

In this issue of Blood, have demonstrated that monocytic cells from patients with chronic myelomonocytic leukemia (CMML) are vulnerable to synergistic inhibition of the antiapoptotic BCL-2 family protein MCL-1 and the RAS/MAPK signaling pathway kinases MEK1 and MEK2.1
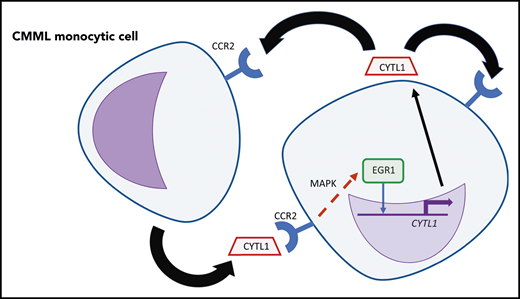
An autocrine and/or paracrine loop of CYTL1 in CMML monocytic cells. CYTL1 binds to CCR2, which results in increased MAPK pathway signaling and then in increased binding of EGR1 near CYTL1, further increasing expression of this cytokine. Professional illustration by Patrick Lane, ScEYEnce Studios.
An autocrine and/or paracrine loop of CYTL1 in CMML monocytic cells. CYTL1 binds to CCR2, which results in increased MAPK pathway signaling and then in increased binding of EGR1 near CYTL1, further increasing expression of this cytokine. Professional illustration by Patrick Lane, ScEYEnce Studios.
CMML is a member of the broader family of myelodysplastic/myeloproliferative syndromes that often affect older patients.2 Unfortunately, clinical options for patients with CMML are limited, resulting in poor prognoses for these individuals.3 As suggested by the name, the defining characteristic of CMML is the clonal expansion of mature monocytic cells arising from dysplastic bone marrow. A previously unresolved question was how these mutated monocytic cells survive longer and accumulate in a greater number, than normal monocytes, which are more tightly regulated.
In recent years, the interrogation of the apoptosis pathway, through the use of peptide mimics that trigger either proapoptotic or antiapoptotic responses (BH3 profiling), has been informative in deciphering mechanisms of evading apoptosis in myeloid malignancies. For example, this investigational approach has resulted in the establishment of venetoclax-based regimens, employing a BCL-2 antagonist, as a standard of care in elderly, unfit patients with acute myeloid leukemia.4,5 Solary et al noted that monocytic cells from patients with CMML were more resistant to apoptosis. Using BH3 profiling with BCL-2 family protein peptides, they demonstrated that CMML monocytic cells have an addiction to the antiapoptotic protein MCL-1 that is not observed in normal monocytes, and they succeeded in using an MCL-1 inhibitor to selectively suppress the survival of CMML monocytic cells.
CMML is frequently associated with aberrant activation of the RAS/RAF/MEK/ERK MAPK signaling pathway, and MEK1 and MEK2 have been identified as potential therapeutic targets in CMML cells carrying RAS mutations.6,7 Activation of MAPK signaling is known to contribute to autocrine or paracrine growth in many different types of myeloid leukemia, supported by cytokines including interleukin-6 in chronic myelogenous leukemia (CML).8 In the present study, Sevin et al have furthered our understanding of the involvement of the MAPK pathway in CMML by identifying the chemokine CYTL1 as an autocrine or paracrine factor responsible for the enhanced survival of CMML cells. By studying a subset of CMML samples that survived in vitro under serum-free conditions, they found that survival was linked to upregulation of the CYTL1 gene. They further showed that plasma from patients with CMML containing CYTL1 was itself sufficient to promote survival of monocytic cells. Acting via the chemokine receptor CCR2, CYTL1 induces phosphorylation of the MAPK protein extracellular signal-regulated kinase, leading to induction of the transcription factor EGR1, which binds to a potential regulatory element upstream of the CYTL1 gene, creating an autocrine feedback loop that can now be targeted by inhibitors of MAPK signaling (see figure).
To develop a novel potential therapeutic strategy for CMML, Sevin et al demonstrated that 2 independent US Food and Drug Administration–approved MEK1 and MEK2 inhibitors (selumetinib and trametinib) can each be used in combination with the MCL-1 inhibitor S63845 to synergistically promote apoptosis of CMML cells, without major impact on normal monocytes. In patient-derived xenografts (PDXs) of CMML cells in mice, the combination of MEK and MCL-1 inhibitors also substantially suppressed the engraftment of CMML. This synergy works, in part, by promoting degradation of the MCL-1 protein, which is not itself upregulated in CMML. Significantly, the treated mice do not develop any significant cytopenias, further supporting the potential use of this combinatorial strategy in further preclinical development. This finding represents an advance over current CMML treatment options, such as hydroxyurea or DNA hypomethylating agents, which also increase apoptosis of normal mature monocytes. Having identified the importance of CYTL1 in CMML, it will now be interesting to see whether this chemokine is also a contributing factor in other myeloid malignancies.
Looking forward, there remains an underlying need to take the clonal architecture of CMML into consideration. Evidence suggests that the mutations responsible for CMML arise in primitive stem progenitor cell populations, and it remains unknown whether drugs effective in mature monocytic CMML cells will also be effective in CMML stem cells. Resolving this issue is likely to require rigorous identification of cells with leukemia-initiating properties in PDX models, as has been done for other myeloid malignancies, but not yet for CMML.9,10 This still requires a further technological advance to confirm the identity of the cell of origin in CMML before we can determine whether MEK/MCL-1–targeted therapy is a long-term treatment option for CMML. In the meantime, this study already suggests an alternative therapeutic strategy to specifically target and debulk the mature myeloid cells that shape the phenotype of CMML.
Conflict-of-interest disclosure: J.L. has received travel funding from Novartis and Daichi-Sankyo, and honoraria from Pfizer, Janssen, and Amgen, not within the scope of this work. P.N.C. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal