

Key Points
Blockade of CD27/CD70 interaction compromises EBV-specific immune control.
CD27 is particularly required for the expansion and effector function of lytic EBV antigen–specific CD8+ T cells.

Abstract
Primary immunodeficiencies in the costimulatory molecule CD27 and its ligand, CD70, predispose for pathologies of uncontrolled Epstein-Barr virus (EBV) infection in nearly all affected patients. We demonstrate that both depletion of CD27+ cells and antibody blocking of CD27 interaction with CD70 cause uncontrolled EBV infection in mice with reconstituted human immune system components. While overall CD8+ T-cell expansion and composition are unaltered after antibody blocking of CD27, only some EBV-specific CD8+ T-cell responses, exemplified by early lytic EBV antigen BMLF1-specific CD8+ T cells, are inhibited in their proliferation and killing of EBV-transformed B cells. This suggests that CD27 is not required for all CD8+ T-cell expansions and cytotoxicity but is required for a subset of CD8+ T-cell responses that protect us from EBV pathology.
Introduction
Epstein-Barr virus (EBV) is a common γ-herpesvirus that infects >95% of the human population.1 At the same time, it was the first human tumor virus discovered in the 1960s.2,3 It is also associated with a spectrum of pathologies in humans,4,5 ranging from B-cell lymphomas and epithelial cell carcinomas to immunopathologies like hemophagocytic lymphohistiocytosis and infectious mononucleosis. The diverse manifestations of EBV pathology are connected to the lytic and 4 latent (latency 0 to III) gene expression programs that EBV can switch between in healthy virus carriers.6 These are also mirrored in EBV-associated malignancies, such as Burkitt lymphoma (latency I) and Hodgkin lymphoma (latency II). Despite this high pathogenic potential and wide distribution of EBV in the human population, the respective tumors are, fortunately, quite rare, with an annual incidence of ∼200 000 cases worldwide.7
A nearly perfect cell-mediated immune control protects us from EBV-associated pathologies. This becomes apparent under conditions of primary or acquired immunodeficiencies, such as HIV coinfection.5,8,9 They identify cytotoxic lymphocytes, mainly CD8+ T cells, as the primary immune compartment for EBV-specific immune control.10-12 Defects in T-cell receptor signaling have been identified in primary immunodeficiencies that predispose for EBV-associated pathologies. These affect among others interleukin-2–inducible T-cell kinase, ZAP70 and PI3K 110δ. Furthermore, development and expansion of cytotoxic lymphocytes is required for EBV-specific immune control and this is compromised by mutations in GATA2, MCM4, XIAP, STK4, and CTPS1. The last group of molecules, which are required for EBV-associated immune control, affect costimulation of CD8+ T cells. Among these, mutations in both CD27 and its ligand, CD70, have been identified as a nearly exclusive cornerstone of EBV-specific immune control. Previous research has shown that nearly all affected patients develop EBV-associated pathologies, with hemophagocytic lymphohistiocytosis–like immunopathologies more often observed in CD27 deficiencies and Hodgkin lymphoma more frequent in CD70 deficiencies.13-18
Therefore, we investigated the requirement for CD27+ lymphocytes and CD27 engagement of CD70 during EBV infection in mice with reconstituted human immune system components. We found that depletion of CD27+ cells and blocking of CD27 compromised EBV-specific immune control, resulting in elevated viral titers and expansion of infected CD39+CD70+ B cells. Overall CD8+ T-cell expansion was not compromised by CD27 blocking. However, the expansion and cytotoxicity of early lytic EBV antigen–specific CD8+ T cells, exemplified by BMLF1-specific cytotoxic lymphocytes, were abolished, suggesting that CD27 is required for an important part of the immune control of EBV.
Methods
Humanized mouse generation and infection
Nonobese diabetic (NOD) severe combined immunodeficiency (scid) γcnull (NSG) mice and HLA-A2 transgenic NSG mice were maintained in ventilated, specific pathogen–free conditions at the Institute of Experimental Immunology, University of Zurich. Newborn pups were reconstituted with human CD34+ hematopoietic progenitor cells (HPCs). Mice with sufficient reconstitution of human immune cells were injected with 105 Raji Green units (RGU) EBV. The detailed procedures for NSG and NSG-A2 reconstitution and virus production are provided in supplemental Methods (available on the Blood Web site). All animal work strictly followed the animal protocols ZH209/2014 and ZH159/17, licensed by the veterinary office of the canton of Zurich, Switzerland.
In vivo anti-CD27 antibody depletion and blocking
CD27-depleting monoclonal antibody (clone: LG.3A10) and the corresponding Armenian hamster immunoglobulin G isotype control (BioLegend) were injected intraperitoneally (IP) at 12.5 μg/g of the mouse weight 2 weeks after EBV infection and continued every 4 days until the termination of experiment. CD27 blocking monoclonal antibody (clone LG.3A10) and the corresponding mouse immunoglobulin G1 isotype control (Absolute Antibody) were injected IP at 6.25 μg/g of the mouse weight 2 weeks after EBV infection and continued every 4 days until the termination of experiment.
ChipCytometry
Splenic tissues harvested at the termination of experiments were embedded in optimum cutting temperature (O.C.T.; Tissue-Tek) compound at −80°C. Cryosections (5-6 μm thick) were fixed and inserted into ZellSafe Chips (Canopy Biosciences) for staining. Samples labeled with fluorescent antibodies were acquired with a Zellkraftwerk ZellScanner One and its ZellExplorer software. After each round of acquisition, the fluorescent signals were photobleached and prepared for the next round of staining to accomplish a 27-marker panel. Additional details are provided in the supplemental Methods.
Laboratory assays
Multiple assays were conducted to analyze the experimental samples, including flow cytometry, in vivo imaging system (IVIS), lymphoblastoid cell line (LCL) generation, cytotoxicity assay, immunohistochemistry and immunofluorescence, serum cytokine quantification, and quantitative reverse transcription polymerase chain reaction (PCR). Assay details are provided in supplemental Methods.
Statistical analysis
Statistical significance was calculated with a (1) Mann-Whitney U test to analyze unpaired data with a non-Gaussian distribution, (2) 1-way analysis of variance (ANOVA) (Kruskal-Wallis test) followed by Dunn’s post hoc test, (3) 2-way ANOVA with Sidak’s (or Tukey’s) multiple comparisons as post hoc test, or (4) 2-tailed, unpaired t test by Prism 7 (GraphPad Software). A D’Agostino-Pearson omnibus normality test was used to determine normality of data. All data points in the graphs are displayed with median and interquartile range, indicated with horizontal lines. "N" represents number of biological replicates unless otherwise stated. High-dimensional analysis details are provided in supplemental Methods.
Results
CD27 shows a similar expression pattern on T cells of humans and humanized mice
To investigate the specific contribution of CD27 to EBV-associated immune control, we have primarily used a mouse model with reconstituted human immune system compartments (humanized mice) that allows establishment of EBV infection, lymphomagenesis, and its cell-mediated immune control in vivo19,20 (Figure 1A). To examine whether humanized mice are a suitable model to study CD27 function, we characterized CD27 expression on single-cell suspensions of peripheral blood mononuclear cells (PBMCs) of healthy donors and humanized mice. The majority of CD27+ cells of both PBMC sources were CD4+ and CD8+ T cells (supplemental Figure 1A; Figure 1B). There was a similarly high level of expression on T cells in individual cell populations (Figure 1C). Slightly higher expression in the naive population (CCR7+CD45RA+) but lower expression in Temra (CCR7−CD45RA+) and Tem (CCR7−CD45RA−) in CD3+CD27+ T cells were observed in humanized mice (Figure 1D-E). Taken together, these data demonstrate that CD27 has a similar distribution pattern on T cells in humanized mice and humans. Thus, we conclude that humanized mice are a suitable model to study the role of CD27 expression on human T cells for EBV infection.

Comparison of CD27 expression between immune cell populations of humans and humanized mice. (A) Humanized mice (huMice) reconstitution scheme. Immunodeficient NOD mice with a loss-of-function mutation in the Prkdc gene and common γ chain deficiency (NOD-scid γcnull, or NSG) were engrafted with human CD34+ HPCs to reconstitute human immune system components and were tested for human immune compartment reconstitution after 3 months. (B) Pie charts show the distribution of CD27+ cells in different immune cell populations examined in huMice blood (n = 3) and human PBMCs (n = 3). (C) Frequency of CD27+ cells in different immune cell populations, comparing huMice peripheral blood (n = 3) and human PBMCs (n = 3). NK, natural killer. (D-E) Representative flow cytometry analysis illustrating the gating strategy to differentiate memory T-cell subsets characterized by CCR7 and CD45RA expression within the CD3+CD27+ population (D) and the frequency of each subset (E), comparing huMice peripheral blood (n = 3) and human PBMCs (n = 3). See also related supplemental Figure 1.
Comparison of CD27 expression between immune cell populations of humans and humanized mice. (A) Humanized mice (huMice) reconstitution scheme. Immunodeficient NOD mice with a loss-of-function mutation in the Prkdc gene and common γ chain deficiency (NOD-scid γcnull, or NSG) were engrafted with human CD34+ HPCs to reconstitute human immune system components and were tested for human immune compartment reconstitution after 3 months. (B) Pie charts show the distribution of CD27+ cells in different immune cell populations examined in huMice blood (n = 3) and human PBMCs (n = 3). (C) Frequency of CD27+ cells in different immune cell populations, comparing huMice peripheral blood (n = 3) and human PBMCs (n = 3). NK, natural killer. (D-E) Representative flow cytometry analysis illustrating the gating strategy to differentiate memory T-cell subsets characterized by CCR7 and CD45RA expression within the CD3+CD27+ population (D) and the frequency of each subset (E), comparing huMice peripheral blood (n = 3) and human PBMCs (n = 3). See also related supplemental Figure 1.
CD27+ cells are important in the immune control of EBV viral loads and tumorigenesis during EBV infection in humanized mice
We next determined how important the CD3+CD27+ cells are to protect against EBV infection. To this end, we adopted the acute (or infectious mononucleosis–like) EBV infection model by injecting humanized mice with 105 RGU B95-8 EBV.21 In order to allow priming of EBV-specific T-cell responses prior to depletion, CD27+ cells were depleted starting at week 2 postinfection (Figure 2A). A depletion effect of CD3+CD27+ T cells was observed and persisted for up to 6 days (supplemental Figure 2A). Therefore, depleting antibody injection was repeated every 4 days. There was a significant drop in CD3+ T cells in the peripheral blood under treatment (Figure 2B). A clear reduction in total CD3+ T-cell count was observed in both blood and spleen samples (Figure 2C-D). There was no significant difference in total CD19+ B-cell counts (Figure 2B), consistent with low CD27 expression in this immune compartment (Figure 1B-C). Loss of T cells in infected animals treated with depleting antibody led to a higher frequency of tumor incidence (Figure 2E). Furthermore, EBV viral burden was significantly elevated in both blood and spleen after CD27 depletion (Figures 2F-H). The detected viral load is primarily cell associated.22 Consistently, the number of EBNA2-expressing cells, which indicates early virus-transformed B cells,23 was significantly increased in spleen (Figure 2I-J). Altogether, these experiments suggest an essential role of CD27+ T cells in providing EBV-specific immunity and preventing development of EBV-associated malignancies.
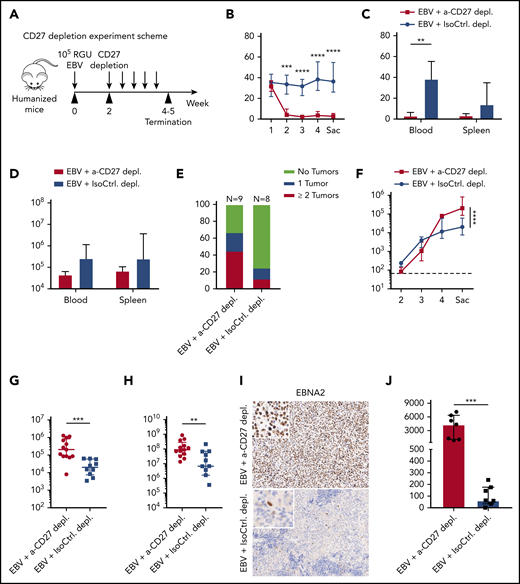
CD27+cells are essential for the immune control of EBV viral loads and tumorigenesis during EBV infection. (A) Workflow of CD27 depletion experiments. HuMice mice were infected (IP) with 105 RGU B95-8 EBV. At week 2 after EBV infection, animals were injected (IP) with 12.5 μg/g of either anti-CD27 depletion antibody or isotype control antibody consecutively every 4 days to ensure the depletion effect until termination of experiment. (B-E) Frequency of CD3+ T cells in anti-CD27 depleting antibody–treated group (α-CD27 depl.) and isotype control antibody–treated group (IsoCtrl. depl.) (B), frequency of CD3+ T cells at the termination of the experiment (C), total CD3+ T-cell count (D), and tumor burden in the respective groups (E). (F-H) EBV viral loads quantified by quantitative PCR over time (F) and at termination of the experiment in peripheral blood (G) and spleen (H). The lower limit of quantification of 122 IU/mL is depicted as horizontal dashed line. (I-J) Immunohistochemistry images of EBNA2 in the respective groups (original magnification ×200) (I), and the quantification of EBNA2+ cells/mm2 in splenic sections (J). Images are representative from 1 of 2 independent experiments. Data (n = 8-13 per group) pooled from 2 independent mouse experiments were graphed (B-H,J) and displayed with median and interquartile range. Two-way ANOVA analysis and Sidak’s multiple comparisons as a post hoc test (B,E-F), and the Mann-Whitney U test (C-H,J) were used to assess P values. **P < .01, ***P < .001, ****P < .0001. p.i., postinfection. See also related supplemental Figure 2.
CD27+cells are essential for the immune control of EBV viral loads and tumorigenesis during EBV infection. (A) Workflow of CD27 depletion experiments. HuMice mice were infected (IP) with 105 RGU B95-8 EBV. At week 2 after EBV infection, animals were injected (IP) with 12.5 μg/g of either anti-CD27 depletion antibody or isotype control antibody consecutively every 4 days to ensure the depletion effect until termination of experiment. (B-E) Frequency of CD3+ T cells in anti-CD27 depleting antibody–treated group (α-CD27 depl.) and isotype control antibody–treated group (IsoCtrl. depl.) (B), frequency of CD3+ T cells at the termination of the experiment (C), total CD3+ T-cell count (D), and tumor burden in the respective groups (E). (F-H) EBV viral loads quantified by quantitative PCR over time (F) and at termination of the experiment in peripheral blood (G) and spleen (H). The lower limit of quantification of 122 IU/mL is depicted as horizontal dashed line. (I-J) Immunohistochemistry images of EBNA2 in the respective groups (original magnification ×200) (I), and the quantification of EBNA2+ cells/mm2 in splenic sections (J). Images are representative from 1 of 2 independent experiments. Data (n = 8-13 per group) pooled from 2 independent mouse experiments were graphed (B-H,J) and displayed with median and interquartile range. Two-way ANOVA analysis and Sidak’s multiple comparisons as a post hoc test (B,E-F), and the Mann-Whitney U test (C-H,J) were used to assess P values. **P < .01, ***P < .001, ****P < .0001. p.i., postinfection. See also related supplemental Figure 2.
Blocking of CD27 diminishes the immune control of EBV infection
We next sought to investigate the effect of CD27 costimulation on EBV-specific immune control using HLA-A2 transgenic humanized mice reconstituted with HLA-A2+ CD34+ HPCs (NSG-A2). To monitor both bulk T-cell subsets and EBV antigen–specific CD8+ T-cell responses, we adoptively transferred recombinant HLA-A2 restricted T-cell receptor (TCR)-transduced human T cells, previously isolated from donor-mate NSG-A2 animals, which are specific for either (1) the early lytic EBV antigen BMLF1 or (2) the latent EBV antigen LMP2 (Figure 3A-B). Both the TCR-transduced (both specificities) and bulk CD8+ T cells showed similar levels of CD27 expression before transfer (Figure 3B). Following transfer, humanized NSG-A2 mice were infected with luciferase-expressing EBV (Luc-EBV) in order to monitor the localization of infection by in vivo imaging and then treated with either CD27 blocking antibody or isotype control starting from week 2 after infection. There was no depletion effect on CD3+ T and CD19+ B cells observed (supplemental Figure 3A-B). The blocking effect of the antibody was checked by flow cytometry using 2 fluorochrome-conjugated anti-CD27 antibodies (supplemental Figure 3C). While the antibody (clone O323) derived from a different clone than the blocking antibody could still detect CD27 (supplemental Figure 3D), the antibody (clone LG.3A10) derived from the same clone as the blocking antibody was inhibited from binding (supplemental Figure 3E).
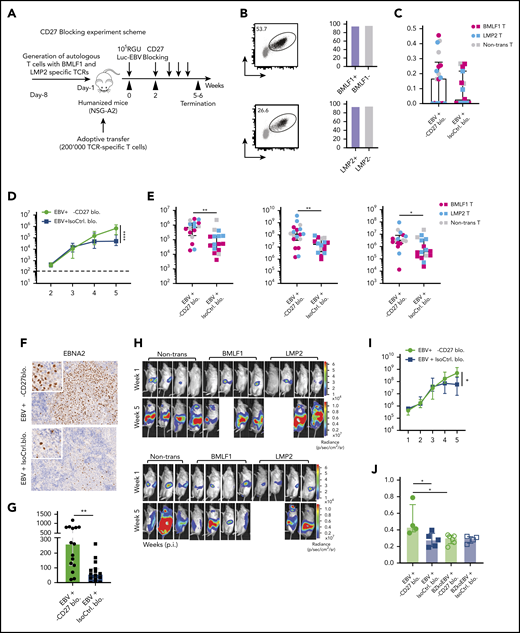

Blocking of CD27 diminishes the immune control of EBV infection. (A) Workflow of CD27 blocking experiments. One day before infection with 105 RGU luciferase encoding B95-8 EBV (Luc-EBV), huNSG-A2 mice (HLA-A2 transgenic NSG mice reconstituted with HLA-A2+ human hematopoietic progenitor cells) were adoptively transferred with 200,000 autologous T cells expressing either BMLF1- or LMP2-specific TCRs that had been transduced ex vivo. The anti-CD27 blocking antibody is the Fc domain re-engineered version of the anti-CD27 depletion antibody of Figure 2 and can no longer engage antibody-directed cellular cytotoxicity. At week 2 after EBV infection, animals were injected (IP) with 6.5 μg/g of either anti-CD27 blocking antibody or isotype control antibody consecutively every 4 days until termination of the experiment. (B) Representative flow cytometry plots of BMLF1- and LMP2-specific TCR-transduced CD8+ T cells (left) using mouse TCRβ-specific antibodies (mTCRb) and BMLF1 and LMP2 peptide plus HLA-A2 pentamers.24 Frequency of CD27 expression on transduced and nontransduced CD8+ T cells (right). (C) Spleen weight of animals treated with either anti-CD27 blocking antibody (α-CD27 blo.) or isotype control antibody (IsoCtrl. blo.) upon different transfer conditions. (D-E) EBV viral loads quantified by quantitative PCR over time during EBV infection (D) at the termination of the experiment in peripheral blood (E, left), spleen (E, middle), and liver (E, right). Mice treated with either anti-CD27 blocking antibody or isotype control antibody in different transfer conditions were compared. (F-G) Representative immunohistochemistry images of EBNA2 in the respective groups (original magnification ×200) (F), and the quantification of EBNA2+ cells/mm2 in splenic sections (G). (H-I) Representative IVIS image analysis at week 1 and week 5 after Luc-EBV infection (H) and quantification of defined region of interest of IVIS images (I). (J) Spleen weight of animals infected with either wild-type EBV or BZLF1 knockout EBV (BZkoEBV), treated with anti-CD27 blocking antibody or isotype control antibody without adoptive transfer. (K-L) EBV viral loads of animals over time during EBV infection (K) at the termination of the experiment in peripheral blood (L, left) and spleen (L, right) in the respective groups. Data (n = 14-16 per group) pooled from 2 independent mouse experiments were graphed (C-E,G,I-L) and displayed with median and interquartile range. Graphs (F-I) (n = 7-8 per group) are representative from 1 out of 2 independent experiments. Graphs (K-L) (n = 4-6 per group) are from 1 experiment. One-way ANOVA analysis (Kruskal-Wallis test) followed by Tukey’s post hoc test (J,L), 2-way ANOVA analysis and Sidak’s multiple comparisons as a post hoc test (D,I,K), and the Mann-Whitney U test (C,E,G) were used to assess P values. *P < .05, **P < .01. See also related supplemental Figure 3.
Blocking of CD27 diminishes the immune control of EBV infection. (A) Workflow of CD27 blocking experiments. One day before infection with 105 RGU luciferase encoding B95-8 EBV (Luc-EBV), huNSG-A2 mice (HLA-A2 transgenic NSG mice reconstituted with HLA-A2+ human hematopoietic progenitor cells) were adoptively transferred with 200,000 autologous T cells expressing either BMLF1- or LMP2-specific TCRs that had been transduced ex vivo. The anti-CD27 blocking antibody is the Fc domain re-engineered version of the anti-CD27 depletion antibody of Figure 2 and can no longer engage antibody-directed cellular cytotoxicity. At week 2 after EBV infection, animals were injected (IP) with 6.5 μg/g of either anti-CD27 blocking antibody or isotype control antibody consecutively every 4 days until termination of the experiment. (B) Representative flow cytometry plots of BMLF1- and LMP2-specific TCR-transduced CD8+ T cells (left) using mouse TCRβ-specific antibodies (mTCRb) and BMLF1 and LMP2 peptide plus HLA-A2 pentamers.24 Frequency of CD27 expression on transduced and nontransduced CD8+ T cells (right). (C) Spleen weight of animals treated with either anti-CD27 blocking antibody (α-CD27 blo.) or isotype control antibody (IsoCtrl. blo.) upon different transfer conditions. (D-E) EBV viral loads quantified by quantitative PCR over time during EBV infection (D) at the termination of the experiment in peripheral blood (E, left), spleen (E, middle), and liver (E, right). Mice treated with either anti-CD27 blocking antibody or isotype control antibody in different transfer conditions were compared. (F-G) Representative immunohistochemistry images of EBNA2 in the respective groups (original magnification ×200) (F), and the quantification of EBNA2+ cells/mm2 in splenic sections (G). (H-I) Representative IVIS image analysis at week 1 and week 5 after Luc-EBV infection (H) and quantification of defined region of interest of IVIS images (I). (J) Spleen weight of animals infected with either wild-type EBV or BZLF1 knockout EBV (BZkoEBV), treated with anti-CD27 blocking antibody or isotype control antibody without adoptive transfer. (K-L) EBV viral loads of animals over time during EBV infection (K) at the termination of the experiment in peripheral blood (L, left) and spleen (L, right) in the respective groups. Data (n = 14-16 per group) pooled from 2 independent mouse experiments were graphed (C-E,G,I-L) and displayed with median and interquartile range. Graphs (F-I) (n = 7-8 per group) are representative from 1 out of 2 independent experiments. Graphs (K-L) (n = 4-6 per group) are from 1 experiment. One-way ANOVA analysis (Kruskal-Wallis test) followed by Tukey’s post hoc test (J,L), 2-way ANOVA analysis and Sidak’s multiple comparisons as a post hoc test (D,I,K), and the Mann-Whitney U test (C,E,G) were used to assess P values. *P < .05, **P < .01. See also related supplemental Figure 3.
Animals treated with CD27 blocking antibody exhibited splenomegaly 5 weeks postinfection (Figure 3C). However, CD27 blocking did not seem to have an effect on animal weight (supplemental Figure 3F) and survival (supplemental Figure 3G). A significant increase in blood viral loads was observed upon CD27 blocking (Figure 3D) as well as in spleen and liver (Figure 3E). Viral loads of the groups transferred with BMLF1- or LMP2-specific T cells or mock-transduced cells were not different (supplemental Figures 3H). Additionally, the number of EBNA2+ cells was significantly higher in splenic sections upon CD27 blocking (Figure 3F-G), and CD27 expression on EBV-infected B cells in blood and spleen was also higher (supplemental Figure 3I). To assess the viral burden and infection progression, mice were imaged with an IVIS Spectrum Imaging System on a weekly basis. The bioluminescent signal showed a significantly higher level due to CD27 blocking at week 5 after EBV infection (Figure 3H-I). Moreover, with respect to individual EBV gene expression, both latent genes (EBER1, EBNA2, LMP1, LMP2a, and EBNA1_Wp) and lytic genes (BMLF1, BMRF1, BGLF5, BNLF2a, and BILF1) were higher expressed after blocking CD27 (supplemental Figure 3J).
In order to examine if CD27 blocking has a selective effect on immune control of latent or lytic EBV infection, humanized NSG mice were infected with wild-type EBV and compared with BZLF1 knockout EBV (BZkoEBV) mice that cannot switch into lytic replication. Interestingly, wild-type EBV-infected animals showed significantly higher spleen weights (Figure 3J) and viral loads in both blood and spleen (Figure 3K-L) upon CD27 blocking, as compared with animals infected with BZkoEBV with and without CD27 inhibition. Additionally, the number of EBNA2+ cells was significantly higher in wild-type EBV-infected animals treated with the anti-CD27 antibody as compared with the other groups (supplemental Figure 3K-L). Taken together, these data reveal that CD27 blockade leads to loss of EBV-specific immune control, primarily against lytic EBV antigen expression.
CD70+CD39+EBNA2+ B cells accumulate upon loss of CD27-mediated immune control of EBV
To better understand which EBV-infected B cells accumulate in the absence of CD27-mediated immune control, we checked their phenotype on sacrifice day (Figure 3A). It has been previously shown that expression of both CD70 and CD39 are upregulated on LCLs, which consist of EBV-transformed B cells.25,26 Both markers were reported to be highly expressed in B-cell lymphomas such as germinal center B-cell–like and activated B-cell–like diffuse large B-cell lymphomas (DLBCLs).24,27 We observed significantly increased expression of CD70 and CD39 in the peripheral blood, spleen, and liver (Figure 4A-C) upon CD27 blocking. No significant increase was seen in CD73+ and CD39+CD73+ populations (Figure 4B). However, CD39 frequency positively correlated with EBV viral loads (Figure 4D). Additionally, we found reduced expression of CXCR5 and CCR7 after blocking CD27, 2 chemokine receptors important for B-cell migration into lymphoid organs (Figure 4E-F).
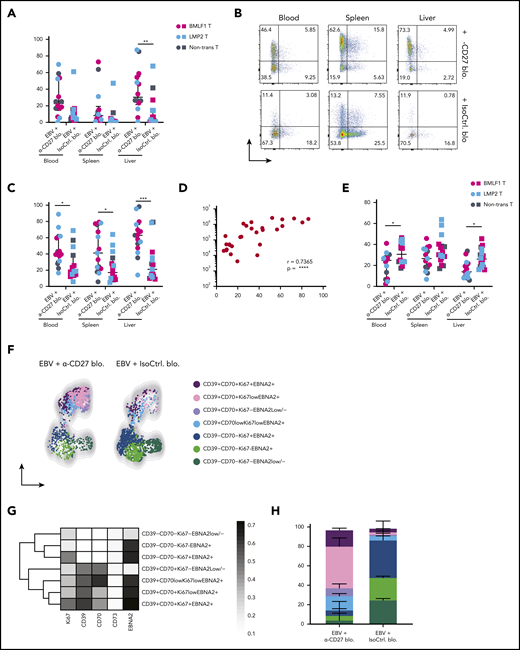
CD70+CD39+EBNA2+B cells accumulate upon loss of CD27-mediated immune control of EBV. (A) Frequency of CD70 expression on CD19+ B cells in multiple organs (blood, spleen, and liver) in anti-CD27 blocking antibody– vs isotype control antibody–treated group. (B) Flow cytometry plots of CD39 and CD73 expression on CD19+ B cells in multiple organs (blood, spleen, and liver) in the indicated experimental groups. (C) Frequency of CD39 expression on CD19+ B cells. (D) Correlation between the CD39 expression on CD19+ B cells and EBV viral loads in blood. (E) Frequency of CXCR5 expression on CD19+ B cells. (F) Representative uniform manifold approximation and projection (UMAP) analysis depicts clusters with coexpression of CD39, CD70, Ki67, and EBNA2 on CD19+ B cells in blood. (G) Data from panel F were transformed and shown in percentile of each population in anti-CD27 blocking antibody– vs isotype control antibody–treated group. (H) Representative heatmap analysis of coexpression of CD39, CD70, Ki67 and EBNA2 on CD19+ B cells in blood. Data (n = 14-16 per group) pooled from 2 independent mouse experiments were graphed (A,C-E) and displayed with median and interquartile range. Graphs (F-H) (n = 7-8 per group) are representative from 1 of 2 independent experiments. The Mann-Whitney U test was used to assess P values (A,C,E), and the Spearman correlation examining rank correlation was used for panel D. *P < .05, **P < .01, ***P < .001. See also related supplemental Figure 4.
CD70+CD39+EBNA2+B cells accumulate upon loss of CD27-mediated immune control of EBV. (A) Frequency of CD70 expression on CD19+ B cells in multiple organs (blood, spleen, and liver) in anti-CD27 blocking antibody– vs isotype control antibody–treated group. (B) Flow cytometry plots of CD39 and CD73 expression on CD19+ B cells in multiple organs (blood, spleen, and liver) in the indicated experimental groups. (C) Frequency of CD39 expression on CD19+ B cells. (D) Correlation between the CD39 expression on CD19+ B cells and EBV viral loads in blood. (E) Frequency of CXCR5 expression on CD19+ B cells. (F) Representative uniform manifold approximation and projection (UMAP) analysis depicts clusters with coexpression of CD39, CD70, Ki67, and EBNA2 on CD19+ B cells in blood. (G) Data from panel F were transformed and shown in percentile of each population in anti-CD27 blocking antibody– vs isotype control antibody–treated group. (H) Representative heatmap analysis of coexpression of CD39, CD70, Ki67 and EBNA2 on CD19+ B cells in blood. Data (n = 14-16 per group) pooled from 2 independent mouse experiments were graphed (A,C-E) and displayed with median and interquartile range. Graphs (F-H) (n = 7-8 per group) are representative from 1 of 2 independent experiments. The Mann-Whitney U test was used to assess P values (A,C,E), and the Spearman correlation examining rank correlation was used for panel D. *P < .05, **P < .01, ***P < .001. See also related supplemental Figure 4.
Next, we assessed the coexpression of CD70 and CD39, together with Ki67 for B-cell proliferation and EBNA2 for EBV infection in blood (supplemental Figure 4A) and spleen (supplemental Figure 4B). Two phenotypically distinct populations were identified with respect to the expression of CD70 and CD39 (Figures 4F and 4G). In animals treated with anti-CD27 blocking antibody, we found the CD70+CD39+ population was more expanded and showed high coexpression of Ki67 and EBNA2 in blood (Figure 4H). The same phenotypic difference was also observed in spleen (supplemental Figure 4C-E). In summary, these results suggest that CD27 blocking allows accumulation of CD39+CD70+ DLBCL-like EBV-infected B cells with possibly more aggressive proliferative behavior.
Early EBV lytic antigen BMLF1-specific CD8+ T cells require CD27 for expansion and cytotoxicity
We next addressed whether CD27 blocking affected particularly EBV-specific CD8+ T-cell responses in vivo. In line with CD27 deficient patient data, which showed normal T-cell differentiation compared with healthy controls in peripheral blood,14 our results demonstrated that CD27 blockade did not lead to a change in bulk T-cell differentiation (Figure 5A), as well as in the memory subset composition of EBV-specific CD8+ T cells (supplemental Figure 5A) in blood. No significant difference was also observed in spleen, liver, and bone marrow (supplemental Figure 5B). We next examined BMLF1- and LMP2-specific CD8+ T cells, which were adoptively transferred into the animals (Figure 5B). In line with CD27 blocking mainly affecting immune control of lytic EBV infection, there was an impaired expansion of BMLF1-specific CD8+ T cells in animals treated with CD27 blocking antibody longitudinally in blood (Figure 5C), while such a difference was not observed for LMP2-specific CD8+ T cells (Figure 5D). Further analysis in spleen and liver showed similar results with selective impairment of BMLF1-specific, but not LMP2-specific, CD8+ T cells (Figure 5E-F). Moreover, significantly increased Ki67 expression on BMLF1-specific CD8+ T cells (depicted as CD8 Pent+ cells), as compared with non-BMLF1-specific T cells (depicted as CD8 Pent− cells), was observed in the isotype antibody–treated group. However, CD27 blockade seemed to suppress proliferation of BMLF1-specific CD8+ T cells, as Ki67 expression was reduced comparable to bulk CD8+ T-cell levels (Figure 5G-H). By contrast, LMP2-specific CD8+ T cells showed no difference (Figure 5I).
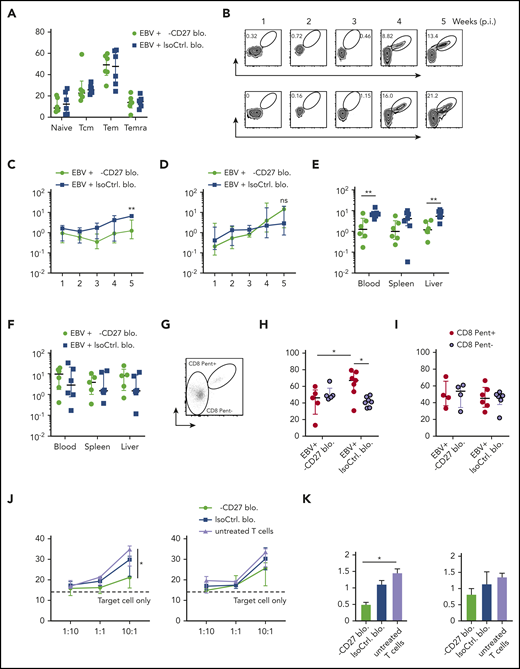
Early EBV lytic antigen BMLF1-specific CD8+T cells require CD27 for expansion and cytotoxicity. (A) CD8+ T-cell memory subsets characterized by CD45RA and CD62L expression and depicted as naive, Tcm, Tem, and Temra in groups treated with either anti-CD27 blocking antibody or isotype control antibody in vivo. (B) Weekly representative flow cytometry plots showing the gating strategy and circulating BMLF1- and LMP2-specific CD8+ T cells in blood in vivo. (C-D) Longitudinal data examining the expansion of BMLF1- (C) and LMP2-specific (D) CD8+ T cells in blood over time after Luc-EBV infection, treated with anti-CD27 blocking antibody vs isotype control antibody. (E-F) Frequency of BMLF1- (E) and LMP2-specific (F) CD8+ T cells in multiple organs (blood, spleen, and liver) at termination of the experiment. (G) Representative flow cytometry plot of EBV-specific TCR-transduced CD8+ T cells (depicted as CD8 Pent+) and the rest of the T cells (depicted as CD8 Pent−) using mouse TCRβ-specific antibodies (mTCRb) and either BMLF1 or LMP2 peptide plus HLA-A2 pentamers.24 (H-I) Frequency of Ki67-expressing cells showing the cell proliferation of EBV-specific BMLF1- (H) and LMP2-specific (I) CD8+ T cells (depicted as CD8 Pent+ cells) vs the rest of CD8+ T cells (depicted as CD8 Pent− cells) in blood in different experimental groups. (J-K) In vitro killing assay with BMLF1- and LMP2-specific T-cell clones generated from healthy EBV carriers ex vivo. T cells were pretreated with either anti-CD27 blocking antibody or isotype control antibody and cocultured with PKH-26 prelabeled autologous LCLs for 21 hours at the indicated effector-to-target ratios (J) and at the ratio of 10:1 (K). Data (n = 5-6 per group) pooled from 2 independent mouse experiments were graphed (A,C-I) and displayed with median and interquartile range. Two-way ANOVA analysis and Sidak’s multiple comparisons as a post hoc test (C-D), and the Mann-Whitney U test (A,E-I) were used. Data (J-K) are pooled from 3 experiments and analyzed using a 2-tailed, unpaired t test; dashed line signifies target cell only. *P < .05; **P < .01. ns, not significant. See also related supplemental Figure 5.
Early EBV lytic antigen BMLF1-specific CD8+T cells require CD27 for expansion and cytotoxicity. (A) CD8+ T-cell memory subsets characterized by CD45RA and CD62L expression and depicted as naive, Tcm, Tem, and Temra in groups treated with either anti-CD27 blocking antibody or isotype control antibody in vivo. (B) Weekly representative flow cytometry plots showing the gating strategy and circulating BMLF1- and LMP2-specific CD8+ T cells in blood in vivo. (C-D) Longitudinal data examining the expansion of BMLF1- (C) and LMP2-specific (D) CD8+ T cells in blood over time after Luc-EBV infection, treated with anti-CD27 blocking antibody vs isotype control antibody. (E-F) Frequency of BMLF1- (E) and LMP2-specific (F) CD8+ T cells in multiple organs (blood, spleen, and liver) at termination of the experiment. (G) Representative flow cytometry plot of EBV-specific TCR-transduced CD8+ T cells (depicted as CD8 Pent+) and the rest of the T cells (depicted as CD8 Pent−) using mouse TCRβ-specific antibodies (mTCRb) and either BMLF1 or LMP2 peptide plus HLA-A2 pentamers.24 (H-I) Frequency of Ki67-expressing cells showing the cell proliferation of EBV-specific BMLF1- (H) and LMP2-specific (I) CD8+ T cells (depicted as CD8 Pent+ cells) vs the rest of CD8+ T cells (depicted as CD8 Pent− cells) in blood in different experimental groups. (J-K) In vitro killing assay with BMLF1- and LMP2-specific T-cell clones generated from healthy EBV carriers ex vivo. T cells were pretreated with either anti-CD27 blocking antibody or isotype control antibody and cocultured with PKH-26 prelabeled autologous LCLs for 21 hours at the indicated effector-to-target ratios (J) and at the ratio of 10:1 (K). Data (n = 5-6 per group) pooled from 2 independent mouse experiments were graphed (A,C-I) and displayed with median and interquartile range. Two-way ANOVA analysis and Sidak’s multiple comparisons as a post hoc test (C-D), and the Mann-Whitney U test (A,E-I) were used. Data (J-K) are pooled from 3 experiments and analyzed using a 2-tailed, unpaired t test; dashed line signifies target cell only. *P < .05; **P < .01. ns, not significant. See also related supplemental Figure 5.
Given that cytotoxicity is the main protective T-cell function during EBV infection, we further assessed T-cell cytolytic response by coculturing LCLs with BMLF1- and LMP2-specific CD8+ T-cell clones generated ex vivo from the autologous healthy human donor.28 We found that BMLF1-specific T cells showed significantly reduced cytotoxic activity against LCLs after blocking CD27 at the effector to target ratio of 10 to 1 (Figure 5J-K). However, the proliferation of LCLs was not influenced by CD27 blocking (Figure 5B). In summary, these data reveal a differential susceptibility of early lytic and latent EBV antigen–specific CD8+ T cells to CD27 blockade during EBV infection.
CD27 blockade compromises CXCR5+EOMES+ CD8+ T-cell accumulation during EBV infection
To interrogate the alterations in proinflammatory cytokine production after CD27 blockade, we examined terminal serum samples. Animals treated with the CD27 blocking antibody exhibited significantly elevated levels of interferon-γ and interleukin-10 and highly increased levels of tumor necrosis factor α (Figure 6A), suggesting that CD27 blocking increases the inflammatory immune responses that might contribute to EBV-associated pathologies. Consistent with a more proinflammatory environment, CD27 blockade also led to a moderately decreased expression of CXCR5 on CD8+ T cells in blood and liver (Figure 6B).

CD27 blockade compromises CXCR5+EOMES+CD8+T-cell accumulation during EBV infection. (A) Cytokine production from serum samples harvested at termination of the experiment. Each individual cytokine is presented in the separate transfer conditions in the upper panel. In the lower panel, pooled data of α-CD27 blocking antibody–treated or isotype control antibody–treated groups is shown. (B) Frequency of CXCR5 expression on CD8+ T cells in the respective groups in blood, spleen, and liver. (C) Frequency of EOMES bright T-bet dim and EOMES dim T-bet bright populations in the respective group in blood. (D) Frequency of CD69 expression on CD8+ T cells in spleen. (E) Frequency of 2B4 (left) and PD1 (right) expression on CD8+ T cells in spleen. (F) Representative ChipCytometry immunofluorescence images for CD20, EBNA2, CD8, CD69, and PD1 in splenic sections of the respective treatment groups. Scale bars, 50 μm. (G) Quantification of the CD8+/EBNA2+CD20+ ratio, as well as the frequency of CD8+CD69+ and CD8+PD1+ cells in 5 to 7 randomly selected fields in splenic sections of isotype control or CD27 blocking antibody–treated animals. Data (n = 14-16 per group) pooled from 2 independent mouse experiments were graphed (A,B,D-F) and displayed with median and interquartile range. The Mann-Whitney U test was used to assess P values (A-E,G). *P < .05, **P < .01, ***P < .001. IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
CD27 blockade compromises CXCR5+EOMES+CD8+T-cell accumulation during EBV infection. (A) Cytokine production from serum samples harvested at termination of the experiment. Each individual cytokine is presented in the separate transfer conditions in the upper panel. In the lower panel, pooled data of α-CD27 blocking antibody–treated or isotype control antibody–treated groups is shown. (B) Frequency of CXCR5 expression on CD8+ T cells in the respective groups in blood, spleen, and liver. (C) Frequency of EOMES bright T-bet dim and EOMES dim T-bet bright populations in the respective group in blood. (D) Frequency of CD69 expression on CD8+ T cells in spleen. (E) Frequency of 2B4 (left) and PD1 (right) expression on CD8+ T cells in spleen. (F) Representative ChipCytometry immunofluorescence images for CD20, EBNA2, CD8, CD69, and PD1 in splenic sections of the respective treatment groups. Scale bars, 50 μm. (G) Quantification of the CD8+/EBNA2+CD20+ ratio, as well as the frequency of CD8+CD69+ and CD8+PD1+ cells in 5 to 7 randomly selected fields in splenic sections of isotype control or CD27 blocking antibody–treated animals. Data (n = 14-16 per group) pooled from 2 independent mouse experiments were graphed (A,B,D-F) and displayed with median and interquartile range. The Mann-Whitney U test was used to assess P values (A-E,G). *P < .05, **P < .01, ***P < .001. IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
We next assessed the differential expression of T-box transcription factors (T-bet) and eomesodermin (EOMES), which are key drivers for effector functions and long-term memory formation of T cells.29,30 We found the frequency of EOMESbright T-betdim CD8+ T cells to be reduced after CD27 blockade (Figures 6C), indicating that cytotoxic effector and effector memory T-cell functions are decreased. In addition, the expression of the activation/tissue residency marker CD69 was diminished (Figure 6D), whereas an upregulation in the expression of costimulatory 2B4 and inhibitory PD1 was observed on CD8+ T cells (Figure 6E).
To obtain spatial information of activated CD8+ T cells in relation to EBV-infected B cells, we investigated splenic sections by ChipCytometry, a multiplexing tissue imaging technology that allows for repeated rounds of immunofluorescence staining and bleaching to assess multiple parameters in histological sections. CD20+ B cells and CD8+ T cells were aggregated in white pulp areas of isotype control–treated spleens (supplemental Figure 6A). We assessed a 27-marker panel plus a nuclear staining (supplemental Table 1; supplemental Figure 6C-F) by ChipCytometry and observed colocalization between CD20 and EBNA2, CD8 and CD69, and CD8 and PD1 in splenic white pulp areas (Figure 6F). Quantification of segmented cells showed higher frequencies of CD8+ T cells and EBNA2+CD20+ cells after CD27 blockade (supplemental Figure 6B). The ratio of CD8+/EBNA2+CD20+ cells was significantly higher upon CD27 blocking, suggesting compromised immune control despite efficient CD8+ T-cell expansion. However, CD8+ T cells were similarly activated as judged by PD1 and CD69 expression (Figure 6G). Thus, these results suggested that CD27 blockade may compromise terminal cytotoxic CD8+ T-cell differentiation with homing capacity to germinal centers in secondary lymphoid organs, where cytotoxic EBV-specific immune control needs to take place.
Discussion
In this study, we performed in vivo experiments using humanized mice as an EBV infection model to address CD27 deficiency during EBV-specific immune responses. Both CD27+ lymphocyte depletion and CD27 blocking compromised EBV-specific immune control. We deciphered that CD27 blockade had a dramatic effect on the protective function of early lytic EBV antigen (BMLF1) specific CD8+ T cells, but to a lesser extent on latent EBV antigen (LMP2) specific CD8+ T cells, despite the elevated gene expression of BMLF1 in B cells of the CD27 blocking antibody–treated group (supplemental Figure 3J), which should have driven BMLF1-specific T-cell expansion more efficiently. In line with our findings, previous studies in CD27-deficient patients showed a detectable level of EBV-specific T cells against LMP2 and autologous LCL restimulation,13,17 suggesting that CD27 signaling may be involved in expansion of early lytic antigen–specific cytotoxic CD8+ T-cell responses. In line with these findings, CD27 was required for the immune control of wild-type, but not lytic replication-deficient, EBV infection.
Although CD27 has been reported to regulate T-cell survival, expansion, and memory generation,31,-33 its blocking did not seem to have an impact on overall T-cell differentiation during EBV infection in humanized mice. This is consistent with clinical observations from both CD27- and CD70-deficient patients,14,16,18 in whom the T-cell repertoire composition also seems largely unaltered compared with healthy controls. Consistent with our findings, only some, but not other EBV-specific CD8+ T-cell responses were diminished in the affected patients.16,18 These CD27 dependent T-cell specificities might be stimulated by and target early EBV-infected B cells in which CD70 peaks at day 5 in mRNA and day 8 in protein expression after infection,34,35 and even earlier leaky lytic EBV antigen expression can be targeted by CD8+ T cells to eliminate EBV-infected B cells.36 These data from both CD27 blocking in humanized mice and CD27- or CD70-deficient patients argue that absence of CD27/CD70 signaling does not alter T-cell repertoire composition or even bulk CD8+ T-cell expansion to EBV infection but that a subset of protective CD8+ T-cell responses against lytic EBV antigens, exemplified by BMLF1-specific CD8+ T cells, depend on CD27 for their expansion and cytotoxicity.
Interestingly, patients with RASGRP1 deficiency, the main activator of the mitogen-activated protein kinase pathway, exhibited a reduced T-cell expansion in a CD27-dependent manner.37 RASGRP1-deficient T cells failed to proliferate upon stimulation with CD70high LCLs.37,38 This result further confirmed the importance of CD27/CD70 and the downstream mitogen-activated protein kinase pathway in the expansion of EBV-specific T cells.
Other costimulatory molecules affecting T-cell interactions with B cells also seem to play a unique role in immunity to EBV. For instance, it has been discovered that in X-linked lymphoproliferative disease type 1, deficiency of the SLAM-associated protein (SAP) compromises costimulatory 2B4 function.39 This leads to a profound effect on cytotoxicity against EBV-infected B cells,40,41 but it still allows expansion of EBV-specific CD8+ T cells.42,43 Accordingly, humanized mice present with similar CD8+ T-cell expansion but less controlled EBV infection after antibody blocking of 2B4, and SAP-deficient patients suffer from EBV pathology despite normal frequencies of EBV-specific CD8+ T cells.41,44 This implies that 2B4 might only be required for cytotoxic CD8+ T-cell recognition of EBV-transformed B cells, while CD27 promotes expansion and cytotoxicity of a subset of protective EBV-specific CD8+ T cells. Possibly compensatory 2B4 upregulation was observed on CD8+ T cells in our experiments upon CD27 blocking, but diminished expression of this coreceptor was reported in CD27- or CD70-deficient patients.18 In contrast to the better-understood role of CD27 and 2B4, the functions of 4-1BB, NKG2D, CTLA-4, and PD-1 that are required for EBV-specific immune control still need to be defined.21,45,,,-49 However, different EBV-associated pathologies were observed in primary immunodeficiencies affecting these molecules, suggesting their nonredundant roles in controlling virus infection. A better understanding of these underlying mechanisms should allow us to harness these costimulatory and coinhibitory functions for immune modulation.
The selective loss of expansion and cytotoxicity of a subset of EBV-specific CD8+ T cells was also associated with a significant upregulation in the coexpression of CD39 and CD70 on the EBV-infected B cells in our study. This indicates more activation of B cells after blocking CD27/CD70 interaction, which might promote EBV-mediated growth transformation and development of persistent infection in B cells, ultimately resulting in DLBCL-like malignancies. This altered phenotype of EBV-infected B cells might be the result of higher EBV viral loads or selective targeting of CD39+CD70+ EBV-infected B cells by the CD27-dependent CD8+ T-cell response. However, in the absence of efficient cytotoxicity against B cells through the CD27/CD70 axis, elevated proinflammatory cytokines might also provoke higher expression of CD70 on EBV-infected B cells. Similar results have also been reported for CD48, the ligand of 2B4, which was upregulated in SAP deficiency during EBV infection.40 Thus, not only does overall EBV infection increase upon CD27 blocking, but also the phenotype of infected B cells seems to change.
These CD39+ B cells might be mainly involved in the expansion of BMLF1-specific CD8+ T cells and in turn be controlled by the subset of EBV-specific cytotoxic CD8+ T-cell responses that fail to expand during CD27 blocking and deficiency, indicating that this subset of EBV-specific CD8+ T cells are particularly sensitive to CD27 deficiency. In contrast, CD8+ T-cell responses to the related β-herpesvirus human cytomegalovirus (HCMV) that has with 50% also a high prevalence in the human population have been shown to be CD27−CD28−.50 This might at least explain why EBV, but not HCMV-specific immune control by T cells, is sensitive to CD27 or CD70 deficiency. The near-exclusive susceptibility of CD27- or CD70-deficient patients to EBV pathology argues that most common infections apart from EBV follow the HCMV example.
Collectively, these results suggest a unique and nonredundant role of CD27 to EBV-specific immunity, mainly emphasizing its function in T-cell expansion and cytotoxicity, primarily of a subset of EBV-specific T cells, such as BMLF1-specific CD8+ T cells. Absence of immune control by these CD27-dependent CD8+ T cells leads to uncontrolled EBV infection. Our results also support a possibly protective role of early antigen-specific CD8+ T-cell responses against EBV infection and lymphomagenesis.1 Moreover, restoration of the CD27/CD70 pathway by CD27 agonistic antibody could be a therapeutic approach for the treatment of EBV-associated lymphomas, especially in patients with a CD70 deficiency.
Reasonable requests for reagents and protocols should be addressed to Christian Münz, Institute of Experimental Immunology, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland; e-mail: muenzc@immunology.uzh.ch.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all members of the Core Facility of the University of Zurich, including the Cytometry Facility and the Center for Microscopy and Image Analysis, for assistance and the Laboratory Animal Services Center for animal husbandry.
Authorship
Contribution: Y.D. designed the study, performed the majority of the experiments, and analyzed the data; B.C., K.Z., and H.Z. helped design and perform experiments; A.M., L.-A.L., and A.B. performed the ChipCyometry and its quantification; P.S., R.C., and A.Z. established the quantification of viral loads; A.H. and H.S. generated the TCR constructs; W.H. provided Luc-EBV; C.M. designed the study with Y.D. and supervised the study; and Y.D. and C.M. prepared the figures and wrote the manuscript.
Conflict-of-interest disclosure: H.S. is a founder of Quell Therapeutics, shareholder of Quell Therapeutics, and shareholder of Kuur Therapeutics. The remaining authors declare no competing financial interests.
Correspondence: Christian Münz, Institute of Experimental Immunology, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland; e-mail: muenzc@immunology.uzh.ch.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal