

Key Points
Ibrutinib, obinutuzumab, and venetoclax combination is well tolerated and can be safely given in relapsed or untreated patients with MCL.
The triple combination provides durable complete molecular responses for treatment-naive or relapsed patients with high-risk genetics.

Abstract
Ibrutinib, obinutuzumab, and venetoclax demonstrate synergy in preclinical models of mantle cell lymphoma (MCL). OAsIs (NCT02558816), a single-arm multicenter prospective phase 1/2 trial, aimed to determine the maximum tolerated dose of venetoclax in combination with fixed doses of ibrutinib and obinutuzumab, in relapsed MCL patients. At the venetoclax MTD, extension cohorts were opened for relapsed and untreated patients. Safety and efficacy were secondary objectives. Minimal residual disease (MRD) was assessed by allele-specific oligonucleotide quantitative polymerase chain reaction. Between 14 October 2015 and 29 May 2018, 48 patients were enrolled. No dose-limiting toxicity was reported, and venetoclax at 400 mg per day was chosen for extension. Eighteen (75%) relapsed and 8 (53%) untreated patients experienced grade 3/4 adverse events. The complete response rate assessed by positron emission tomography at the end of cycle 6 was 67% in relapsed and 86.6% in untreated patients. MRD clearance for evaluable patients was seen in 71.5% of relapsed (10/14 patients) and 100% of untreated MRD-evaluable patients (n = 12) at the end of 3 cycles. The median follow-up for relapsed patients was 17 months (range, 10-35 months). The 2-year progression-free survival (PFS) was 69.5% (95% confidence interval [CI], 52.9%-91.4%) and 68.6% (95% CI, 49.5%-95.1%) for overall survival. The median follow-up was 14 months (range, 5-19) for untreated patients, the 1-year PFS was 93.3% (95% CI, 81.5%-100%). The combination of obinutuzumab, ibrutinib, and venetoclax is well tolerated and provides high response rates, including at the molecular level, in relapsed and untreated MCL patients. This trial was registered at www.clinicaltrials.gov as #NCT02558816.
Introduction
Mantle cell lymphoma (MCL) is a hematological malignancy that is characterized by recurrent genetic alterations that affect cell cycle, DNA damage, and cell survival pathways. B-cell receptor signaling via Bruton tyrosine kinase (BTK), together with microenvironmental cues that modulate BCL2-dependent survival pathways, have also been identified as key functional dependencies in MCL disease pathogenesis and as potential therapeutic targets.1
Newly diagnosed patients with MCL are commonly treated with immuno-chemotherapy plus rituximab maintenance as ongoing therapy of residual lymphoma B cells, with autologous stem-cell transplant consolidation in younger patients. However, most patients experience relapse and response duration decreases from one salvage therapy to the next.2-4
Ibrutinib is a first-in-class BTK inhibitor (BTKi) and has been approved in relapsed MCL, where an overall response rate of 68% was seen.5 However, the complete response rate was 23%, with a progression-free survival (PFS) and overall survival (OS) of 31% and 47% at 2 years, respectively. Resistance to BTKi can occur by diverse mechanisms, including acquired BTK mutations,6 activation of the alternative NF-κB pathway,7 activation of alternate kinase signaling networks,8 and in some instances metabolic reprogramming.9 Venetoclax is a Bcl-2 inhibitor that has shown single agent efficacy in MCL.10 Here again, resistance is encountered, in particular through overexpression of BCL-2 family proteins MCL-1 and BCL-xL via MCL microenvironmental cues.11 Other mechanisms of primary or acquired resistance to venetoclax have been described in MCL, including imbalance in BCL-2 protein expression.12,13 Dual targeting of BTK and BCL-2 signaling pathways by combination therapy with ibrutinib plus venetoclax has recently demonstrated high efficacy in relapsed MCL.14
Preclinical investigations in primary MCL cells have shown that microenvironment-dependent long-term expansion and drug resistance to venetoclax can be counteracted by obinutuzumab, a type II glycoengineered humanized anti-CD20 antibody.15 Mechanistically, this occurs through downregulation of BCL-xL expression via inhibition of both classical and alternative NF-κB signaling. These findings, together with reports describing favorable clinical results in chronic lymphocytic leukemia, provided a rationale for investigation of combination therapy by ibrutinib, obinutuzumab, and venetoclax in MCL. Treatment by obinutuzumab, ABT-199 (venetoclax) plus ibrutinib in relapsed/refractory MCL (OAsIs) trial was initiated to investigate the maximum tolerated dose (MTD) of venetoclax in combination with fixed doses of ibrutinib plus obinutuzumab and assess the tolerability, safety, and efficacy in both relapsed and untreated patients with MCL.
Methods
Study design and participants
The OAsIs study (NCT02558816) was a prospective, open-label, multicenter phase 1/2 trial done in 5 centers in France and 1 in the United Kingdom. OAsIs was designed to assess the safety, tolerability, and efficacy of ibrutinib, obinutuzumab, and venetoclax. Patients were enrolled at the time of relapse or progression (cohort A or B, respectively) or when newly diagnosed (cohort C). Full descriptions of treatment regimens are given in the supplemental Appendix (available on the Blood Web site). This was the first time that obinutuzumab plus ibrutinib had been used in MCL. Therefore, 9 relapsed patients were treated with fixed doses of ibrutinib (560 mg/d for ≥2 years or until progression) and obinutuzumab (1 g IV, cycle 1 days 1, 8, and 15; cycles 2-6 day 1; and every 2 months until cycle 24) to ensure safety (cohort A). Cohort A was completed before the second part of the trial (cohort B) for relapsed or refractory patients commenced. The primary objective of cohort B was to determine the MTD of venetoclax when combined with ibrutinib and obinutuzumab. Treatment consisted of fixed doses of both obinutuzumab (1 g IV cycle 1 days 1, 8, and 15; cycle 1b-8 day 1; and every 2 months until cycle 23) and ibrutinib (560 mg/d for ≥2 years) combined with escalating doses of venetoclax. There were 3 predetermined doses of venetoclax: 400 mg/d, 600 mg/d, and 800 mg/d. Cohort C was open to previously untreated MCL patients. This used the cohort B regimen, but with a fixed dose of venetoclax as defined from that part of the study and in accordance with the data and safety monitoring committee (DSMC) recommendation. Dose-limiting toxicities (DLTs) were assessed during the first cycle for cohort A and during cycle 2 for cohorts B and C.
To prevent tumor lysis syndrome (TLS), the protocol was amended to include an additional venetoclax ramp-up cycle, called cycle 1-b, where venetoclax was given as follows: cycle 1-b week 1, 20 mg/d; week 2, 50 mg/d; week 3, 100 mg/d; and week 4, 200 mg/d.14 At the start of cycle 2, venetoclax was further escalated as follows, C2W1 400 mg/d, C2W2 400, 600 or 800 mg/d. A risk stratification for TLS classified patients into 3 risk groups (supplemental Appendix, section S4).
Patients >18 years with either relapsed or recently diagnosed and previously untreated MCL were eligible for inclusion in the OAsIs trial. HIV-positive patients or patients presenting major comorbidities not related to lymphoma at diagnosis were excluded. The key eligibility criteria are listed in the supplemental Appendix. MCL diagnosis was established by local expert pathologists according to the World Health Organization classification.
All patients provided written informed consent. The study was performed according to the Declaration of Helsinki and approved by the following national competent authorities: Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM) (on 11 September 2015) plus National Ethics Committees (Comité de Protection des Personnes [CPP] Ouest VI) (18 August 2015) (France); Medicines & Healthcare products Regulatory Agency (MHRA)/Health Research Authority (HRA) (on 22 July 2016 and 07 October 2016) and Research Ethics Committee (REC) (18 July 2016) for (United Kingdom).
Procedures
At baseline, disease characteristics were assessed through clinical examination, standard biological parameters, bone marrow biopsy, computed tomography scan (neck, chest, abdomen and pelvis) and/or positron emission tomography scan. Response to treatment was assessed using clinical examination at each visit, computed tomography scan (Cheson 99 criteria), positron emission tomography scan (Lugano criteria) (supplemental Appendix) and at the molecular level (see below). The MCL international index (MIPI) was calculated at time of inclusion in the trial16 (Appendix).
Testing for minimal residual disease (MRD) was performed by allele-specific oligonucleotide quantitative polymerase chain reaction (ASO-qPCR) assays targeting either the clonal immunoglobulin H (IgH) rearrangement or the t(11;14)(q13;q32) translocation, designed to reach a sensitivity of 10−5, in blood and bone marrow.17,18 The local investigators were blinded to the MRD results. TP53 mutation status was assessed by targeted capture sequencing on pretreatment blood or bone marrow DNA with demonstrated clonal tumor B cells, as assessed by flow cytometry or ASO-qPCR. TP53 deletion status was assessed by array comparative genomic hybridization (CGH) on pretreatment blood or bone marrow DNA samples (500 ng DNA) when tumor B-cell infiltration was ≥20% (lower sensitivity limit of the assay), as assessed by either 4-color flow cytometry or ASO-qPCR.18 IGHV mutation status was assessed by sequencing of clonal framework region 1 IGH sequences and analysis for percent identity to the nearest germline VH sequence by using V-quest: >98% identity is scored as nonmutated IGHV.19
Outcomes
The primary end point was the occurrence of unacceptable toxicity defined as an adverse event (AE) or an abnormal laboratory value assessed as unrelated to disease progression, intercurrent illness, or concomitant medications occurring during the first cycle for cohort A or during cycle 2 for cohorts B and C. This had to meet any of the following criteria: grade 4 hematological toxicity for >10 days (except lymphopenia) or any grade ≥3 nonhematological toxicity lasting >10 days or leading to a treatment delay of >2 weeks, as defined in retreatment requirements. All grade 4 life-threatening events were considered as a DLT regardless of the duration of the event. Toxicity grades were classified according to the National Cancer Institute, Common Terminology Criteria for Adverse Events v 4.0. Overall response rate, complete response rate, partial response rate, OS, and PFS were secondary end points. Data were updated on 3 March 2020.
Statistical analyses
The aim of the trial was to assess the MTD of venetoclax when combined with fixed doses of ibrutinib and obinutuzumab. The MTD in cohort B was defined as the dose at which a DLT occurred in 33% of the patients during cycle 2. In the event of DLT, a modified continual reassessment method (CRM) was used to allocate doses of venetoclax from patient 1 to patient 12.20-22 We used a design with grouped inclusion of 3 patients per dose level (400, 600, and 800 mg/d). The model used for the CRM estimation was an exponential model. After assessment of MTD in cohort B, cohort C could be opened for inclusion using a fixed dose of venetoclax, according to the results from cohort B and following independent data monitoring committee review and approval. Twelve additional patients were enrolled in cohort B to assess safety and efficacy. We planned to enroll 48 patients (9 in cohort A, 24 in cohort B, and 15 in cohort C). Safety and efficacy analyses were conducted on all subjects receiving ≥1 dose of study treatment, and response rates were described by number and percent. PFS and OS were analyzed using the Kaplan-Meier method. All outputs were produced using R version 4.0.0. This trial was registered at www.clinicaltrials.gov as #NCT02558816.
Role of the funding source
Roche SAS supplied obinutuzumab, Janssen-Cilag supplied ibrutinib and AbbVie supplied venetoclax. Roche SAS, Janssen-Cilag and AbbVie funded the trial and contributed neither to protocol design, trial execution, data collection, data analysis nor writing of the present manuscript. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication. The trial was sponsored by the CHU de Nantes (Nantes, France).
Results
Characteristics of the patients
Forty-eight patients were enrolled in the study between 14 October 2015 and 29 May 2018. Patient characteristics at baseline according to each cohort are shown in Table 1. MIPI score at inclusion was high in 12 patients, including 4 of the 15 in cohort C. Screening for TP53 alterations was performed by NGS and/or array CGH in blood or bone marrow with demonstrable clonal B-cell infiltration for a total of 37 patients. Overall, 13 cases of TP53 alteration were identified of which 4 had biallelic alterations. IGHV mutation status was assessed in a total of 35 of 48 patients. In 25 of these 35 patients, the clonal IGHV rearrangement comprised an unmutated IGHV gene.
Patient characteristics at inclusion
| Cohort A: relapsed patients, ibrutinib and obinutuzumab (n = 9) | Cohort B: relapsed patients, ibrutinib, obinutuzumab, and venetoclax (n = 24) | Cohort C: untreated patients, ibrutinib, obinutuzumab, and venetoclax (n = 15) | |
|---|---|---|---|
| Age (y), median (range) | 64 (58-82) | 66 (45-76) | 65 (51-77) |
| Sex | |||
| Female | 3 (33) | 5 (21) | 9 (60) |
| Male | 6 (67) | 19 (79) | 6 (40) |
| No. of previous lines, median (range) | 1 (1, 4) | 1 (1, 3) | — |
| Median time from previous line of treatment (months) | 31 (1-65) | 24 (1-172) | — |
| Prior stem-cell transplantation | |||
| Yes | 7 (78) | 13 (54) | — |
| Stage of MCL at inclusion | |||
| II | 1 (11) | 3 (12) | 0 (0) |
| III-IV | 8 (89) | 21 (88) | 15 (100) |
| Bone marrow involved | |||
| No | 4 (44) | 13 (59) | 7 (47) |
| Yes | 5 (56) | 9 (41) | 8 (53) |
| Missing | — | 2 | — |
| MIPI score at inclusion | |||
| High risk | 1 (11) | 7 (29) | 4 (27) |
| Intermediate risk | 4 (44) | 8 (33) | 11 (73) |
| Low risk | 4 (44) | 9 (38) | 0 (0) |
| MIPIb score at inclusion | |||
| High risk | 5 (62%) | 11 (61%) | 6 (55%) |
| Intermediate risk | 3 (38%) | 7 (39%) | 5 (45%) |
| Missing | 1 | 6 | 4 |
| Tumor size >5 cm | |||
| No | 5 (56) | 15 (62) | 9 (60) |
| Yes | 4 (44) | 9 (38) | 6 (40) |
| ECOG performance-status | |||
| 0-1 | 8 (89) | 22 (92) | 15 (100) |
| 2 | 1 (11) | 2 (8) | 0 (0) |
| Blastic or pleomorphic | |||
| Blastic | 2 (22) | 4 (17) | 0 (0) |
| Pleomorphic MCL | 0 (0) | 1 (4) | 1 (7) |
| Missing | 0 (0) | 1 (4) | 0 (0) |
| Cytogenetic and molecular features | |||
| TP53 mutated (NGS) | 1 (11) | 5 (21) | 2 (13) |
| Not evaluable* | 2 | 7 | 1 |
| 17p deletion | 1 (11) | 2 (8) | 6 (40) |
| Not evaluable* | 5 | 18 | 5 |
| IGHV mutated (NGS) | 1 (11) | 7 (29) | 2 (13) |
| Not evaluable* | 2 | 8 | 3 |
| Cohort A: relapsed patients, ibrutinib and obinutuzumab (n = 9) | Cohort B: relapsed patients, ibrutinib, obinutuzumab, and venetoclax (n = 24) | Cohort C: untreated patients, ibrutinib, obinutuzumab, and venetoclax (n = 15) | |
|---|---|---|---|
| Age (y), median (range) | 64 (58-82) | 66 (45-76) | 65 (51-77) |
| Sex | |||
| Female | 3 (33) | 5 (21) | 9 (60) |
| Male | 6 (67) | 19 (79) | 6 (40) |
| No. of previous lines, median (range) | 1 (1, 4) | 1 (1, 3) | — |
| Median time from previous line of treatment (months) | 31 (1-65) | 24 (1-172) | — |
| Prior stem-cell transplantation | |||
| Yes | 7 (78) | 13 (54) | — |
| Stage of MCL at inclusion | |||
| II | 1 (11) | 3 (12) | 0 (0) |
| III-IV | 8 (89) | 21 (88) | 15 (100) |
| Bone marrow involved | |||
| No | 4 (44) | 13 (59) | 7 (47) |
| Yes | 5 (56) | 9 (41) | 8 (53) |
| Missing | — | 2 | — |
| MIPI score at inclusion | |||
| High risk | 1 (11) | 7 (29) | 4 (27) |
| Intermediate risk | 4 (44) | 8 (33) | 11 (73) |
| Low risk | 4 (44) | 9 (38) | 0 (0) |
| MIPIb score at inclusion | |||
| High risk | 5 (62%) | 11 (61%) | 6 (55%) |
| Intermediate risk | 3 (38%) | 7 (39%) | 5 (45%) |
| Missing | 1 | 6 | 4 |
| Tumor size >5 cm | |||
| No | 5 (56) | 15 (62) | 9 (60) |
| Yes | 4 (44) | 9 (38) | 6 (40) |
| ECOG performance-status | |||
| 0-1 | 8 (89) | 22 (92) | 15 (100) |
| 2 | 1 (11) | 2 (8) | 0 (0) |
| Blastic or pleomorphic | |||
| Blastic | 2 (22) | 4 (17) | 0 (0) |
| Pleomorphic MCL | 0 (0) | 1 (4) | 1 (7) |
| Missing | 0 (0) | 1 (4) | 0 (0) |
| Cytogenetic and molecular features | |||
| TP53 mutated (NGS) | 1 (11) | 5 (21) | 2 (13) |
| Not evaluable* | 2 | 7 | 1 |
| 17p deletion | 1 (11) | 2 (8) | 6 (40) |
| Not evaluable* | 5 | 18 | 5 |
| IGHV mutated (NGS) | 1 (11) | 7 (29) | 2 (13) |
| Not evaluable* | 2 | 8 | 3 |
ECOG, Eastern Cooperative Oncology Group; MIPIb, biologic Mantle Cell Lymphoma International Prognostic Index; NGS, next generation sequencing.
Not evaluated; IGHV mutation status and TP53 screening by NGS is assessed in blood and/or bone marrow only if clonal B cells are detectable by flow or PCR. Assessment is not evaluated if no clonal infiltration; array CGH is performed only when patients show >20% infiltration by flow the sample (blood and/or bone marrow) and is not evaluated if <20% infiltration.
Safety
Since no DLT was observed in cohort A (n = 9), cohort B was opened. The first 6 patients who received the triple combination (3 at venetoclax 400 mg/d and 3 at venetoclax 600 mg/d) did not experience any DLT. Among the first 3 patients who received venetoclax at 800 mg/d, 1 was not eligible for toxicity assessment because of grade 3 neutropenia that occurred during cycle 1, before the patient started venetoclax. Following DSMC recommendation, 3 additional patients were enrolled at that dose level. None experienced DLT. The CRM model was thus not needed. Following DSMC recommendation (see “Discussion”), a dose of venetoclax 400 mg/d, together with obinutuzumab and ibrutinib, was chosen to complete the trial. Cohort C was then opened for untreated patients (n = 15), and 12 additional patients were enrolled in cohort B, as preplanned. Thus, 24 relapsed patients were enrolled in cohort B, including 15 patients who received venetoclax at 400 mg/d.
No DLT was reported in any cohort. The most frequent unexpected AEs and serious AEs (SAEs) between cycles 1 and 6 are shown in Table 2. Eight (89%) patients in cohort A, 18 (75%) patients in cohort B, and 8 (53%) patients in cohort C had grade 3 or higher AEs during this period. The most frequent grade 3 or 4 AEs in all cohorts were thrombocytopenia and neutropenia. Grade 3 or 4 thrombocytopenia was only reported in relapsed patients, as was neutropenia grade 3. Two patients in cohort A and 2 in cohort B received blood transfusions between cycles 1 and 6. One patient in cohort A, 5 in cohort B, and no patients in cohort C required platelet transfusions. One patient (cohort B, cycle 5) presented with grade 3 atrial fibrillation. One biological TLS grade 3 (cohort A, cycle 1) and 1 biological TLS grade 1 (cohort C, cycle 1) occurred. Three patients in cohort A, 8 in cohort B, and 2 in cohort C received <90% of planned treatment (see supplemental Appendix). In brief, ibrutinib was temporally stopped or dose reduced because of toxicity in 3 patients in cohort A (neutropenia in 2 patients and chronic graft-versus-host disease [cGVHD] in 1 patient), 5 patients in cohort B (thrombocytopenia in 2 cases, palpitation and atrial fibrillation in 1 patient, diarrhea in 2 cases, and neutropenia in 2 cases), and 2 patients in cohort C (rash in 1 case and alanine aminotransferase increase in 1 case). Venetoclax was temporally stopped or dose reduced only in cohort B (neutropenia in 4 cases).
AEs and SAEs from C1 to C6
| Cohort A Relapsed patients, ibrutinib, obinutuzumab (n = 9) | Cohort B Relapsed patients, ibrutinib, obinutuzumab, venetoclax (n = 24) | Cohort C Untreated patients, ibrutinib, obinutuzumab, venetoclax (n = 15) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Grade 1-2 (>20% patients) | All grade 3 | All grade 4 | Grade 1-2 (>20% patients) | All grade 3 | All grade 4 | Grade 1-2 (>20% patients) | All grade 3 | All grade 4 | |
| Any AE | 9 (100) | 8 (89) | 3 (33) | 23 (96) | 17 (71) | 12 (50) | 15 (100) | 5 (33) | 3 (20) |
| Thrombocytopenia | 8 (89) | 2 (22) | 0 | 11 (46) | 10 (42) | 3 (12) | 3 (20) | 0 | 0 |
| Neutropenia | 0 | 5 (56) | 3 (33) | 4 (17) | 9 (38) | 8 (33) | 3 (20) | 2 (13) | 1 (7) |
| Musculoskeletal pain | 6 (67) | 0 | 0 | 10 (42) | 0 | 0 | 5 (33) | 0 | 0 |
| Diarrhea | 1 (11) | 0 | 0 | 7 (29) | 3 (12) | 0 | 6 (40) | 0 | 0 |
| Nausea | 4 (44) | 0 | 0 | 7 (29) | 0 | 0 | 3 (20) | 0 | 0 |
| Headache | 3 (33) | 0 | 0 | 8 (33) | 0 | 0 | 3 (20) | 0 | 0 |
| Upper respiratory infection | 5 (56) | 0 | 0 | 4 (17) | 0 | 0 | 3 (20) | 0 | 0 |
| Asthenia | 2 (22) | 0 | 0 | 5 (21) | 0 | 0 | 4 (27) | 0 | 0 |
| Pyrexia | 3 (33) | 0 | 0 | 5 (21) | 0 | 0 | 2 (13) | 0 | 0 |
| Lymphopenia | 0 | 2 (22) | 1 (11) | 1 (4) | 3 (12) | 3 (12) | 0 | 1 (7) | 0 |
| Hypokalemia | 2 (22) | 1 (11) | 0 | 5 (21) | 0 | 0 | 1 (7) | 0 | 0 |
| Hypophosphatemia | 0 | 3 (33) | 1 (11) | 0 | 5 (21) | 2 (8) | 0 | 0 | 0 |
| Epistaxis | 1 (11) | 0 | 0 | 5 (21) | 0 | 0 | 0 | 0 | 0 |
| Infusion-related reaction | 1 (11) | 0 | 0 | 2 (8) | 1 (4) | 0 | 2 (13) | 0 | 0 |
| Upper respiratory disorders | 3 (33) | 0 | 0 | 2 (8) | 0 | 0 | 1 (7) | 0 | 0 |
| Rash | 1 (11) | 0 | 0 | 4 (17) | 0 | 0 | 1 (7) | 1 (7) | 0 |
| Gastroesophageal reflux | 2 (22) | 0 | 0 | 2 (8) | 0 | 0 | 1 (7) | 0 | 0 |
| Discomfort | 3 (33) | 0 | 0 | 2 (8) | 0 | 0 | 0 | 0 | 0 |
| Neuropathy, peripheral | 2 (22) | 0 | 0 | 1 (4) | 0 | 0 | 2 (13) | 0 | 0 |
| Hypertension | 1 (11) | 0 | 0 | 2 (8) | 1 (4) | 0 | 1 (7) | 0 | 0 |
| Anemia | 2 (22) | 0 | 0 | 1 (4) | 2 (8) | 0 | 0 | 0 | 0 |
| Abdominal pain, upper | 3 (33) | 0 | 0 | 0 | 0 | 0 | 1 (7) | 0 | 0 |
| Vomiting | 2 (22) | 0 | 0 | 2 (8) | 0 | 0 | 0 | 0 | 0 |
| Edema, peripheral | 4 (44) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Bronchitis/lung infection | 2 (22) | 0 | 0 | 2 (8) | 0 | 0 | 0 | 0 | 0 |
| Constipation | 2 (22) | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 | 0 |
| GGT increased | 0 | 0 | 0 | 1 (4) | 2 (8) | 0 | 0 | 0 | 0 |
| ALAT increased | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 | 1 (7) |
| ASAT increased | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 1 (7) | 0 |
| Hepatic cytolysis | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 | 1 (7) |
| Soft tissue infection | 2 (22) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hyperglycemia | 1 (11) | 2 (22) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hyperkalemia | 1 (11) | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| Hyperuricemia | 1 (11) | 1 (11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Tumor compression | 0 | 1 (11) | 0 | 1 (4) | 0 | 0 | 0 | 0 | 0 |
| Hyperlymphocytosis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (7) | 0 |
| Alkaline phosphatase increased | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| cGVHD | 0 | 1 (11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Malnutrition | 0 | 1 (11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Any SAE | 0 | 3 (33) | 0 | 1 (4) | 4 (17) | 1 (4) | 2 (13) | 1 (7) | 0 |
| Febrile neutropenia | 0 | 1 (11) | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| TLS | 0 | 1 (11) | 0 | 0 | 0 | 0 | 1 (7) | 0 | 0 |
| Neutropenia | 0 | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 |
| Atrial fibrillation | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| Palpitations | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| Pyrexia | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 | 0 |
| Appendicitis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (7) | 0 |
| Neutropenic sepsis | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| Staphylococcal sepsis | 0 | 1 (11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Urosepsis | 0 | 1 (11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Gouty arthritis | 0 | 1 (11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Basal cell carcinoma | 0 | 0 | 0 | 0 | 0 | 0 | 1 (7) | 0 | 0 |
| Squamous cell carcinoma of the tongue | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| Lung disorder | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| Pleural effusion | 0 | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 |
| Cohort A Relapsed patients, ibrutinib, obinutuzumab (n = 9) | Cohort B Relapsed patients, ibrutinib, obinutuzumab, venetoclax (n = 24) | Cohort C Untreated patients, ibrutinib, obinutuzumab, venetoclax (n = 15) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Grade 1-2 (>20% patients) | All grade 3 | All grade 4 | Grade 1-2 (>20% patients) | All grade 3 | All grade 4 | Grade 1-2 (>20% patients) | All grade 3 | All grade 4 | |
| Any AE | 9 (100) | 8 (89) | 3 (33) | 23 (96) | 17 (71) | 12 (50) | 15 (100) | 5 (33) | 3 (20) |
| Thrombocytopenia | 8 (89) | 2 (22) | 0 | 11 (46) | 10 (42) | 3 (12) | 3 (20) | 0 | 0 |
| Neutropenia | 0 | 5 (56) | 3 (33) | 4 (17) | 9 (38) | 8 (33) | 3 (20) | 2 (13) | 1 (7) |
| Musculoskeletal pain | 6 (67) | 0 | 0 | 10 (42) | 0 | 0 | 5 (33) | 0 | 0 |
| Diarrhea | 1 (11) | 0 | 0 | 7 (29) | 3 (12) | 0 | 6 (40) | 0 | 0 |
| Nausea | 4 (44) | 0 | 0 | 7 (29) | 0 | 0 | 3 (20) | 0 | 0 |
| Headache | 3 (33) | 0 | 0 | 8 (33) | 0 | 0 | 3 (20) | 0 | 0 |
| Upper respiratory infection | 5 (56) | 0 | 0 | 4 (17) | 0 | 0 | 3 (20) | 0 | 0 |
| Asthenia | 2 (22) | 0 | 0 | 5 (21) | 0 | 0 | 4 (27) | 0 | 0 |
| Pyrexia | 3 (33) | 0 | 0 | 5 (21) | 0 | 0 | 2 (13) | 0 | 0 |
| Lymphopenia | 0 | 2 (22) | 1 (11) | 1 (4) | 3 (12) | 3 (12) | 0 | 1 (7) | 0 |
| Hypokalemia | 2 (22) | 1 (11) | 0 | 5 (21) | 0 | 0 | 1 (7) | 0 | 0 |
| Hypophosphatemia | 0 | 3 (33) | 1 (11) | 0 | 5 (21) | 2 (8) | 0 | 0 | 0 |
| Epistaxis | 1 (11) | 0 | 0 | 5 (21) | 0 | 0 | 0 | 0 | 0 |
| Infusion-related reaction | 1 (11) | 0 | 0 | 2 (8) | 1 (4) | 0 | 2 (13) | 0 | 0 |
| Upper respiratory disorders | 3 (33) | 0 | 0 | 2 (8) | 0 | 0 | 1 (7) | 0 | 0 |
| Rash | 1 (11) | 0 | 0 | 4 (17) | 0 | 0 | 1 (7) | 1 (7) | 0 |
| Gastroesophageal reflux | 2 (22) | 0 | 0 | 2 (8) | 0 | 0 | 1 (7) | 0 | 0 |
| Discomfort | 3 (33) | 0 | 0 | 2 (8) | 0 | 0 | 0 | 0 | 0 |
| Neuropathy, peripheral | 2 (22) | 0 | 0 | 1 (4) | 0 | 0 | 2 (13) | 0 | 0 |
| Hypertension | 1 (11) | 0 | 0 | 2 (8) | 1 (4) | 0 | 1 (7) | 0 | 0 |
| Anemia | 2 (22) | 0 | 0 | 1 (4) | 2 (8) | 0 | 0 | 0 | 0 |
| Abdominal pain, upper | 3 (33) | 0 | 0 | 0 | 0 | 0 | 1 (7) | 0 | 0 |
| Vomiting | 2 (22) | 0 | 0 | 2 (8) | 0 | 0 | 0 | 0 | 0 |
| Edema, peripheral | 4 (44) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Bronchitis/lung infection | 2 (22) | 0 | 0 | 2 (8) | 0 | 0 | 0 | 0 | 0 |
| Constipation | 2 (22) | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 | 0 |
| GGT increased | 0 | 0 | 0 | 1 (4) | 2 (8) | 0 | 0 | 0 | 0 |
| ALAT increased | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 | 1 (7) |
| ASAT increased | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 1 (7) | 0 |
| Hepatic cytolysis | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 | 1 (7) |
| Soft tissue infection | 2 (22) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hyperglycemia | 1 (11) | 2 (22) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hyperkalemia | 1 (11) | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| Hyperuricemia | 1 (11) | 1 (11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Tumor compression | 0 | 1 (11) | 0 | 1 (4) | 0 | 0 | 0 | 0 | 0 |
| Hyperlymphocytosis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (7) | 0 |
| Alkaline phosphatase increased | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| cGVHD | 0 | 1 (11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Malnutrition | 0 | 1 (11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Any SAE | 0 | 3 (33) | 0 | 1 (4) | 4 (17) | 1 (4) | 2 (13) | 1 (7) | 0 |
| Febrile neutropenia | 0 | 1 (11) | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| TLS | 0 | 1 (11) | 0 | 0 | 0 | 0 | 1 (7) | 0 | 0 |
| Neutropenia | 0 | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 |
| Atrial fibrillation | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| Palpitations | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| Pyrexia | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 | 0 |
| Appendicitis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (7) | 0 |
| Neutropenic sepsis | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| Staphylococcal sepsis | 0 | 1 (11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Urosepsis | 0 | 1 (11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Gouty arthritis | 0 | 1 (11) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Basal cell carcinoma | 0 | 0 | 0 | 0 | 0 | 0 | 1 (7) | 0 | 0 |
| Squamous cell carcinoma of the tongue | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| Lung disorder | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 0 |
| Pleural effusion | 0 | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 |
All grade 1 and 2 AEs reported in ≥20% of patients in each cohort (≥2 patients in cohort A, ≥5 patients in cohort B, and ≥3 patients in cohort C); all grade 3 and 4 events.
ALAT, alanine aminotransferase; ASAT, aspartate aminotransferase; GGT, γ-glutamyl-transferase.
At the time of the present analysis, 3 patients stopped treatment because of AE after cycle 6. One patient in cohort B (cycle 18) stopped treatment because of grade 3 thrombocytopenia and neutropenia. One patient stopped treatment because of acute peripheral neuropathy (cohort C), which led to treatment discontinuation after cycle 9. This patient was in complete remission (CR) at the molecular level at the end of cycle 6 and still remains in CR (6 months after end of treatment). One allografted patient stopped treatment because of cGVHD that led to ibrutinib discontinuation. Controlled grade 1 cGVHD was preexisting at the time of inclusion and progressed to grade 3 after cycle 1, when the patient had only received obinutuzumab and ibrutinib. Upon stopping ibrutinib, cGVHD returned to grade 1 but returned to grade 3 when ibrutinib was restarted. Two patients in cohort B presented atrial fibrillation, 1 at cycle 10 (grade 2) and 1 at cycle 12 (grade 1). These last 2 patients remained on therapy. All AEs and SAEs and treatment compliance are presented in the supplemental Appendix.
Responses and patient outcome
Responses according to Cheson 99 criteria, Lugano criteria, and MRD status
| Cohort A: relapsed patients, ibrutinib and obinutuzumab (n = 9) | Cohort B: relapsed patients, ibrutinib, obinutuzumab, and venetoclax (n = 24) | Cohort C: untreated patients, ibrutinib, obinutuzumab, and venetoclax (n = 15) | |
|---|---|---|---|
| Response at end of cycle 2 (Cheson 99) | |||
| CR/CRu | 3 (34) | 9 (38) | 8 (53) |
| PR | 2 (22) | 11 (46) | 7 (47) |
| SD | 2 (22) | 1 (4) | — |
| PD | 1 (11) | 3 (12) | — |
| Missing | 1 (11) | — | — |
| Response at end of cycle 4 (Cheson 99) | |||
| CR/CRu | 4 (44) | 15 (62) | 12 (80) |
| PR | 3 (34) | 4 (17) | 2 (13) |
| SD | 1 (11) | 1 (4) | — |
| PD | 1 (11) | 4 (17) | 1 (7) |
| Response at end of cycle 6 (Cheson 99) | |||
| CR/CRu | 7 (78) | 16 (67) | 12 (80) |
| PR | 0 (0) | 2 (8) | 2 (13) |
| SD | 1 (11) | — | — |
| PD | 1 (11) | 4 (17) | 1 (7) |
| Not done | — | 2 (8)† | — |
| Response at end of cycle 6 (Lugano) | |||
| CR | 7 (78) | 16 (67) | 13 (86) |
| PR | 1 (11) | 1 (4) | 1 (7) |
| PD | 1 (11) | 5 (21) | 1 (7) |
| Not done | — | 2 (8)† | — |
| MRD in blood at end of cycle 3 | |||
| Negative | 4 (44) | 10 (42) | 12 (80) |
| Positive | 2 (22) | 4 (17) | — |
| Not evaluable for MRD* | 2 (22) | 7 (29) | 3 (20) |
| Progression before end of cycle 3 | 1 (11) | 3 (12) | — |
| MRD in blood at end of cycle 6 | |||
| Negative | 4 (44) | 10 (42) | 11 (73) |
| Positive | 2 (22) | 3 (12) | — |
| Not evaluable for MRD* | 2 (22) | 7 (29)† | 2 (13) |
| Progression before end cycle 6 | 1 (11) | 4 (17) | 1 (7) |
| Missing sample | — | — | 1 (7) |
| MRD in BM at end of cycle 6 | |||
| Negative | 4 (44) | 9 (38) | 10 (67) |
| Positive | 1 (11) | 3 (12) | — |
| Not evaluable for MRD* | 2 (22) | 7 (29)† | 2 (13) |
| Progression before end cycle 6 | 1 (11) | 4 (17) | 1 (7) |
| Missing sample | 1 (11) | 1 (4) | 2 (13) |
| Cohort A: relapsed patients, ibrutinib and obinutuzumab (n = 9) | Cohort B: relapsed patients, ibrutinib, obinutuzumab, and venetoclax (n = 24) | Cohort C: untreated patients, ibrutinib, obinutuzumab, and venetoclax (n = 15) | |
|---|---|---|---|
| Response at end of cycle 2 (Cheson 99) | |||
| CR/CRu | 3 (34) | 9 (38) | 8 (53) |
| PR | 2 (22) | 11 (46) | 7 (47) |
| SD | 2 (22) | 1 (4) | — |
| PD | 1 (11) | 3 (12) | — |
| Missing | 1 (11) | — | — |
| Response at end of cycle 4 (Cheson 99) | |||
| CR/CRu | 4 (44) | 15 (62) | 12 (80) |
| PR | 3 (34) | 4 (17) | 2 (13) |
| SD | 1 (11) | 1 (4) | — |
| PD | 1 (11) | 4 (17) | 1 (7) |
| Response at end of cycle 6 (Cheson 99) | |||
| CR/CRu | 7 (78) | 16 (67) | 12 (80) |
| PR | 0 (0) | 2 (8) | 2 (13) |
| SD | 1 (11) | — | — |
| PD | 1 (11) | 4 (17) | 1 (7) |
| Not done | — | 2 (8)† | — |
| Response at end of cycle 6 (Lugano) | |||
| CR | 7 (78) | 16 (67) | 13 (86) |
| PR | 1 (11) | 1 (4) | 1 (7) |
| PD | 1 (11) | 5 (21) | 1 (7) |
| Not done | — | 2 (8)† | — |
| MRD in blood at end of cycle 3 | |||
| Negative | 4 (44) | 10 (42) | 12 (80) |
| Positive | 2 (22) | 4 (17) | — |
| Not evaluable for MRD* | 2 (22) | 7 (29) | 3 (20) |
| Progression before end of cycle 3 | 1 (11) | 3 (12) | — |
| MRD in blood at end of cycle 6 | |||
| Negative | 4 (44) | 10 (42) | 11 (73) |
| Positive | 2 (22) | 3 (12) | — |
| Not evaluable for MRD* | 2 (22) | 7 (29)† | 2 (13) |
| Progression before end cycle 6 | 1 (11) | 4 (17) | 1 (7) |
| Missing sample | — | — | 1 (7) |
| MRD in BM at end of cycle 6 | |||
| Negative | 4 (44) | 9 (38) | 10 (67) |
| Positive | 1 (11) | 3 (12) | — |
| Not evaluable for MRD* | 2 (22) | 7 (29)† | 2 (13) |
| Progression before end cycle 6 | 1 (11) | 4 (17) | 1 (7) |
| Missing sample | 1 (11) | 1 (4) | 2 (13) |
BM, bone marrow; CRu, complete remission unconfirmed; PD; progression disease; PR, partial response; SD, stable disease.
Not evaluable; MRD assays are performed only in patients with clonal disease that is detectable by ASO-qPCR to sensitivity of 10−4/10−5. Assay is not evaluated if this is not possible.
Two patients were allografted before cycle 6.
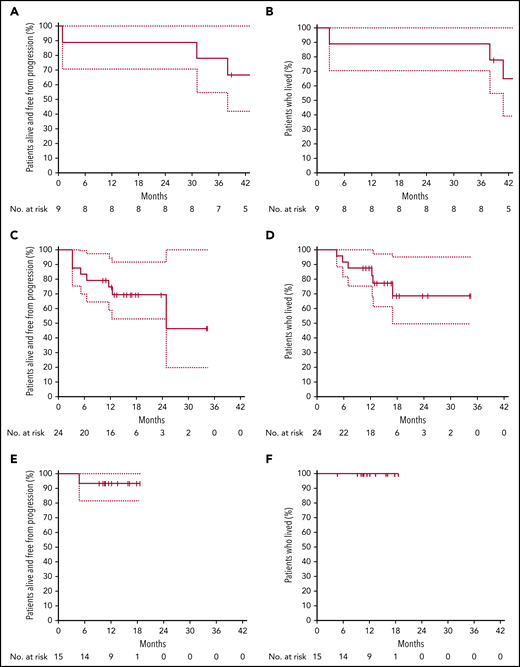
PFS and OS according to cohort. PFS (A) and OS (B) in cohort A (ibrutinib plus obinutuzumab), relapse patients. PFS (C) and OS (D) in cohort B (ibrutinib, obinutuzumab, and venetoclax), relapse patients. PFS (E) and OS (F) in cohort C (ibrutinib, obinutuzumab plus venetoclax), untreated patients.
PFS and OS according to cohort. PFS (A) and OS (B) in cohort A (ibrutinib plus obinutuzumab), relapse patients. PFS (C) and OS (D) in cohort B (ibrutinib, obinutuzumab, and venetoclax), relapse patients. PFS (E) and OS (F) in cohort C (ibrutinib, obinutuzumab plus venetoclax), untreated patients.
Relapsed patients treated with ibrutinib plus obinutuzumab (cohort A)
Of the 9 patients treated, 7 achieved CR, according to Cheson or Lugano criteria, at the end of cycle 6. Six patients completed the 24 cycles of treatment, while 3 had discontinued therapy (progression of disease in 1 patient who was TP53 wild-type and cGVHD reactivation in 1 patient; 1 patient underwent allograft upon the local investigator’s decision). One out of 2 patients with blastoid variant reached CR at the end of cycle 6 (this patient is alive and in CR at last contact), and 1 died because of progression. With a median follow-up of 45 months range, 39-49 months), the 1- and 2-year PFS and OS were 89% (95% confidence interval [CI], 70.6%-100.0%), respectively. Median duration of response was not reached.
Relapsed patients treated with triple combination (cohort B) (n = 24)
After completing 2 cycles of therapy, 20 patients (84%) had an objective response, including 9 CR/CRu according to Cheson criteria. At the end of cycle 6, 16 patients (67%) were in CR/CRu, and 4 had progressed, one of whom was TP53 mutated (others were not informative for TP53 status). Two out of 4 patients with blastoid variant reached CR at end of cycle 6, 1 underwent allograft after cycle 4, and 1 progressed according to Lugano criteria. Three out of 4 patients with blastoid variant were alive and in CR at last contact. One patient with pleomorphic variant progressed at cycle 2. At the last follow-up, 3 patients have completed the full program and 12 have discontinued therapy. The reasons for treatment discontinuation were progression of disease (7 patients), investigator decision (3 patients) (all 3 underwent allograft), AE (1 patient) (thrombocytopenia and neutropenia grade 3), and treatment of an unrelated solid tumor (which, after review, was present at the time of inclusion). Nine patients (venetoclax 400 mg/d) remain on the protocol. At the time of this analysis, 18 patients were alive, and all were disease-free. With a median follow-up of 17 months (range, 10-35 months), the 1-year PFS was 74.5% (95% CI, 58.8%-94.5%) and 87.5% (95% CI, 75.2%-100%) for OS. Median duration of response was not reached.
Untreated patients treated with triple combination (cohort C) (n = 15)
One patient progressed at cycle 4, while the 14 remaining patients have responded and remain disease-free under treatment. One patient with pleomorphic variant reached CR and was on therapy at last contact (cycle 11). The only patient who progressed was not TP53 mutated nor 17p deleted. With a median follow-up of 14 months (range, 5 to 19), OS and PFS at one year are 100% and 93.3% (95% CI, 81.5%- 100%), respectively. Median duration of response was not reached.
MRD assessment and TP53 alteration
MRD assessment was possible in 32 of 48 study patients (included all patients with TP53 alteration) (Figure 2). Molecular MRD analysis was not possible in 16 patients for the following reasons; no MRD marker because of no initial clonal infiltration, 11 of 48 trial patients (cohort A, n = 2; B, n = 7; C, n = 2), assay failure in 2 of 48 trial patients (cohort B, n = 1; C, n = 1), or lack of follow-up samples in 3 of 48 trial patients (cohort A, 1 patient in early progression before cycle 3; cohort B, n = 2 for either insufficient DNA or early progression before cycle 3 in 1 patient each).
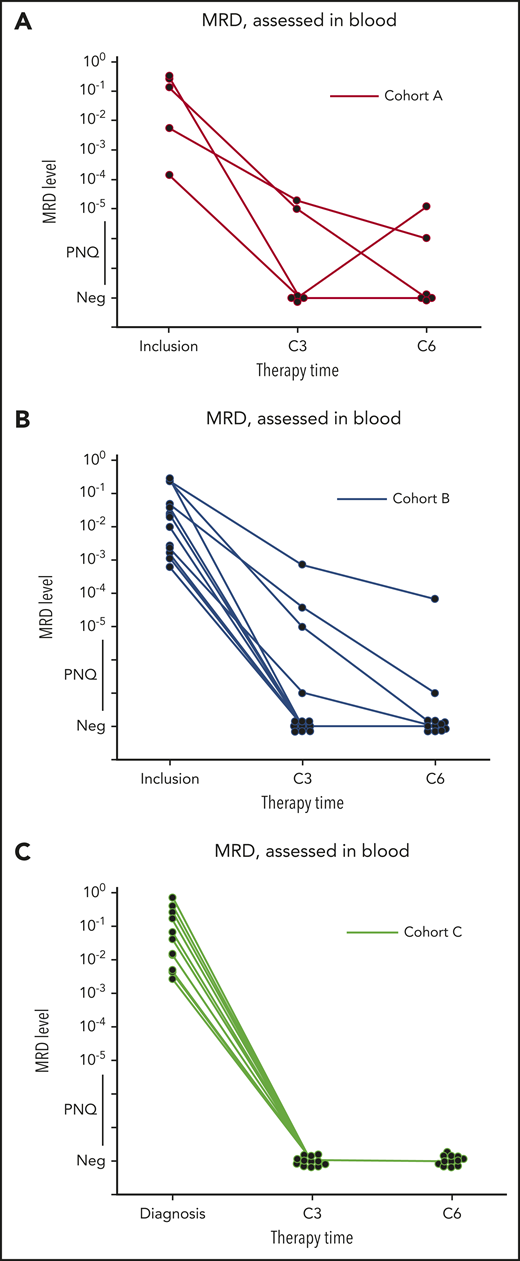
Clearance of MRD. (A-C) Absolute changes in the level of MRD in peripheral blood, as assessed by ASO-qPCR, in 32 patients who had data that could be evaluated from cohorts A (n = 6), B (n = 14), and C (n = 12), as indicated. Therapy time points C3 (end of cycle 3) and C6 (end of cycle 6) of ibrutinib, obinutuzumab, and venetoclax are shown. MRD levels that are detectable, but not quantifiable, are shown as positive nonquantifiable (PNQ). The threshold for MRD negativity in the blood varied according to the sample. Neg, negative values.
Clearance of MRD. (A-C) Absolute changes in the level of MRD in peripheral blood, as assessed by ASO-qPCR, in 32 patients who had data that could be evaluated from cohorts A (n = 6), B (n = 14), and C (n = 12), as indicated. Therapy time points C3 (end of cycle 3) and C6 (end of cycle 6) of ibrutinib, obinutuzumab, and venetoclax are shown. MRD levels that are detectable, but not quantifiable, are shown as positive nonquantifiable (PNQ). The threshold for MRD negativity in the blood varied according to the sample. Neg, negative values.
Among MRD-evaluable patients (n = 32), 26 (4 out of 6 in cohort A; 10 out of 14 in cohort B, and 12 out of 12 in cohort C) were MRD negative (81%) by ASO-qPCR in the peripheral blood after cycle 3 (including 11 patients with TP53 alterations), while 6 were MRD positive (including 1 with TP53 mutation in cohort B). Of those patients who were MRD negative at cycle 3, all remained negative at cycle 6 when tested in both blood and bone marrow by ASO-qPCR, except for 1 patient (treated in cohort A) who converted to MRD positivity (this patient had a TP53 alteration). Of the 6 patients who were MRD positive in the blood at cycle 3, 2 became negative at cycle 6 in both blood and bone marrow (1 in cohort A and 1 in cohort B). A third MRD-positive patient (in cycle 3 and cycle 6 blood) was reported MRD negative in the bone marrow at cycle 6. In total, 4 patients showed persistent MRD positivity at cycle 6, of whom 1 with TP53 alteration (cohort B). Molecular response rates at C3 across cohorts were 4 out of 6 MRD negative in cohort A (66%), 10 out of 14 MRD negative in cohort B (71.4%), and 12 out of 12 MRD negative in cohort C (100%).
IGHV mutational status did not appear to be correlated with MRD response. Seven out of 8 IGHV-mutation and 20 of 25 IGHV-nonmutated cases, respectively, were MRD negative in blood by cycle 3 (for response rates of 87.5% and 80%, respectively).
Discussion
The triple combination of obinutuzumab, ABT-199 (venetoclax) plus ibrutinib is well tolerated, with no DLTs reported, and the MTD (maximum tested dose was 800 mg/d) for venetoclax was not reached. A dose of 400 mg/d of venetoclax was taken forward into the expansion phase of this study. There were several lines of evidence that supported this decision. First, patients treated with venetoclax doses of 600 to 800 mg/d were more frequently transfused over time, and the observed response rates were not higher than with the 400-mg/d dose, suggesting that the venetoclax dose-effect was limited, as already seen in in vitro experiments.15 Also, using a lower dose of venetoclax has the advantage of reducing the pill burden (venetoclax is formulated in 100-mg tablets and ibrutinib in 140-mg tablets), which is important in the setting of lengthy therapies for elderly patients with MCL. Furthermore, a comparison between obinutuzumab and ibrutinib (cohort A) with obinutuzumab ibrutinib and venetoclax (cohort B) in relapsed patients shows that the addition of venetoclax increases toxicity, in particular hematological toxicities such as thrombocytopenia and neutropenia. The number of AEs was much lower when the triple combination (with venetoclax 400 mg/d) was given to treatment-naive patients (cohort C). In particular, hematological AEs were rare in untreated patients who had not been previously exposed to hematotoxic agents such as chemotherapy.
The OAsIs and AIM (ibrutinib plus venetoclax in relapsed patients) trials raise the question of the best treatment option for relapsed MCL, even if such a cross-study comparison should be interpreted with caution because patients numbers are small, follow-ups are short, and patient populations are different. With these limitations in mind, we observe that the overall response rate for the triple combination in relapsed patients according to Cheson criteria was 79%, with 62% achieving a CR at the end of cycle 4, as compared with 42% in CR with ibrutinib plus venetoclax at the same time point.14 According to Lugano criteria, 62% reached CR at week 16 with ibrutinib plus venetoclax compared with 67% at cycle 6 with triple combination. MRD evaluation differed between the trials and was not possible by ASO-PCR in all patients. The MRD clearance rate for MRD-evaluable patients with ibrutinib plus venetoclax at week 16 was 15% (2 patients out of 13 evaluable) compared with 71.5% (10 patients out of 14 evaluable) at the same time point in the present trial. In an intention-to-treat analysis taking into account all included patients, MRD clearance rates at week 16 are were 41.7% (10 patients out of 24) for relapsed patients treated with the triple combination vs 8% (2 out of 24) with ibrutinib plus venetoclax, increasing to 9 out of 24 (38%) with ongoing therapy. In OAsIs, all MRD-evaluable, untreated patients (12 out of 15) reached MRD negativity in the blood at cycle 3 and remained MRD negative at the end of cycle 6. Thus, the triple combination should be positioned early in the course of the disease, including frontline and upstream of chimeric antigen receptor T-cell therapy, which has recently shown high efficacy in relapsed MCL patients previously exposed to BTKi.23
OAsIs was not designed or powered to compare ibrutinib plus obinutuzumab (cohort A) with the addition of venetoclax (cohort B) in relapsed patients, and we cannot conclude what additional benefit or toxicity is derived from the addition of venetoclax. However, results in the OAsIs trial show that triple combination is effective, achieving deep responses, even in high-risk patients with TP53 alterations or blastoid MCL.24 Indeed, the only refractory patient in the untreated cohort encountered was TP53 wild-type. Preclinical data showed that obinutuzumab and ibrutinib could overcome microenvironment-driven venetoclax resistance via blockade of NF-κB–dependent Bcl-xL induction.15 Interestingly, Bcl-xL has recently been implicated in resistance to ibrutinib plus venetoclax in MCL patients.25 It will be important to check these findings in the current trial, particularly since despite very encouraging results, some patients progress rapidly. A caveat is the relatively small numbers of patients studied. Further investigations in the clinical setting are required to fully explore this.
In conclusion, the OAsIs trial demonstrates that the combination of ibrutinib, obinutuzumab, and venetoclax is well tolerated and highly active in MCL. It provides an early and high percentage of MRD negativity with CR and sustained clinical and molecular responses in both relapsed and untreated patients. Known high-risk features (such as TP53 alteration) appear not to impact efficacy, but numbers are small. OAsIs strongly supports further clinical investigations of the combination of obinutuzumab, venetoclax, and ibrutinib for untreated MCL patients.
Requests for access to the study data can be e-mailed to the corresponding author.
Additional information is available online, including a list of investigators, inclusion and exclusion criteria, treatment regimens, risk stratification for the TLS and prevention, tumor status according to 1999 International Working Group criteria, tumor status according to Lugano criteria, MIPI and biological MIPI, all AEs from cycle 1 by SOCNAME and PTNAME according to cohorts, all AEs from cycle 1 to cycle 6 according to venetoclax doses in cohort B, drug administration during the first 6 cycles in all cohorts, and an amendments list.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors would like to thank the patients who participated in this study and their families, as well as investigators and staff at all OAsIs clinical sites. They also acknowledge Maelle Ningre (senior manager of the OAsIs trial, Délégation à la Recherche Clinique et à l’Innovation (DCRI) CHU de Nantes, Nantes, France) Lucie Planche (statistician, DRCI CHU de Nantes), Alexandra Jobert (pharmacovigilance department, DRCI CHU de Nantes), and Caroline Chapusot for supervising the NGS work; Cyril Fournier for bioinformatics; and all of the research teams and nurses in participating centers.
M.B.C. acknowledges additional funding from the Fondation ARC and the Région Bourgogne-Franche Comté and FEDER programs. Roche SAS supplied obinutuzumab and funded the trial. Janssen-Cilag supplied ibrutinib and funded the trial. AbbVie funded the trial and supplied venetoclax.
Authorship
Contribution: S.L.G. and S. Rule designed the trial; D.C. provided the rationale; M.B.C. designed and performed the MRD and genetic analyses; data were collected by the investigators and analyzed by the sponsor; all authors had access to the data; S.L.G., S. Rule, and M.B.C. wrote the initial draft of the manuscript; all authors contributed to the subsequent drafts, reviewed them, and jointly decided to submit the manuscript for publication; and all authors vouch for the integrity, accuracy, and the completeness of the data and the analysis and that the trial was conducted according to the protocol.
Conflict-of-interest disclosure: S.L.G. reports grants, personal fees, and nonfinancial support from Roche Genentech, Janssen-Cilag, and AbbVie during the conduct of the study and reports personal fees from Celgene, Jazz Pharmaceutical, Gilead-Kite, Loxo, and Daiichi-Sankyo outside the submitted work. F.M. has received honoraria from Bristol-Myers Squibb and Janssen and served as a consultant or advisor to Celgene, Bayer, AbbVie, Verasteem, Gilead, Servier, Roche/Genentech, and Epizyme; G.C. has received honoraria from Janssen, Sanofi, AbbVie, Gilead, Roche, and Celgene and served as a consultant or Celgene and Roche/Genentech. O.C. reports grants, personal fees, and nonfinancial support from Roche, Genentech, and AbbVie during the conduct of the study; reports personal fees from MSD, BMS, Amgen, and Celgene; and reports grants and personal fees from Takeda, Gilead-Kite, outside the submitted work. C.H. research fundings from Takeda and AbbVie and honoraria and nonfinancial support from Roche, Janssen-Cilag, AbbVie, and Takeda. E.T. reports consultancy for Janssen-Cilag and AbbVie. C.R. reports research grants from Roche and personal fees and nonfinancial support from Janssen, Roche, and Takeda outside the submitted work. S. Rule received research support from Janssen-Cilag and personal fees from Janssen, Gilead, Astra Zeneca, Kite, Sunesis, and Roche outside the submitted work. The remaining authors declare no competing financial interests.
A list of all investigators can be found in the supplemental Appendix.
Correspondence: Steven Le Gouill, University Hospital Hôtel-Dieu, 44093 Nantes cedex 01, France; e-mail: steven.legouill@chu-nantes.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal