of several naturally occurring NDMs in the context of a c-myc rearrangement as the driving oncogene, reflecting another frequent genomic alteration in DLBCL development and treatment responsiveness. Collectively, this approach demonstrated a pathogenic contribution via prosurvival signaling of all 4 moieties, that is, mutant versions of MyD88, CARD11, CD79B, and NFKBIZ, tested. Two of the mutants, CARD11-L244P and MyD88-L265P, selectively engaged in noncanonical NF-κB p52 signaling and macrophage-attracting chemokine secretion, evoking in turn a macrophage-relayed release of prosenescent TGF-β. As a consequence, if carrying 1 of these 2 moieties, lymphomas stood out as strongly senescence-prone. Importantly, our investigations pinpointed the very same mutants as drivers of immune-blunting and therapeutically exploitable PD ligand expression in mutant-expressing lymphoma cells, and, presumably via their senescence-associated secretome, in lymphoma-infiltrating macrophages as well. Notably, additional macrophage-presented checkpoint mechanisms such as CD47, for instance, may further contribute to immune evasion.61 Although sole overexpression of PD-L1 in Myc-driven lymphomas without CARD11-L244P or MyD88-L265P mutations was not accompanied by an increased fraction of senescent cells, we found both mutants to create a unique cell-autonomous and non–cell-autonomous ecosystem of a partly senescent, distinctly chemokine-secreting and PD-L1/2+ lymphoma cell population, PD-L1+ and TGF-β–secreting bystander macrophages, and PD1+ cytotoxic T cells. Checkpoint-derepressed T cells preferentially but not exclusively killed – like an “immunological cell-based senolytic”: senescent lymphoma cells whose B-cell–genuine role as rather weak antigen-presenting cells may have been boosted by the proimmunogenic impact of their SASP. Importantly, human MyD88-L265P DLBCL only retain the immune evasion phenotype if they are senescence-prone, that is, high in Suvarness, a mouse-derived Suv39h1 H3K9 methyltransferase status-based transcript signature indicating the capability to senesce. Notably, this subgroup represents nearly one-quarter of all MyD88-L265P–mutant DLBCL patients. However, if MyD88-mutant human lymphomas had, as a frequently observed event, lost the 9p21-encoded INK4a/ARF locus and simultaneously their prosenescent phenotype in the course of lymphoma development, immune evasion is no longer a typical feature, and, hence, those MyD88-L265P DLBCL with low Suvarness tend not to maintain high PD-L1/2 expression (data not shown). Importantly, senescent-preferential killing by cytotoxic T cells may not just deplete biologically inert cells, but those promoting aggressive tumor biology, via secretion of bystander-nursing SASP factors, or via stemness reprogramming and spontaneous cell-cycle reentry.58,62
Senescent cells are recognized and cleared by cells of the innate immune system,61 and CD4+ T cells were shown to prime engulfment of Ras-senescent cells by macrophages.63 To the best of our knowledge, we report here first-time evidence of senescence-triggered and directly senescence-targeting adaptive immune control, a fundamental finding with pivotal implications for immune-checkpoint blockade certainly going well beyond the role of distinct NF-κB–hyperactivating mutations in aggressive B-cell lymphomagenesis. Future investigations will address the nature of this CD8+ T-cell–mediated immune surveillance with respect to driving immunogens, boosting cytokines, and its efficacy toward nonsenescent target cells, even in adoptively transferred or vaccine-like settings.
Lastly, we like to emphasize the power and suitability of mouse model–based studies of complex biological processes that involve the interplay of several cell compartments during tumor development, therapy, and progression. The widely used, aggressive B-cell lymphoma biology-elucidating, although certainly not ideally DLBCL-recapitulating Eµ-myc transgenic mouse lymphoma model,23,44-47,58,64,65 with strengths and weaknesses compared with more recently developed models of DLBCL,56,57,66-76 proved particularly instrumental in addressing those dynamic processes in vivo, which are virtually unapproachable in standard patient biopsies taken at diagnosis. Importantly, our systems biology-based, signature-driven cross-species investigations confirmed the applicability of critical mouse-derived discoveries to human DLBCL, and highlighted the idea of ascribing transcriptome-based signatures to tumors with a distinct mutational status in order to include additional signature-positive but nonmutated tumors to the same subset of individual malignancies that may benefit from a specifically targeting treatment strategy. Marking, for instance, MyDness+ or CARDness+ individual DLBCL samples as functionally related to MyD88-L265P– or CARD11-L244P–mutant cases, respectively, is a conceptually novel approach (albeit remotely reminiscent of the genome-defined “BRCAness” status assigned to homologous repair-defective tumors harboring distinct mutations outside BRCA1/277 ) to address the possibility that extragenic pathway mutations or nonmutational signaling deregulation may functionally mimic the typical biological properties of distinct genetic lesions. We exploited this strategy here to group genomically heterogeneous tumor samples via a shared, functionally, and therapeutically relevant transcriptional signature. Further investigations, retrospective reanalyses of anti-PD1–exploring trials, and piloting clinical studies are now required to clarify the role for immune-checkpoint–blocking therapies in such newly defined DLBCL patient subsets.
The RNA-seq data generated for this study have been deposited in the Gene Expression Omnibus database (accession number GSE141454). Analysis scripts are available from L.C. (anna.dolnik@charite.de) and A.D. (liam_childs@hotmail.com) upon request.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the late A. Harris, T. Jenuwein, and M. Serrano for mice; J. Ruland for materials; N. Burbach and A. Lau for technical assistance; and members of the C.A.S. laboratory for discussions and editorial advice.
J.S. is an awardee of the Berlin Institute of Health (BIH)-Charité Junior Clinician Scientist Program funded by the Charité–Universitätsmedizin Berlin and the BIH. This work was supported by grants (to C.A.S.) from the Deutsche Krebshilfe (110678), the Bundesministerium für Bildung und Forschung (BMBF) e:Med program project SeneSys (031L0189A), the Deutsche Forschungsgemeinschaft (DFG; GO 2688/1-1| SCHM 1633/11-1, SCHM 1633/9-1, and the research training group “CompCancer” GRK 2424), and the Förderverein Hämatologie und internistische Onkologie (Tyle Private Foundation, Linz, Austria). This interdisciplinary work was further made possible by the Berlin School of Integrative Oncology (BSIO) graduate program funded within the German Excellence Initiative (with A.-M.G.-S. as a member of this program), and the German Cancer Consortium (GCC).
Authorship
Contribution: M.R., J.S., S.M., and A.-M.G.-S. performed experiments; P.L. performed histopathology; P.R.-P., A.D., T.P.H., K.S., G.K., R.F., D.C., and L.C. performed bioinformatic analysis of RNA-seq data; M.R. and C.A.S. designed the research and wrote the paper; and X.C., D.N.Y.F., S.D., A.B., L.B., A.R., and H.C.R. critically reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Clemens A. Schmitt, Hematology, Oncology, and Tumor Immunology, Charité – Universitätsmedizin Berlin and Max-Delbrück-Center for Molecular Medicine, D-13353 Berlin, Germany; e-mail: clemens.schmitt@charite.de or Kepler Universitätsklinikum, Klinik für Interne 3–Schwerpunkt Hämatologie und Onkologie, Med Campus III, Johannes Kepler University, 4021 Linz, Austria; e-mail: clemens.schmitt@kepleruniklinikum.at.
