

Key Points
Monocytes that accumulate in patients with CMML have a defective apoptosis through addiction to MCL1.
Combined MCL1 and MEK inhibition restores monocyte apoptosis and decreases leukemic burden in xenografted animals.

Abstract
Mouse models of chronic myeloid malignancies suggest that targeting mature cells of the malignant clone disrupts feedback loops that promote disease expansion. Here, we show that in chronic myelomonocytic leukemia (CMML), monocytes that accumulate in the peripheral blood show a decreased propensity to die by apoptosis. BH3 profiling demonstrates their addiction to myeloid cell leukemia-1 (MCL1), which can be targeted with the small molecule inhibitor S63845. RNA sequencing and DNA methylation pattern analysis both point to the implication of the mitogen-activated protein kinase (MAPK) pathway in the resistance of CMML monocytes to death and reveal an autocrine pathway in which the secreted cytokine-like protein 1 (CYTL1) promotes extracellular signal-regulated kinase (ERK) activation through C-C chemokine receptor type 2 (CCR2). Combined MAPK and MCL1 inhibition restores apoptosis of monocytes from patients with CMML and reduces the expansion of patient-derived xenografts in mice. These results show that the combined inhibition of MCL1 and MAPK is a promising approach to slow down CMML progression by inducing leukemic monocyte apoptosis.
Introduction
Chronic myeloid malignancies are diseases of the hematopoietic stem cell (HSC) in which lineage differentiation is preserved. Mature myeloid cells of the malignant clone contribute to disease development, as demonstrated in a mouse model of chronic myeloid leukemia in which BCR/ABL tyrosine kinase activity promotes overproduction of interleukin-6 (IL-6) by mature cells of the clone. In turn, IL-6 skews immature cell differentiation toward the myeloid lineage.1 Neutralization of IL-6 blocks disease installation and progression in this model,2 suggesting that targeting cytokine-dependent feed forward loops may constitute a new efficient therapeutic approach in chronic myeloid malignancies.
Chronic myelomonocytic leukemia (CMML) is a prototypic chronic myeloid malignancy in that it associates features of myelodysplastic syndromes and myeloproliferative neoplasms.3 This clonal disorder, mostly observed in the elderly, is associated with the stepwise accumulation of somatic mutations in epigenetic, splicing, and signaling genes in an HSC.4 Feedback loops involving mutation-induced cytokine overproduction may exist in CMML. The TET2 gene mutation, the most frequent somatic event in this disease, compromises the ability of the protein to downregulate the IL6 gene expression in monocytes and macrophages at the resolution phase of inflammation, thus generating an inflammatory state that drives excessive myelopoiesis.5,6
Because of their advanced age and comorbidities, few patients with CMML are eligible for allogeneic stem cell transplantation.7 Hypomethylating agents transiently reverse the disease phenotype in responding patients, but their genomic impact remains limited.8 Other conventional therapies aim at attenuating symptoms by a personalized strategy of minimizing cytopenia-induced or proliferation-associated symptoms.9 These conventional therapies do not alter the natural course of this severe disease, which indicates an unmet need for curative approaches. Currently tested strategies target recurrent mutations in actionable genes4 or the unique dependence of CMML on granulocyte-macrophage colony-stimulating factor.10 Theoretically, a therapeutic strategy that eliminates mature differentiation products of the malignant clone might complement these approaches.
The hallmark of CMML is a persistent peripheral blood monocytosis of ≥1 × 109/L with monocytes accounting for ≥10% of the white blood cells.3 Monocytes that accumulate in the peripheral blood of patients with CMML are predominantly classic monocytes with a CD14+CD16− phenotype.11 Virtually all of these cells are generated by the leukemic clone.12
Here, we identify defective apoptosis of circulating classic monocytes in CMML. Resistance to apoptosis involves the BCL2-related protein myeloid cell leukemia-1 (MCL1), together with the overproduction of cytokine-like protein 1 (CYTL1),13 a cytokine that activates the mitogen-activated protein kinase (MAPK) signaling pathway by interacting with C-C chemokine receptor type 2 (CCR2) in an autocrine or paracrine manner. Inhibition of MCL1 using the small molecule S63845, combined with adenosine triphosphate–independent inhibitors of MAPKs (MEK1 and MEK2), either selumetinib or trametinib, restores monocyte apoptosis and decreases tissue infiltration by leukemic cells in xenografted mice. These results show that the combined inhibition of MCL1 and MEK as a therapeutic opportunity should be evaluated for the treatment of CMML.
Patients and methods
Patient samples
Peripheral blood samples were collected from patients with CMML and age-matched healthy donors in the context of a noninterventional study validated by the ethical committee Ile de France 1 (DC-2014-2091). Buffy coats were collected from young healthy blood donors (Etablissement Français du Sang, Rungis, France). Patients with CMML were diagnosed according to the last iteration of the World Health Organization classification of myeloid malignancies.3 Clinico-biological characteristics of the patients, summarized in Table 1, were obtained from our registered database (DR-2016-256). Peripheral blood mononuclear cells were sorted by using density centrifugation Pancoll (Pan-Biotech, Dutscher, Brumath, France) and CD14+ monocytes through negative selection with magnetic beads and the AutoMacs system (Miltenyi Biotech, Paris, France). Gene mutations were screened as described.8,12
Characteristics of patients with CMML included in the trial
| Characteristic | N | % |
|---|---|---|
| No. of patients | 190 | |
| Mean age (range), y | 74.3 (28-100) | |
| Sex | ||
| Male | 116 | |
| Female | 74 | |
| CMML (World Health Organization classification) | ||
| 0 | 82 | |
| 1 | 60 | |
| 2 | 48 | |
| Proliferative | 99 | |
| Dysplastic | 91 | |
| Mean no. of leukocytes (range), ×109/L | 20.8 (3.8-137.3) | |
| Mean no. of monocytes (range), ×109/L | 4.7 (0.4-29.9) | |
| Mean platelet count (range), ×109/L | 156.3 (14-996) | |
| Mean hemoglobin level (range), g/dL | 11.6 (6.8-16.8) | |
| Karyotype | ||
| Normal | 116 | |
| Abnormal | 33 | |
| Not available | 41 | |
| Main genetic alterations, positive/tested | ||
| TET2 | 124/173 | 72 |
| SRSF2 | 63/161 | 39 |
| ASXL1 | 72/173 | 42 |
| NRAS | 29/172 | 17 |
| KRAS | 24/172 | 14 |
| CBL | 15/172 | 9 |
| Characteristic | N | % |
|---|---|---|
| No. of patients | 190 | |
| Mean age (range), y | 74.3 (28-100) | |
| Sex | ||
| Male | 116 | |
| Female | 74 | |
| CMML (World Health Organization classification) | ||
| 0 | 82 | |
| 1 | 60 | |
| 2 | 48 | |
| Proliferative | 99 | |
| Dysplastic | 91 | |
| Mean no. of leukocytes (range), ×109/L | 20.8 (3.8-137.3) | |
| Mean no. of monocytes (range), ×109/L | 4.7 (0.4-29.9) | |
| Mean platelet count (range), ×109/L | 156.3 (14-996) | |
| Mean hemoglobin level (range), g/dL | 11.6 (6.8-16.8) | |
| Karyotype | ||
| Normal | 116 | |
| Abnormal | 33 | |
| Not available | 41 | |
| Main genetic alterations, positive/tested | ||
| TET2 | 124/173 | 72 |
| SRSF2 | 63/161 | 39 |
| ASXL1 | 72/173 | 42 |
| NRAS | 29/172 | 17 |
| KRAS | 24/172 | 14 |
| CBL | 15/172 | 9 |
Samples from a given patient were used for several independent experiments (supplemental Table 2), and serial sampling was performed in some patients. A total of 246 samples collected from 190 patients were tested.
Reagents
Intracellular BH3 (iBH3) peptides were made by Proteogenix (Schiltigheim, France). Venetoclax, navitoclax, and selumetinib (AZD-6244) were purchased from Selleck Chemicals (Munich, Germany), and trametinib was purchased from Euromedex (Souffelweyersheim, France). S63845 was synthesized by Servier.14 Recombinant human CYTL1 (rhCYTL1) was purchased from CliniSciences (Nanterre, France), CAS 445479 and RS 504393 from Sigma-Aldrich (Saint-Quentin-Fallavier, France) and Tocris (Noyal-Châtillon-sur-Seiche, France), respectively. Antibodies are listed in the supplemental Data, available on the Blood Web site.
Cell death detection
Sorted CD14+ monocytes were cultured overnight in RPMI 1640 medium supplemented or not with fetal calf serum (Thermo Fisher Scientific). Cell death was identified by Trypan blue staining or by flow cytometry analysis of cells stained with annexin V-fluorescein isothiocyanate (AnV-FITC) and propidium iodide (PI) antibodies before flow cytometry analysis using Canto X cytometer (BD Biosciences) and FlowJo (FlowJo LLC, Ashland, OR).
iBH3 profiling
Peripheral blood mononuclear cells were stained with Live/Dead Blue (Invitrogen, Cergy Pontoise, France) for 20 minutes on ice and then with CD45, CD24, CD14, CD16, CD2, and CD56 antibodies in magnetic-activated cell sorting buffer. They were pelleted and suspended in derived from trehalose experimental buffer (135 mM trehalose, 50 mM KCl, 20 μM EDTA, 20 μM EGTA, 5 mM succinate, 0.1% bovine serum albumin, 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [HEPES]-KOH, pH 7.5) before being exposed to peptides with 0.002% w/v digitonin as described.15 The fraction of cytochrome c released was calculated as 100 × (1 – [(MFIsample – MFIFMO)/(MFIPUMA2A – MFIFMO)]), where MFI is mean fluorescence intensity and FMO is fluorescence minus one.
Confocal analysis
CD14+ monocytes seeded on cover glasses were fixed in 4% paraformaldehyde, permeabilized with ethanol 70%, and labeled with mouse anti-γH2AX (1:500, Millipore, Molsheim, France), rabbit anti-active caspase-3 (1:150, Cell Signaling Technology), then with Alexa Fluor 488 goat anti-mouse immunoglobulin G and Alexa Fluor 555 goat anti-rabbit immunoglobulin G, respectively before being analyzed as described.16
Gene expression analysis
Total RNA (0.5 µg) from CD14+ monocytes was reverse transcribed using SuperScript VILO complementary DNA synthesis kit (Thermo Fisher Scientific). Reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR) was performed with SyBR Green master mix in an Applied Biosystems 7500 thermocycler as described.17 Data were normalized to 4 housekeeping genes (PPIA, GAPDH, HPRT, and RPL32; primer sequences in supplemental Table 1).
Immunoblotting
Cells lysed with Laemmli buffer (5% sodium dodecyl sulfate, 10% glycerol, 32.9 mM tris(hydroxymethyl)aminomethane [Tris]-HCl, pH 6.8) supplemented with dithiothreitol 0.1 M and protease and phosphatase inhibitors (Roche) were subjected to migration, transfer, and analysis according to standard protocols.
DNA methylation by enhanced reduced representation bisulfite sequencing (ERRBS)
High-molecular-weight genomic DNA (25 ng) was used to perform ERRBS, was sequenced on an Illumina HiSeq 2500, and was analyzed as described.8
RNA sequencing (RNA-seq)
CD14+ monocyte RNA (integrity score ≥7.0) was used to prepare libraries with Illumina TruSeq RNA Sample Prep Kit V2 and sequence on a 75-bp pair-end run using a NextSeq 500 High Output Kit as described.16 Transcripts from Gencode V24 were quantified in transcripts per million using Kallisto software. Gene-level expression was the sum of transcripts per million values for each gene transcript. Raw reads were mapped to the hg19 genome with Tophat2 (v2.0.14)/Bowtie2 (v2.1.0). The number of reads per gene was counted using HTSeq (0.5.4p5) and GENECODE (v24lift37), and differential gene expression analysis was performed by using DESeq2 package (v1.10.1). Genes with an absolute log2 fold change >1 and P < .05 were reported.
Chromatin immunoprecipitation sequencing (ChIP-seq)
ChIP experiments were performed with a ChIP-IT kit (Active Motif). Cells were cross-linked with the addition of 1% formaldehyde to the culture medium for 10 minutes, and the reaction was stopped with glycine. After lysis in sodium dodecyl sulfate lysis buffer and sonication, antibodies (5 µg) were incubated overnight. Chromatin immunoprecipitated with early growth response 1 (EGR1) antibody (sc110, Santa Cruz Biotechnology) was eluted from magnetic beads before reversing cross-links. Sequencing and analysis were performed as described.16 ChIP-PCR experiments are described in the supplemental Data.
Cytokine dosage
Human plasma was obtained by peripheral blood centrifugation (180g for 10 minutes and then at 21 000g for 5 minutes). CYTL1 was quantified using an enzyme-linked immunosorbent assay (ELISA) (CliniSciences). Cytokines were quantified in mouse plasma using V-PLEX (Human Proinflammatory Panel II, MSD, Rockville, MD) and custom mouse interleukin-1β (mIL-1β), mIL-6, and mouse tumor necrosis factor α (mTNF-α) (MSD).
Patient-derived xenografts (PDXs)
Experiments approved by the Ethical Committee (2016-104-7171) were performed following 2 previously described methods.18,19 Research was conducted in accordance with the Declaration of Helsinki. Details are provided in the supplemental Data.
Statistics
Data are displayed as means ± standard error of the mean. Statistically significant differences were assessed using the Mann-Whitney U test or Student t test (2 groups) or an analysis of variance (>2 groups). Similarity of variance was tested using GraphPad PRISM (San Diego, CA) before any statistical analysis. P < .05 was considered significant.
Results
Defective apoptosis of CMML peripheral blood monocytes
CD14+ monocytes were isolated from the peripheral blood of 190 patients with CMML whose characteristics are summarized in Table 1 and age-matched healthy donors. These cells (CMML, 16; healthy donors, 27; supplemental Table 2, cohort 1) were cultured in the absence of serum. After 4 days, virtually all the healthy donor monocytes stained positively with Trypan blue, whereas a variable fraction of CMML monocytes remained unstained, indicating better survival (Figure 1A). The death of healthy donor monocytes cultured for 24 hours in the absence or presence of serum was associated with AnV labeling (supplemental Figure 1A), a decrease in the mitochondrial membrane potential (supplemental Figure 1B), cleavage of the fluorogenic caspase substrates Ac-DEVD-AMC (supplemental Figure 1C) and Ac-IETD-AFC (supplemental Figure 1D), and cleavage of poly adenosine diphosphate (ADP)-ribose polymerase (PARP) (supplemental Figure 1E), indicating caspase-dependent apoptosis that could be prevented by the addition of 1 µM Q-VD-OPh (supplemental Figure 1A-E). The resistance of CMML monocytes to apoptosis was validated by analysis of AnV-FITC and PI-stained monocytes collected from 21 patients with CMML (supplemental Table 2, cohort 2) and 30 age-matched healthy donors that were cultured for 4 days in serum-free medium (Figure 1B). This resistance to apoptosis could be detected in a shorter assay in which monocytes were cultured in serum-containing medium for 24 hours. Again, monocytes from patients with CMML (n = 78; supplemental Table 2, cohort 3) demonstrated a decreased frequency of apoptosis compared with age-matched control monocytes (n = 102) (Figure 1C). The resistance of CMML monocytes to apoptosis was further confirmed by a decreased immunofluorescence detection of nuclear γH2AX and active caspase-3 (supplemental Figure 1F) and a decreased cleavage of Ac-DEVD-AMC fluorogenic substrate (supplemental Figure 1G). Of note, when explored immediately after cell sorting, no difference between cells from healthy donors and CMML monocytes could be detected (supplemental Figure 1H). Clinically, no correlation was found between CMML monocyte resistance to apoptosis and World Health Organization classification (Figure 1D). The increased resistance of CMML monocytes to death correlated with a higher number of NRAS mutations, without a significant relationship with other recurrent mutations (Figure 1E). No correlation was detected between monocyte sensitivity to apoptosis and blood cell parameters (supplemental Figure 1I), with only a trend toward an increased resistance of monocytes to apoptosis with their increasing count in the peripheral blood (supplemental Figure 1J). Monocyte viability was similar in patients who received hydroxyurea treatment and untreated patients (supplemental Figure 1K), even when monocytes of the same patients were tested before and after having initiated hydroxyurea treatment (not shown). In contrast, patients who responded to a demethylating drug demonstrated a therapy-induced increase in monocytic apoptosis (supplemental Figure 1L), which was not observed in nonresponding patients or those with a stable disease (supplemental Figure 1M). Of note, we did not detect any resistance to apoptosis by CMML CD34+ cells in liquid culture (supplemental Figure 1N).

Defective apoptosis of CMML peripheral blood monocytes. (A) Cell viability of CD14+ monocytes sorted from the peripheral blood of patients with CMML (n = 16) and healthy donors (n = 27) was analyzed by Trypan blue dye exclusion after 4 days in culture without serum. (B) CD14+ monocytes were sorted from the peripheral blood of patients with CMML (n = 21) and healthy donors (n = 30) and stained with AnV-FITC and PI after 4 days in culture without serum; the AnV+/PI– fraction was measured by flow cytometry. (C) AnV+/PI– fraction of CD14+ monocytes sorted from the peripheral blood of patients with CMML (n = 78) and healthy donors (n = 102) as determined by flow cytometry after 24 hours in culture in the presence of 10% fetal calf serum. (D-E) Patient samples tested in panel C were classified according to patient characteristics, including the 2016 iteration of the World Health Organization classification separating CMML-0 from CMML-1 and CMML-2 (D) and the presence or absence of mutations in the indicated genes (E). Error bars are mean ± standard error of the mean (SEM). Mann-Whitney U test: *P < .05; ***P < .001; ****P < .0001. MUT, mutant; WT, wild-type.
Defective apoptosis of CMML peripheral blood monocytes. (A) Cell viability of CD14+ monocytes sorted from the peripheral blood of patients with CMML (n = 16) and healthy donors (n = 27) was analyzed by Trypan blue dye exclusion after 4 days in culture without serum. (B) CD14+ monocytes were sorted from the peripheral blood of patients with CMML (n = 21) and healthy donors (n = 30) and stained with AnV-FITC and PI after 4 days in culture without serum; the AnV+/PI– fraction was measured by flow cytometry. (C) AnV+/PI– fraction of CD14+ monocytes sorted from the peripheral blood of patients with CMML (n = 78) and healthy donors (n = 102) as determined by flow cytometry after 24 hours in culture in the presence of 10% fetal calf serum. (D-E) Patient samples tested in panel C were classified according to patient characteristics, including the 2016 iteration of the World Health Organization classification separating CMML-0 from CMML-1 and CMML-2 (D) and the presence or absence of mutations in the indicated genes (E). Error bars are mean ± standard error of the mean (SEM). Mann-Whitney U test: *P < .05; ***P < .001; ****P < .0001. MUT, mutant; WT, wild-type.
CMML monocytes are addicted to MCL1
To determine the molecular mechanisms through which CMML monocytes resist apoptosis, we investigated their functional dependence on specific BCL2 family members by using a BH3 profiling assay.15 Briefly, we measured the propensity of monocytes exposed to peptides that mimic the BH3 domain of proapoptotic BCL2 family members (supplemental Table 3) to initiate mitochondrial apoptosis, as indicated by the cytosolic release of cytochrome c. The tested peptides were selected to measure the dependency of CMML monocytes on antiapoptotic BCL2 family members that include BCL2, BCLXL, MCL1, BFL1, and BCLW (supplemental Figure 2A). Results obtained in 21 patients with CMML (supplemental Table 2, cohort 4) compared with 18 age-matched healthy donors indicated that healthy donor and CMML monocytes were equally sensitive to BIM and PUMA-derived BH3 peptides. In contrast, CMML monocytes were more sensitive to NOXA- and HRK-derived BH3 peptides and their combination, suggesting an increased dependency on MCL1 and BCLXL (Figure 2A). Cells that were sensitive to the NOXA-derived BH3 peptide were also sensitive to the HRK-derived BH3 peptide (supplemental Figure 2B). RT-qPCR analysis of BCL2 family gene expression (CMML, up to 30 patients; supplemental Table 2, cohort 5) did not detect any significant change in CMML cells compared with age-matched healthy donor monocytes (Figure 2B; supplemental Figure 2C). Immunoblotting of sorted CMML monocyte proteins (supplemental Table 2, cohort 6) compared with those of age-matched healthy donors did not detect any significant difference in BCL2, MCL1, and BCLXL protein expression (supplemental Figure 2D-E). These results indicated that the dependency of CMML monocytes on MCL1 and BCLXL could not be explained by the upregulation of the corresponding genes and proteins. We then explored the ability of BCL2 family protein inhibitors to restore CMML monocyte apoptosis. Compared with age-matched healthy donor monocytes, CMML cells showed an increased sensitivity to the selective MCL1 inhibitor S63845 (CMML, 89; supplemental Table 2, cohort 7), but not to the specific BCL2 inhibitor venetoclax or to the BCL2, BCLXL, and BCLW inhibitor navitoclax (Figure 2C). As observed in other cell types,14 exposure to S63845 increased MCL1 expression in sorted monocytes (Figure 2D). S63845 compound also triggered PARP cleavage (Figure 2D), further supporting its ability to restore caspase-dependent apoptosis in CMML monocytes. Altogether, CMML monocytes seemed to be mostly addicted to the antiapoptotic effects of MCL1.
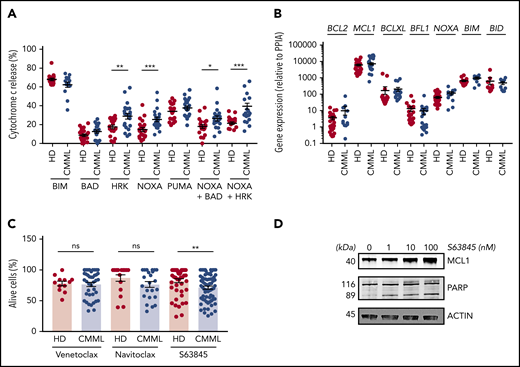
CMML monocyte addiction to MCL1. (A) iBH3 profiling: CD14+ peripheral blood monocytes collected from patients with CMML (n = 14-21) and healthy donors (n = 17-22) were incubated with the indicated peptides (80 μM) before measuring the flow of the fraction of cytochrome c released. (B) RT-qPCR analysis of indicated gene ex-pression in sorted peripheral blood monocytes collected from patients with CMML (n = 9-30) and healthy donors (n = 9-28) using PPIA as a housekeeping gene. (C) Peripheral blood monocytes from patients with CMML (n = 24-71) and healthy donors (n = 12-49) were treated with venetoclax (1 µM), navitoclax (100 nM), or S63845 (10 nM) for 24 hours at 37°C before measuring the fraction of AnV–/PI– cells by flow cytometry. (D) Immunoblot analysis of indicated proteins in sorted monocytes treated with indicated doses of S63845 for 24 hours. Data are shown as mean ± SEM. Mann-Whitney U test: *P < .05; **P < .01; ***P < .001. ns, not significant.
CMML monocyte addiction to MCL1. (A) iBH3 profiling: CD14+ peripheral blood monocytes collected from patients with CMML (n = 14-21) and healthy donors (n = 17-22) were incubated with the indicated peptides (80 μM) before measuring the flow of the fraction of cytochrome c released. (B) RT-qPCR analysis of indicated gene ex-pression in sorted peripheral blood monocytes collected from patients with CMML (n = 9-30) and healthy donors (n = 9-28) using PPIA as a housekeeping gene. (C) Peripheral blood monocytes from patients with CMML (n = 24-71) and healthy donors (n = 12-49) were treated with venetoclax (1 µM), navitoclax (100 nM), or S63845 (10 nM) for 24 hours at 37°C before measuring the fraction of AnV–/PI– cells by flow cytometry. (D) Immunoblot analysis of indicated proteins in sorted monocytes treated with indicated doses of S63845 for 24 hours. Data are shown as mean ± SEM. Mann-Whitney U test: *P < .05; **P < .01; ***P < .001. ns, not significant.
MAPK signaling pathway activation may contribute to resistance to apoptosis
To further explore the molecular mechanisms involved in monocyte resistance to apoptosis, we segregated a cohort of 19 patients with CMML into 2 groups based on the fraction of dead monocytes (>50% or ≤50%) after 24 hours in serum-containing medium. We used ERRBS to explore DNA methylation patterns in sorted monocytes collected from 10 patients with the most resistant cells (defective apoptosis) and 9 patients with less resistant cells (nondefective apoptosis) (supplemental Figure 3A; supplemental Table 2, cohort 8), showing a limited number of differentially methylated regions between the 2 groups (Figure 3A). Pathway analysis using Gene Ontology Molecular Function identified the mitogen-activated protein 3 kinase (MAP3K) pathway as the most significantly distinct between defective and nondefective CMML monocytes (Figure 3B). We also sequenced the RNA of these sorted monocytes, identifying only 24 differentially expressed genes (Figure 3C; supplemental Figure 3B; supplemental Table 2, cohort 8; supplemental Table 4). RT-qPCR analysis of samples used for RNA-seq (compared with 11 healthy donor monocytes) validated CYTL1 and EREG overexpression as well as ITGA2B, PTK2, and TUBB1 downregulation in CMML monocytes with defective apoptosis compared with the 2 other groups (supplemental Figure 3C). GLTSCR2 was the only gene whose deregulation in monocytes with defective apoptosis was not validated by RT-qPCR analysis (supplemental Figure 3C). We did not detect a significant overlap between differentially expressed genes and differentially methylated regions, even when focusing on topologically associating domains.

Overexpression of CYTL1 in CMML monocytes. (A) Volcano plot analysis of differentially methylated regions identified by ERRBS by comparing CMML monocytes with defective (n = 10) and nondefective (n = 9) apoptosis, based on their survival after 24 hours in serum-containing medium (>50% or ≤50%). (B) Gene Ontology (GO) pathway analysis of ERRBS data, identifying MAP3K as the most significant differentially regulated pathway. (C) Volcano plot analysis of differentially expressed genes identified by RNA-seq of samples analyzed in panel A. (D) Healthy donor (HD) monocytes were incubated for 24 hours in culture medium with 10% peripheral blood plasma collected from healthy donors (n = 4) or patients with CMML (n = 19). The fraction of surviving cells (AnV–/PI–) was normalized to that of control monocytes in serum-free medium. (E) CYTL1 gene expression measured by RT-qPCR in healthy donor and CMML monocytes, using RPL32 as a housekeeping gene. (F) CYTL1 gene expression analyzed in RNA-seq data from Franzini et al.20 Kruskal-Wallis test, Dunn’s multiple comparison: **P < .01. (G) CYTL1 protein level measured in the peripheral blood plasma of healthy donors and patients with CMML. Error bars are mean ± SEM. Mann-Whitney U test (D-E,G): *P < .05; **P < .01; ****P < .0001.
Overexpression of CYTL1 in CMML monocytes. (A) Volcano plot analysis of differentially methylated regions identified by ERRBS by comparing CMML monocytes with defective (n = 10) and nondefective (n = 9) apoptosis, based on their survival after 24 hours in serum-containing medium (>50% or ≤50%). (B) Gene Ontology (GO) pathway analysis of ERRBS data, identifying MAP3K as the most significant differentially regulated pathway. (C) Volcano plot analysis of differentially expressed genes identified by RNA-seq of samples analyzed in panel A. (D) Healthy donor (HD) monocytes were incubated for 24 hours in culture medium with 10% peripheral blood plasma collected from healthy donors (n = 4) or patients with CMML (n = 19). The fraction of surviving cells (AnV–/PI–) was normalized to that of control monocytes in serum-free medium. (E) CYTL1 gene expression measured by RT-qPCR in healthy donor and CMML monocytes, using RPL32 as a housekeeping gene. (F) CYTL1 gene expression analyzed in RNA-seq data from Franzini et al.20 Kruskal-Wallis test, Dunn’s multiple comparison: **P < .01. (G) CYTL1 protein level measured in the peripheral blood plasma of healthy donors and patients with CMML. Error bars are mean ± SEM. Mann-Whitney U test (D-E,G): *P < .05; **P < .01; ****P < .0001.
The same cutoff value (>50% or ≤50% monocyte survival after 24 hours in serum-containing medium) was applied to an extended cohort of patients and age-matched controls, indicating that the sensitivity to apoptosis of nondefective monocytes was similar to that of controls (supplemental Figure 3D). Adding 10% peripheral blood plasma collected from patients with CMML to culture medium could improve healthy donor monocyte survival, which was not observed with healthy donor plasma (Figure 3D; supplemental Table 2, cohort 9). This observation suggested that 1 or several soluble factors present in the plasma of patients with CMML could inhibit monocyte apoptosis. On the basis of this observation, we selected CYTL1 among differentially expressed genes between CMML monocytes with and without defective apoptosis (Figure 3C). CYTL1 encodes a small secreted protein that acts as a chemo-attractant activating the ERK pathway in human monocytes.13 By further exploring CYTL1 gene expression in an independent cohort of 35 patients with CMML (supplemental Table 2, cohort 10) compared with 19 healthy donors, we validated CYTL1 gene overexpression in patients with CMML compared with healthy donor monocytes (Figure 3E). CYTL1 was es-pecially, but not exclusively, associated with apoptosis resistance (supplemental Figure 3E). We also examined CYTL1 gene expression in a recently published20 cohort of monocytes sorted from the peripheral blood patients with CMML (n = 12) and young (n = 16) and age-matched (n = 12) healthy donors. Again, we observed CYTL1 overexpression of monocytes from patients with CMML compared with that of healthy donors of any age (Figure 3F). This overexpression was not observed in sorted CD34+ cells, plasmacytoid dendritic cells,21 granulocytes, and bone marrow mesenchymal stromal cells (not shown). In accordance with the overabundance of CYTL1 messenger RNA in CMML monocytes, CYTL1 protein measured by ELISA was significantly increased in the peripheral blood plasma of patients with CMML compared with healthy donors (Figure 3G; supplemental Table 2, cohort 11), and this was especially significant for patients with defective apoptosis (supplemental Figure 3F). CYTL1 gene expression in peripheral blood monocytes or CYTL1 protein plasma levels did not correlate with any clinical or biological CMML characteristics (not shown), indicating that CYTL1 overexpression may be independent of the other disease parameters.
CYTL1 protects human monocytes from apoptosis
We further investigated the upregulation of CYTL1 in monocytes of patients with CMML, and we analyzed ChIP-seq data obtained from sorted human monocytes (H3K27me3, H3K27ac, H3K4me1, H3K4me3). In healthy donor monocytes and 2 monocytes from patients with CMML, we also immunoprecipitated the EGR1 transcription factor, a well-identified target of phosphorylated ERK (p-ERK).22 The combined deposition of H3K4me1, H3K4me3, and H3K27ac suggested the presence of an active enhancer upstream of the CYTL1 gene to which EGR1 was recruited in CMML monocytes (Figure 4A), which was supported by genome-wide integration of enhancers and target genes in the framework of GeneCards (GH04J005032).23 ChIP-PCR using 2 distinct sets of primers further validated the enrichment of EGR1 to this enhancer in CMML samples with increased CYTL1 gene expression compared with healthy donor cells (Figure 4B). Small interfering RNA–mediated downregulation of EGR1, performed in U937 cells, decreased CYTL1 expression correlating with the decrease in EGR1 (supplemental Figure 4A). Exposure of healthy donor monocytes to 100 ng/mL rhCYTL1 induced the phosphorylation of ERK in a time- and dose-dependent manner (Figure 4C; supplemental 4B), and this effect could be prevented with a 30-minute preincubation and co-incubation with the CCR2 antagonist CAS 445479-97-0 (8 nM) (Figure 4C). Incubation of healthy donor monocytes in serum-free medium in the absence or presence of 100 ng/mL rhCYTL1 for 24 hours confirmed that this cytokine is capable of preventing apoptosis induced by serum deprivation (Figure 4D), even in low-binding plates that prevent their adhesion (supplemental Figure 4C). Addition of CAS 445479-97-0 (8 nM) or another CCR2 antagonist RS 504598 (10 µM) suppressed the cytoprotective effect of rhCYTL1 (Figure 4E). Altogether, these results suggest an autocrine or paracrine feedback loop in which CYTL1, through CCR2-mediated ERK phosphorylation, inhibits monocyte death accompanied by EGR1 recruitment to the CYTL1 gene enhancer. Expression of the CCL2 gene, which encodes another ligand for CCR2, was similar in healthy donor and CMML monocytes (supplemental Figure 4D-E).

Increased survival of monocytes through a CYTL1/CCR2/MEK pathway. (A) ChIP-seq analysis of the CYTL1 gene in healthy donor monocytes. H3K27me3, H3K4me1, H3K4me3, and H3K27ac ChIP-seq data were obtained in silico. ChIP-seq of EGR1 was performed in monocyte samples from 2 healthy donors and 2 patients with CMML. Dotted rectangle indicates a suspected gene enhancer, based on histone marks repartition. (B) ChIP-qPCR analysis of EGR1 enrichment on CYTL1 enhancer using 2 sets of primers (Enh Set 1 and Set 2) in CMML monocytes with low (similar to controls) or high (compared with control; see Figure 3E) CYTL1 gene expression; CMML#1 and CMML#2 shown in panel A. (C) Immunoblot analysis of indicated proteins (p-ERK: ERK phosphorylated on thr202 and tyr204) in cells treated with 100 ng/mL rhCYTL1 for 15 minutes or 8 nM CAS 445 for 30 minutes or both. Actin was used as a loading control. Lower panel: quantification of the above immunoblot. (D) Healthy donor monocytes were incubated for 24 hours with or without 100 ng/mL CYTL1 before measuring the surviving fraction of cells by flow cytometry (AnV–/PI–, results normalized to control monocytes). Mann-Whitney U test: ****P < .0001. (E) Healthy donor monocytes were incubated for 24 hours with 100 ng/mL CYTL1 in the absence or presence of CAS 445479-97-0 (8 nM) or RS 504393 (10 µM) before measuring the surviving fraction of cells by flow cytometry (AnV–/PI–, results normalized to CYTL1 alone). Error bars are mean ± SEM. Kruskal-Wallis test, Dunn’s multiple comparison: *P < .05; **P < .01. AU, arbitrary units; HD_bed, healthy donor browser extensible data; IgG, immunoglobulin G.
Increased survival of monocytes through a CYTL1/CCR2/MEK pathway. (A) ChIP-seq analysis of the CYTL1 gene in healthy donor monocytes. H3K27me3, H3K4me1, H3K4me3, and H3K27ac ChIP-seq data were obtained in silico. ChIP-seq of EGR1 was performed in monocyte samples from 2 healthy donors and 2 patients with CMML. Dotted rectangle indicates a suspected gene enhancer, based on histone marks repartition. (B) ChIP-qPCR analysis of EGR1 enrichment on CYTL1 enhancer using 2 sets of primers (Enh Set 1 and Set 2) in CMML monocytes with low (similar to controls) or high (compared with control; see Figure 3E) CYTL1 gene expression; CMML#1 and CMML#2 shown in panel A. (C) Immunoblot analysis of indicated proteins (p-ERK: ERK phosphorylated on thr202 and tyr204) in cells treated with 100 ng/mL rhCYTL1 for 15 minutes or 8 nM CAS 445 for 30 minutes or both. Actin was used as a loading control. Lower panel: quantification of the above immunoblot. (D) Healthy donor monocytes were incubated for 24 hours with or without 100 ng/mL CYTL1 before measuring the surviving fraction of cells by flow cytometry (AnV–/PI–, results normalized to control monocytes). Mann-Whitney U test: ****P < .0001. (E) Healthy donor monocytes were incubated for 24 hours with 100 ng/mL CYTL1 in the absence or presence of CAS 445479-97-0 (8 nM) or RS 504393 (10 µM) before measuring the surviving fraction of cells by flow cytometry (AnV–/PI–, results normalized to CYTL1 alone). Error bars are mean ± SEM. Kruskal-Wallis test, Dunn’s multiple comparison: *P < .05; **P < .01. AU, arbitrary units; HD_bed, healthy donor browser extensible data; IgG, immunoglobulin G.
S63845 synergizes with MEK inhibitors to restore CMML monocyte apoptosis
Knowing that CYTL1 is overproduced in CMML and activates the MAPK/ERK (MEK) pathway in monocytes,13 we explored the possibility that 2 US Food and Drug Administration–approved orally bioavailable MEK inhibitors, namely selumetinib and trametinib, could increase CMML monocyte susceptibility to the MCL1 inhibitor S63845. The 2 combinations (selumetinib plus S63845 or trametinib plus S63845) exhibited a synergistic ability to promote CMML monocyte apoptosis, which was more pronounced in CMML monocytes than in healthy donor monocytes (Figure 5A-B; supplemental Table 2, cohort 12). This effect could be prevented by Q-VD-OPh, suggesting caspase-dependent apoptosis, whereas necrostatin-1, which targets the pronecrotic kinase receptor interacting protein-1, did not show any protective activity (supplemental Figure 5A). Importantly, the 2 combinations, in addition to decreasing p-ERK, induced a decrease in MCL1 protein levels (Figure 5C-D) that was not observed with each of these drugs tested alone (Figure 5D-E). These drugs showed limited effects on the survival of enriched healthy donor and CMML CD34+ cells (supplemental Figure 5B).
![S63845/MEK inhibitor combination induced CMML monocyte apoptosis. (A-B) Sorted monocytes from healthy donor (gray) or patients with CMML (colors) were treated with 10 nM S63845, 100 nM AZD6244 (AZD, selumetinib) (A), 10 nM trametinib (TRAM) (B), or their combination for 24 hours before measuring the surviving fraction of cells (AnV–/PI–) by flow cytometry (results are normalized to cells treated with dimethyl sulfoxide [DMSO] alone). (C) Immunoblot analysis of indicated proteins in monocytes from patients with CMML treated for 24 hours as in panels A and B. (D) Quantification of the p-ERK/ERK and MCL1/ACTIN ratio using imageQuant LAS 4000 camera and ImageJ software on indicated numbers of immunoblots. (E) Immunoblot analysis of indicated proteins in monocytes from 4 patients with CMML treated for 4 hours as in panels A and B (using imageQuant LAS 4000 camera and ImageJ software). For multiple sample analyses, data are presented as mean ± SEM. Mann-Whitney U test: *P < .05; **P < .01; ***P < .001; ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/24/10.1182_blood.2020008729/2/m_bloodbld2020008729f5.png?Expires=1764970478&Signature=qOgNAOx40l-TOP3L7sR7Js1pvdIZ4UrwQN8jLekXVu3q9a736NwIKwCrt4cXuRyR2maqCKdg7UmLGjzqrlxaMgjlwsrY0FO5Zne4n3XBAqHgya~NPExR~ezi6R7c10piIJYPoVBzCZbIw3LGNIa54v-7JdUk1DUZL7NRUacw6mKudXhoBPli6iEKpiVZ-8hDfENC2~IhC-dl5xG4QlFnuOjXlb88XBDEn63h32PmloXZFiWDvm3q5X6PA0xeWQmRXgn6tlfvePOrLR~7jDylgcUmOSJiWxE1SX1G-FoV09sNP8CWHBjYvwk3J-BUb3Lvxh3VgPq3QvwPksTzD56-LA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
S63845/MEK inhibitor combination induced CMML monocyte apoptosis. (A-B) Sorted monocytes from healthy donor (gray) or patients with CMML (colors) were treated with 10 nM S63845, 100 nM AZD6244 (AZD, selumetinib) (A), 10 nM trametinib (TRAM) (B), or their combination for 24 hours before measuring the surviving fraction of cells (AnV–/PI–) by flow cytometry (results are normalized to cells treated with dimethyl sulfoxide [DMSO] alone). (C) Immunoblot analysis of indicated proteins in monocytes from patients with CMML treated for 24 hours as in panels A and B. (D) Quantification of the p-ERK/ERK and MCL1/ACTIN ratio using imageQuant LAS 4000 camera and ImageJ software on indicated numbers of immunoblots. (E) Immunoblot analysis of indicated proteins in monocytes from 4 patients with CMML treated for 4 hours as in panels A and B (using imageQuant LAS 4000 camera and ImageJ software). For multiple sample analyses, data are presented as mean ± SEM. Mann-Whitney U test: *P < .05; **P < .01; ***P < .001; ****P < .0001.
S63845/MEK inhibitor combination induced CMML monocyte apoptosis. (A-B) Sorted monocytes from healthy donor (gray) or patients with CMML (colors) were treated with 10 nM S63845, 100 nM AZD6244 (AZD, selumetinib) (A), 10 nM trametinib (TRAM) (B), or their combination for 24 hours before measuring the surviving fraction of cells (AnV–/PI–) by flow cytometry (results are normalized to cells treated with dimethyl sulfoxide [DMSO] alone). (C) Immunoblot analysis of indicated proteins in monocytes from patients with CMML treated for 24 hours as in panels A and B. (D) Quantification of the p-ERK/ERK and MCL1/ACTIN ratio using imageQuant LAS 4000 camera and ImageJ software on indicated numbers of immunoblots. (E) Immunoblot analysis of indicated proteins in monocytes from 4 patients with CMML treated for 4 hours as in panels A and B (using imageQuant LAS 4000 camera and ImageJ software). For multiple sample analyses, data are presented as mean ± SEM. Mann-Whitney U test: *P < .05; **P < .01; ***P < .001; ****P < .0001.
S63845 combined with MEK inhibitors decreases CMML cells in vivo
We used PDX models18,19 to test the efficacy of S63845 in combination with MEK inhibitors. PDXs of CMML hardly allow amplification through serial transplantation without oncogenic modification.24 First, PDXs generated from 2 patients (Figure 6A) were treated 7 weeks after intravenous injection of CD34+ cells with vehicle (n = 12) or intravenous S63845 (20 mg/kg) once per week and once-per-day oral AZD6244 (10 mg/kg) 5 days per week (n = 15) for 3 weeks (Figure 6B), which induced a significant decrease in splenic size (Figure 6B) and weight (Figure 6C), as well as in the total number of human CD45+ (hCD45+) and hCD45+CD14+ cells infiltrating the spleen and hCD45+ cells in the peripheral blood (Figure 6C). The drug combination did not affect mouse hemoglobin levels or white blood cell counts, but it significantly decreased the level of inflammatory human cytokines, including IL-1β, IL-6, IL-8, and TNF-α, in the peripheral blood (vehicle, n = 7; combination, n = 8) (Figure 6D). In independent experiments, mice intravenously injected with patient cells were treated with the combination of intravenous S63845 (once per week) or oral trametinib (1.0 mg/kg 5 days per week) or their combination for 3 weeks (Figure 6E; supplemental Figure 6). The combination induced a significant decrease in splenic weight and the total number of human cells infiltrating the spleen and in peripheral blood (Figure 6E). This PDX model mimics monocyte patient sensitivity to the tested drugs ex vivo, that is, S63845 efficacy was not increased in these patient cells by the combination with trametinib (not shown). Analysis of 2 additional PDX models showed a better efficacy of the combination in decreasing bone marrow hCD45+ cells (supplemental Figure 6). Together, these results demonstrate the ability of the MCL1/MEK inhibitor combination to reduce CMML leukemic cell burden in vivo.

The S63845/MEK inhibitor combination decreases leukemic infiltration in CMML PDX models. (A) Experimental plan of PDX models obtained by IV (i.v.) injection of 0.4 × 106 CD34+ sorted cells (B-C) or 3.5 × 106 bone marrow mononucleated cells (E) at 7 weeks before starting treatment with S63845 (20 mg/kg IV once per week for 3 weeks) and gavage with either selumetinib 10 mg/kg (B-C) or trametinib 1 mg/kg (E) 5 days per week for 3 weeks. (B) Photographs of spleen size in 4 mice treated with vehicle (Veh) or the S63845/selumetinib combo. (C) Impact of the S63845/selumetinib combo on spleen weight, the absolute number of human CD45+ and CD45+CD14+ cells detected in the spleen, and the number of human cells per μL of peripheral blood. Results of 2 independent PDX models are mixed (vehicle, n = 13; combo, n = 15). (D) Plasma level of indicated human cytokine measured in the peripheral blood plasma of mice treated as in panel C. (E) Impact of S63845, trametinib, and their combination (n = 8 per group) on spleen weight, the absolute number of human cells detected in the spleen or the bone marrow, and the number of human cells per μL of peripheral blood. Error bars are mean ± SEM. Mann-Whitney U test: *P < .05; **P < .01; ***P < .001. p.o., by mouth.
The S63845/MEK inhibitor combination decreases leukemic infiltration in CMML PDX models. (A) Experimental plan of PDX models obtained by IV (i.v.) injection of 0.4 × 106 CD34+ sorted cells (B-C) or 3.5 × 106 bone marrow mononucleated cells (E) at 7 weeks before starting treatment with S63845 (20 mg/kg IV once per week for 3 weeks) and gavage with either selumetinib 10 mg/kg (B-C) or trametinib 1 mg/kg (E) 5 days per week for 3 weeks. (B) Photographs of spleen size in 4 mice treated with vehicle (Veh) or the S63845/selumetinib combo. (C) Impact of the S63845/selumetinib combo on spleen weight, the absolute number of human CD45+ and CD45+CD14+ cells detected in the spleen, and the number of human cells per μL of peripheral blood. Results of 2 independent PDX models are mixed (vehicle, n = 13; combo, n = 15). (D) Plasma level of indicated human cytokine measured in the peripheral blood plasma of mice treated as in panel C. (E) Impact of S63845, trametinib, and their combination (n = 8 per group) on spleen weight, the absolute number of human cells detected in the spleen or the bone marrow, and the number of human cells per μL of peripheral blood. Error bars are mean ± SEM. Mann-Whitney U test: *P < .05; **P < .01; ***P < .001. p.o., by mouth.
Discussion
This study identifies an MCL1/MEK-dependent inhibition of monocyte apoptosis in CMML, which involves a CYTL1- and CCR2-mediated autocrine or paracrine loop. Simultaneous inactivation of MCL1 and MEK efficiently decreases leukemic burden and the generation of inflammatory cytokines in PDX models, suggesting a novel therapeutic approach to slow down CMML progression.
Monocytes that accumulate in patients with CMML are frequently addicted to MCL1, an anti-apoptotic protein of the BCL2 family that was initially detected in leukemic cells undergoing monocyte differentiation.25,26 Although MCL1 gene expression is increased in many tumor types,27,28 CMML monocytes that resist apoptosis do not show an overexpression of the gene or the protein, yet are more dependent on MCL1 than healthy donor monocytes. This resistance is not an intrinsic property of the classical monocyte subset whose increased fraction characterizes the disease and may be the consequence of genetic and epigenetic events involved in disease generation and progression.29
S63845 is a selective and potent small-molecule MCL1 antagonist that demonstrated impressive preclinical activity against selected tumors and little toxicity in mouse models.14 The activity of BH3 mimetics is commonly optimized by combining it with other agents.29-36 Analysis of DNA methylation patterns and gene expression in CMML monocyte samples pointed to MAP3K pathway activation in apoptosis-resistant cells, suggesting that MEK inhibitors could enforce the ability of MCL1 inhibitors to restore CMML monocyte apoptosis.
Among the genes whose expression was increased in apoptosis-resistant CMML monocytes, CYTL1 overexpression was validated in several independent cohorts of patients. The CYTL1 gene could be slightly more expressed in CMML compared with reactive monocytes but the small size of the cohort precluded any definitive conclusion. Initially cloned in human CD34+ cells,37 this conserved gene encodes a small secreted protein38 with a cytokine-like structure38,39 that resembles CCL2. CYTL1 could bind with high affinity to the receptor of CCL2 and other chemokines known as CCR2 at the surface of monocytes and macrophages.13 Although CMML monocytes do not overexpress the CCL2 gene, the cytokine can be increased in the plasma of patients with CMML40 and may further promote the resistance of CMML monocytes to apoptosis. Antagonizing CCR2 with inhibitors that are currently tested in the treatment of diabetes mellitus,41 cancer,42 and HIV1 infection43 could be an alternative to MEK inhibitors to promote the efficacy of MCL1 inhibition in CMML.
Increasing evidence indicates a causative role of inflammation in the pathogenesis of myeloid malignancies.44 Anti-inflammatory drugs targeting the IL6/STAT3 pathway inhibit the survival advantage provided by Tet2 gene deletion in mouse HSCs.6 In PDX models of CMML, the combination of MCL1 and MEK inhibitors decreased the secretion of human inflammatory cytokines IL-1β, IL-6, IL-8, and TNF-α, which are part of the inflammatory secretome identified in patients with CMML.40 Restoring apoptosis of CMML monocytes might inhibit the production of inflammatory cytokines that contribute to disease progression through interaction with normal and leukemic stem cells.20
Recovery of apoptosis could account for the clinical impact of hypomethylating agents that otherwise fail to decrease mutational burden.8 In contrast, hydroxyurea does not improve monocyte apoptosis, indicating a distinct mode of action compared with that of MCL1 inhibitors. Because Mcl1+/− heterozygous mice tolerate cytotoxic drugs at clinically relevant doses,45 the combination of MCL1 inhibitors with standard chemotherapy remains a therapeutic option.
Altogether, defective apoptosis in CMML monocytes involves MCL1 and MEK pathway activation through the cytokine-like CYTL1 interacting with CCR2 in an autocrine or paracrine loop. These results, which further validate the MEK pathway as a potential therapeutic target in CMML,24,46 suggest innovative therapeutic options to explore in this disease. As soon as ongoing trials can demonstrate the innocuousness of these drugs, such a combination will deserve to be clinically tested.
Data are available under accession number GSE165305 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE165305).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by a grant from Servier laboratories in the context of the Molecular Medicine in Oncology program led by Gustave Roussy Institute (Agence Nationale de la Recherche). E.S. and G.K. received funding from the Ligue contre le Cancer (Équipes Labellisées). E.S. received funding from the Fonds Amgen France pour la Science et l’Humain and the Institut National du Cancer (INCa). G.K. received funding from Agence National de la Recherche (ANR), Projets Blancs, Cancéropôle Ile-de-France, INCa, INSERM, Institut Universitaire de France, LabEx Immuno-Oncology (ANR-18-IDEX-0001), Recherche Hospitalo-Universitaire Torino Lumière, Site de Recherche Intégrée contre le Cancer (SIRIC) Stratified Oncology Cell DNA Repair and Tumor Immune Elimination, and SIRIC Cancer Research and Personalized Medicine. Several research groups are part of the IdEx Université de Paris ANR-18-IDEX-0001.
Authorship
Contribution: E.S. conceptualized the study; F.D., G.K., E.P., M.E.F., N.D., and E.S. created the methodology; M.S., F.D., S. Badel, M.M., H.L.N., M.T.-M., Q.Y., D.S.-B., O.K., and N.D. performed the investigation; L.L., M.T.-M., M.E.F., N.D., and E.S. performed the formal analysis; E.S. acquired the funding; B.B., O.W.-B., V.S., L.K.-B., S. Banquet, A.D., P.F., R.I., T.B., G.E., C.B., and S.T. provided various resources; M.S. and E.S. curated the data; E.S. validated and visualized the data and wrote the original draft of the manuscript; G.K., M.E.F., N.D., and E.S. wrote, reviewed, and edited the manuscript; and E.S. supervised the study.
Conflict-of-interest disclosure: L.K.-B., S. Banquet, and A.D. are employees of Servier Laboratories. The remaining authors declare no competing financial interests.
Correspondence: Eric Solary, INSERM U1287, Gustave Roussy Cancer Campus, 114 Rue Edouard Vaillant, 94805 Villejuif, France; e-mail: eric.solary@gustaveroussy.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal