

Key Points
Severe COVID-19 is associated with increased antibody-mediated procoagulant platelets.
Procoagulant platelets and platelet apoptosis in severe COVID-19 is correlated with D-dimer and higher incidence of thromboembolisms.

Abstract
The pathophysiology of COVID-19–associated thrombosis seems to be multifactorial. We hypothesized that COVID-19 is accompanied by procoagulant platelets with subsequent alteration of the coagulation system. We investigated depolarization of mitochondrial inner transmembrane potential (ΔΨm), cytosolic calcium (Ca2+) concentration, and phosphatidylserine (PS) externalization. Platelets from COVID-19 patients in the intensive care unit (ICU; n = 21) showed higher ΔΨm depolarization, cytosolic Ca2+, and PS externalization compared with healthy controls (n = 18) and non-ICU COVID-19 patients (n = 4). Moreover, significant higher cytosolic Ca2+ and PS were observed compared with a septic ICU control group (ICU control; n = 5). In the ICU control group, cytosolic Ca2+ and PS externalization were comparable with healthy controls, with an increase in ΔΨm depolarization. Sera from COVID-19 patients in the ICU induced a significant increase in apoptosis markers (ΔΨm depolarization, cytosolic Ca2+, and PS externalization) compared with healthy volunteers and septic ICU controls. Interestingly, immunoglobulin G fractions from COVID-19 patients induced an Fcγ receptor IIA–dependent platelet apoptosis (ΔΨm depolarization, cytosolic Ca2+, and PS externalization). Enhanced PS externalization in platelets from COVID-19 patients in the ICU was associated with increased sequential organ failure assessment score (r = 0.5635) and D-dimer (r = 0.4473). Most importantly, patients with thrombosis had significantly higher PS externalization compared with those without. The strong correlations between markers for apoptosic and procoagulant platelets and D-dimer levels, as well as the incidence of thrombosis, may indicate that antibody-mediated procoagulant platelets potentially contributes to sustained increased thromboembolic risk in ICU COVID-19 patients.
Introduction
Accumulating evidence indicates an association between SARS-CoV-2–associated pneumonia and hypercoagulable state of patients with COVID-19 who require intensive care (severe infection).1 It is becoming more and more clear that infections with SARS-CoV-2 do not meet the criteria of disseminated intravascular coagualation (DIC) according to the International Society of Thrombosis and Haemostasis, and the patterns of coagulation seem to be different from severe infections.2 Only in patients with severe aggravation of disease did an overt DIC occur.3 At the same time, however, the incidence of acute pulmonary embolism (PE), deep vein thrombosis, ischemic stroke, myocardial infarction, and/or systemic arterial embolism in COVID-19 patients admitted to the intensive care unit (ICU) is as high as 49%.4 The pathophysiology of COVID-19–associated thromboembolic events seems to be complex and multifactorial, involving interplay between cellular and plasma elements of the hemostatic system and components of the innate immune response to the infecting pathogen. The phosphatidylserine (PS) externalization in apoptotic platelets, as a substrate, might be an initiation for multiple coagulation factors.5 Therefore, a combination of several activation events initiated by exposure of the endothelium, platelets, and leukocytes to pathogen- and damage-associated molecular patterns might be responsible for the uncontrolled activation of the coagulation system in severely ill COVID-19 patients.6,7
To date, most clinical reports on COVID-19–associated coagulopathy focused on an increased activation of the plasma coagulation system.1-3,7 Elevated D-dimer levels were consistently reported, whereas their gradual increase during disease course is particularly associated with disease worsening.8 Other coagulation abnormalities such as prothrombin time (PT) and activated partial thromboplastin time (aPTT) prolongation, together with severe thrombocytopenia, have been found to be associated with life-threatening DIC.9 Platelets have been recently recognized as a mediator of inflammation and sensor of infectious agents through the interaction of surface receptors and pathogens or immune system derivatives.10 Viral infections elicit the systemic inflammatory response that affects platelets, which become activated on antigen specific recognition and interaction with white blood cells.11 While platelet activation plays a critical role in the procoagulatory effect of viral infections.12,13 Platelets derived from patients with HIV and secondary dengue virus infection have been shown to have increased upregulation of the intrinsic pathway of apoptosis.14-16 In addition, it is well known that the survival and lifespan of platelets is finely regulated by the intrinsic or mitochondrial apoptosis pathway.17,18
We hypothesized that coagulation disturbances observed in COVID-19 patients in the ICU are accompanied by platelet apoptosis with subsequent alterations of the coagulation system. In platelets, apoptosis is mediated by mitochondrial outer membrane permeabilization, which is regulated by the members of the Bcl2 protein family that either promote (proapoptotic proteins: Bak, Bax, and Bim) or inhibit (antiapoptotic proteins: Bcl-xL and Bcl-w) the apoptosis.19 The mitochondrial inner membrane potential (ΔΨm) collapse is followed by the efflux of cytochrome c into the cytoplasm, which forms a multiprotein complex called apoptosome. The latter triggers the activation of caspase 9 and the following downstream caspase cascade including caspase 3 and 7. Finally, the PS externalization on the extracellular membrane represents one of the late stages of the apoptosis pathway.20 In this study, we found evidence that platelets from patients with severe COVID-19 in the ICU have an upregulation of apoptotic markers. Most importantly, sera and immunoglobulin (IgG) fractions isolated from COVID-19 patients were able to induce apoptosis in platelets from healthy donors. Furthermore, our data indicate that platelet apoptosis might be associated with thromboembolic complications and increased mortality in severe COVID-19.
Methods
Study cohort and evaluation of the clinical data
During the SARS-CoV-2 outbreak in Tuebingen (between 1 March and 30 April 2020), 27 COVID-19 patients in the ICU and non-ICU patients were referred to our laboratory for extended investigations of the coagulation system. Blood samples from healthy donors (n = 18) and septic non–COVID-19 patients in the ICU (n = 5) were collected to serve as controls. Surgical patients who developed postoperative sepsis and needed passive ventilation because of respiratory failure were included in the non–COVID-19 patients in the ICU in the control group. Among them, 2 patients had pneumonia, 2 developed a systemic infection after trauma, and 1 patient had a septic reaction after perforation of the appendix.
Electronic medical records were used to collect demographic data, clinical treatments, and outcome. Diagnosis of thromboembolic complications was made when clinically or laboratory indicated (ie, unexpected increase in D-dimer levels) based on computer tomography, ultrasound imaging, or, in case of death, by postmortem pathology. To estimate the status of critical illness, the sequential organ failure assessment (SOFA) score system was used as previously described.21 Data were independently reviewed by 2 physicians (K.A. and S.H.). In case of disagreements, a third physician was consulted (M.M.). For more details, see supplemental Material available on the Blood Web site.
Determination of apoptosis in patients’ platelets
Platelet isolation
Platelets were isolated from citrated blood and tested within 3 hours. In brief, after 1 centrifugation step (120g, 20 minutes at room temperature, without brake), platelet-rich plasma (PRP) was gently separated and used for further analysis.
Assessment of the inner ΔΨm
To detect changes in ΔΨm, the tetramethylrhodamine ethyl ester (TMRE) assay kit (Abcam, Cambridge, UK) was used as previously described, with minor modifications.22,23 In brief, PRP was stained with 10 µM TMRE (30 minutes at room temperature) and directly measured by flow cytometry (FC) (Navios, Beckman-Coulter). The complete depolarization of platelet mitochondrial potential was induced using the uncoupler of mitochondrial oxidative phosphorylation carbonyl cyanide 4-trifluoromethoxy phenylhydrazone (10 µM, 1 hour at 37°C).16 Changes in the ΔΨm were determined as ratio of the mean of fluorescence intensity (MFI) signal from TMRE from healthy donor PRP compared with the MFI signal of platelets from COVID-19 and non–COVID-19 patients in the ICU.
Quantification of cytosolic calcium concentration
Cytosolic calcium (Ca2+) concentration was determine using Fluo-3/AM (Sigma-Aldrich, St. Louis, MO) as previously described,24 with minor modifications. In brief, PRP was incubated with Fluo3/AM (3 µM) in trisaminomethane (TRIS) buffer (10 mM TRIS, 0.9% NaCl, 1 mM CaCl2, pH 7.4) for 30 minutes at 37°C. After washing once with TRIS buffer, cells were resuspended in TRIS containing 1 mM CaCl2. Ca2+-dependent fluorescence intensity of samples was measured with an excitation wavelength of 488 nm and an emission wavelength of 530 nm by FC. PRP treated with ionomycin (2 μM, 30 minutes at 37°C; Sigma-Aldrich) served as a positive control. Test results were normalized to the mean MFI of healthy donors tested in parallel.
Detection of PS externalization
PRP was stained with Annexin V-fluorescein isothiocyanate (Immunotools, Friesoyhte, Germany) and CD41-PE-Cy5 (Beckman Coulter) in TRIS buffer (30 minutes at room temperature) and directly analyzed by FC, as previously described.25 Cells incubated with 60 µM ionomycin (30 minutes at room temperature) served as a positive control. Test results were determined as fold increase of the percentage of PS-positive events in PRP from COVID-19 or non–COVID-19 patients in the ICU compared with platelets from healthy donors tested in parallel.
Assessment of antibody-mediated apoptosis
Sera and IgG incubation with washed platelets
To exclude unspecific effects like the activation of platelets via complement or nonspecific immune complexes, all sera were heat inactivated (56°C for 30 minutes), followed by a sharp centrifugation step at 5000g. The supernatant was collected.
All experiments involving patient sera were performed after incubation of 5 µL serum with 25 µL washed platelets (7.5 × 106; see supplemental Material for more information on the preparation of washed platelets) for 1.5 hours under rotating conditions at room temperature. Where indicated, platelets were pretreated with 50 µM cyclosprorin-A (Novartis Pharma GmbH, Basel, Switzerland) or Q-VD-OPh (caspase inhibitor; Sigma-Aldrich) for 30 minutes at room temperature before serum was added. Afterward, samples were washed once (7 minutes, 650g, room temperature, without brake) and gently resuspended in 75 µL of phosphate-buffered saline (Dulbecco; Biochrom GmbH, Berlin, Germany).
When indicated, IgG fractions were isolated from serum (for more details, see supplemental Material).
Detection of ΔΨm changes induced by patient sera
Washed platelets were stained with 10 µM TMRE (30 minutes at room temperature) and directly measured by FC. As a positive control, cells were preincubated with carbonyl cyanide 4-trifluoromethoxy phenylhydrazone (10 µM for 30 minutes at 37°C). Serum-mediated apoptosis was quantified as ratio of ΔΨm depolarization, comparing the MFI of washed platelets incubated with serum from healthy donors with sera from (non-ICU and ICU) COVID-19 or non–COVID-19 patients in the ICU. Test results were normalized to the mean MFI of patients’ sera compared with sera from healthy donors tested in parallel.
Quantification of cytosolic Ca2+ concentration after patient sera incubation
On incubation with patient sera, washed platelets were incubated with Fluo3/AM (3 µM) in TRIS buffer (30 minutes at 37°C). After washing once with TRIS buffer, cells were resuspended in TRIS supplemented with 1 mM CaCl2. Subsequently, Ca2+-dependent fluorescence intensity of samples was measured by FC. Ionomycin (5 µM, 15 minutes at room temperature) was used as positive control. Test results were normalized to the mean MFI of patient sera compared with sera from healthy donors tested in parallel.
Assessment of PS externalization induced by patient sera
Washed platelets (1 × 106) were resuspended in Hank´s balanced salt solution (137 mM NaCl, 1.25 mM CaCl2, 5.5 mM glucose; Carl-Roth, Karlsruhe, Germany), stained with Annexin V-fluorescein isothiocyanate (Immunotools), and directly analyzed by FC. As a positive control, washed platelets were incubated with ionomycin (5 µM, 15 minutes at room temperature). Test results were determined as fold increase of the percentage of PS-positive events in platelets on incubation with patient sera compared with cells incubated with healthy donors tested in parallel.
Western blot analysis
Protein levels of cleaved caspase 9 were determined by western blot. In brief, washed platelet were centrifuged (5 minutes, 700g at 4°C), and the pellet was resuspended in Radio-ImmunoPrecipitation Assay (RIPA) lysis buffer (ThermoFisher Scientific, Paisley, UK). The proteins were separated by electrophoresis using 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. See the supplemental Material for further details.
Ethics statement
The study was conducted in accordance with the declaration of Helsinki. Written informed consent was obtained from all volunteers before any study-related procedures. All tests were performed with leftover blood samples from routine testing. The study protocol of patient material was approved by the Institutional Review Board of the University of Tuebingen.
Statistical analyses
Statistical analysis was performed using GraphPad Prism, Version 7.0 (GraphPad, La Jolla, CA) and SPSS version 26.0 (SPSS Inc., Chicago, IL). Because potential daily variations in FC measurements might result in bias in data analysis, test results were normalized to 2 healthy donors tested in parallel at the same time point (raw data are available in the supplemental Data). For further information, see the supplemental Material.
Results
Study cohort
Blood samples were collected from 27 consecutive COVID-19 patients who were admitted to our hospital with severe acute respiratory distress symptoms (ARDS) requiring intensive care (ICU, n = 23) and hospitalized patients (non-ICU n = 4) without ARDS. Two blood samples were excluded because of insufficient material. In total, 21 COVID-19 patients in the ICU were enrolled in the study, of whom 18 of 21 (86%) patients were male (Table 1). The mean age was 60 years (range, 29-88 years; Table 1). Fifteen of 21 (71%) patients had known risk factors for severe COVID-19 infection as described previously,26 including hypertension in 14 of 21 (67%), obesity in 4 of 21 (19%), coronary artery disease in 4 of 21 (19%), and diabetes mellitus in 5 of 21 (24%). Four patients had only minor symptoms of COVID-19 and did not require admission to the ICU. Samples from patients in the ICU with sepsis were used as a control group. Detailed information on the clinical manifestations of the 25 (non-ICU, n = 4; ICU, n = 21) COVID-19 patients and the 5 non–COVID-19 patients in the ICU (ICU control) are available in the supplemental Data (supplemental Table 1).
Baseline parameters of 21 COVID-19 patients and non–COVID-19 control patients in the ICU
| ICU COVID-19 | Control ICU (n = 5) | ||||
|---|---|---|---|---|---|
| Total (n = 21) | Survivors (n = 14) | Nonsurvivors (n = 7) | P | ||
| Age (mean ± SD), y | 60 ± 17 | 58 ± 18 | 63 ± 16 | .359 | 61 ± 28 |
| Male (%), n | 18 (85.7) | 11 (78.6) | 7 (100) | .186 | 4 (80) |
| Body mass index (mean ± SD) | 28.6 ± 5 | 28 ± 5 | 29 ± 4 | .543 | 23 ± 6 |
| Cardiovascular risk factors, n (%) | |||||
| Arterial hypertension | 14 (66.7) | 9 (64.3) | 5 (71.4) | .743 | 1 (20%) |
| Dyslipidemia | 5 (23.8) | 3 (21.4) | 2 (28.6) | .717 | 2 (40%) |
| Diabetes mellitus | 5 (23.8) | 4 (28.6) | 1 (14.3) | .469 | 1 (20%) |
| Current smokers | 0 (0.0) | 0 (0.0) | 0 (0.0) | NA | NA |
| Obesity* | 4 (19.0) | 3 (21.4) | 1 (14.3) | .807 | 1 (20%) |
| Atrial fibrillation | 4 (19.0) | 4 (28.6) | 0 (0.0) | .116 | 0 (0.0) |
| Known CAD | 4 (19.0) | 2 (14.3) | 2 (28.6) | .597 | 1 (20%) |
| Chronic kidney disease | 0 (0.0) | 0 (0.0) | 0 (0.0) | NA | NA |
| Echocardiography | |||||
| Left ventricular function, % (mean ± SD) | 56 ± 6 | 57 ± 7 | 54 ± 6 | .516 | NA |
| RV pressure, mmHg (mean ± SD) | 26 ± 3 | 23 ± 6 | 33 ± 15 | .401 | NA |
| Admission laboratory, median (25th-75th percentile) | |||||
| Leukocytes, 1000/µL | 6.5 (4.2-9.7) | 6.7 (4.3-11.0) | 5.6 (3.0-6.8) | .233 | 13.3 (8.2-28.1) |
| Lymphocytes, 1000/µL | 0.6 (0.3-1.0) | 0.7 (0.5-1.1) | 0.3 (0.3-0.8) | .044 | NT |
| PLT, ×109/L | 179 (152-262) | 165 (87-218) | 182 (154-336) | .154 | 237 (73.5-486.5) |
| INR | 1.0 (1.0-1.1) | 1.0 (1.0-1.1) | 1.1 (1.0-1.1) | .474 | 1.3 (1.2-1.5) |
| aPTT, s | 26 (24-30) | 25 (22-30) | 28 (24-31) | .2609 | 30 (23.5-56.5) |
| Fibrinogen, mg/dL | 574 (482-689) | 613 (495-730) | 494 (463-574) | .2242 | NT |
| D-dimer, µg/mL | 1.7 (1.0-5.5) | 1.8 (1.0-4.4) | 1.2 (0.9-18.0) | 1.000 | NT |
| GFR, mL/m2 | 73.9 (42.4-93.2) | 65.8 (43.1-87.8) | 81.6 (26.7-139.4) | .296 | 72.7 (17.0-91.4) |
| Creatinin, mg/dL | 1.0 (0.8-1.7) | 1.1 (0.9-1.7) | 0.9 (0.6-2.5) | .331 | 1.8 (0.85-4.7) |
| C-reactive protein, mg/dL | 17.1 (11.8-27.6) | 15.0 (12.1-19.1) | 28.3 (7.7-29.5) | .136 | 9.7 (0.1-20.6) |
| Procalcitonin, ng/mL | 0.7 (0.2-1.8) | 0.7 (0.2-2.8) | 0.2 (0.2-2.0) | .823 | 3.13 (0.45-20.24) |
| Troponin I, ng/dL | 26 (13.8-111.3) | 29 (16-210) | 22 (6-34) | .284 | 27.5 (17-38) |
| NT pro-BNP, ng/L | 749 (385-3148) | 1169 (239-4543) | 537 (385-2324) | .661 | NT |
| CK, U/L | 312 (181-1166) | 300 (159-1416) | 414 (280-1057) | .602 | 222 (40-2227) |
| AST, U/L | 62 (45-118) | 63 (43-134) | 61 (47-108) | .881 | 59 (15-390) |
| ALT, U/L | 45 (28-76) | 46 (29-68) | 34 (19-88) | .455 | NT |
| LDH, U/L | 415 (375-498) | 426 (382-504) | 394 (312-497) | .502 | 219 (199-410) |
| Medication at admission to hospital, n (%) | |||||
| Oral anticoagulation | 1 (4.8) | 1 (7.1) | 0 (0.0) | .424 | 0 (0.0) |
| ACEi/ARB | 9 (42.9) | 5 (35.7) | 4 (57.1) | .515 | 2 (40) |
| Aldosterone inhibitors | 2 (9.5) | 0 (0.0) | 2 (28.6) | .051 | 0 (0.0) |
| Diuretics | 2 (9.5) | 1 (7.1) | 1 (14.3) | .696 | 3 (5) |
| Calcium channel blockers | 3 (14.3) | 2 (14.3) | 1 (14.3) | .869 | 0 (0.0) |
| Beta blockers | 7 (33.3) | 3 (21.4) | 4 (57.1) | .152 | 2 (40) |
| Statins | 5 (23.8) | 2 (14.3) | 3 (42.9) | .210 | 1 (20) |
| ASA | 5 (23.8) | 2 (14.3) | 3 (42.9) | .210 | 2 (40) |
| P2Y12 blockers | 0 (0.0) | 0 (0.0) | 0 (0.0) | NA | 0 (0.0) |
| ICU COVID-19 | Control ICU (n = 5) | ||||
|---|---|---|---|---|---|
| Total (n = 21) | Survivors (n = 14) | Nonsurvivors (n = 7) | P | ||
| Age (mean ± SD), y | 60 ± 17 | 58 ± 18 | 63 ± 16 | .359 | 61 ± 28 |
| Male (%), n | 18 (85.7) | 11 (78.6) | 7 (100) | .186 | 4 (80) |
| Body mass index (mean ± SD) | 28.6 ± 5 | 28 ± 5 | 29 ± 4 | .543 | 23 ± 6 |
| Cardiovascular risk factors, n (%) | |||||
| Arterial hypertension | 14 (66.7) | 9 (64.3) | 5 (71.4) | .743 | 1 (20%) |
| Dyslipidemia | 5 (23.8) | 3 (21.4) | 2 (28.6) | .717 | 2 (40%) |
| Diabetes mellitus | 5 (23.8) | 4 (28.6) | 1 (14.3) | .469 | 1 (20%) |
| Current smokers | 0 (0.0) | 0 (0.0) | 0 (0.0) | NA | NA |
| Obesity* | 4 (19.0) | 3 (21.4) | 1 (14.3) | .807 | 1 (20%) |
| Atrial fibrillation | 4 (19.0) | 4 (28.6) | 0 (0.0) | .116 | 0 (0.0) |
| Known CAD | 4 (19.0) | 2 (14.3) | 2 (28.6) | .597 | 1 (20%) |
| Chronic kidney disease | 0 (0.0) | 0 (0.0) | 0 (0.0) | NA | NA |
| Echocardiography | |||||
| Left ventricular function, % (mean ± SD) | 56 ± 6 | 57 ± 7 | 54 ± 6 | .516 | NA |
| RV pressure, mmHg (mean ± SD) | 26 ± 3 | 23 ± 6 | 33 ± 15 | .401 | NA |
| Admission laboratory, median (25th-75th percentile) | |||||
| Leukocytes, 1000/µL | 6.5 (4.2-9.7) | 6.7 (4.3-11.0) | 5.6 (3.0-6.8) | .233 | 13.3 (8.2-28.1) |
| Lymphocytes, 1000/µL | 0.6 (0.3-1.0) | 0.7 (0.5-1.1) | 0.3 (0.3-0.8) | .044 | NT |
| PLT, ×109/L | 179 (152-262) | 165 (87-218) | 182 (154-336) | .154 | 237 (73.5-486.5) |
| INR | 1.0 (1.0-1.1) | 1.0 (1.0-1.1) | 1.1 (1.0-1.1) | .474 | 1.3 (1.2-1.5) |
| aPTT, s | 26 (24-30) | 25 (22-30) | 28 (24-31) | .2609 | 30 (23.5-56.5) |
| Fibrinogen, mg/dL | 574 (482-689) | 613 (495-730) | 494 (463-574) | .2242 | NT |
| D-dimer, µg/mL | 1.7 (1.0-5.5) | 1.8 (1.0-4.4) | 1.2 (0.9-18.0) | 1.000 | NT |
| GFR, mL/m2 | 73.9 (42.4-93.2) | 65.8 (43.1-87.8) | 81.6 (26.7-139.4) | .296 | 72.7 (17.0-91.4) |
| Creatinin, mg/dL | 1.0 (0.8-1.7) | 1.1 (0.9-1.7) | 0.9 (0.6-2.5) | .331 | 1.8 (0.85-4.7) |
| C-reactive protein, mg/dL | 17.1 (11.8-27.6) | 15.0 (12.1-19.1) | 28.3 (7.7-29.5) | .136 | 9.7 (0.1-20.6) |
| Procalcitonin, ng/mL | 0.7 (0.2-1.8) | 0.7 (0.2-2.8) | 0.2 (0.2-2.0) | .823 | 3.13 (0.45-20.24) |
| Troponin I, ng/dL | 26 (13.8-111.3) | 29 (16-210) | 22 (6-34) | .284 | 27.5 (17-38) |
| NT pro-BNP, ng/L | 749 (385-3148) | 1169 (239-4543) | 537 (385-2324) | .661 | NT |
| CK, U/L | 312 (181-1166) | 300 (159-1416) | 414 (280-1057) | .602 | 222 (40-2227) |
| AST, U/L | 62 (45-118) | 63 (43-134) | 61 (47-108) | .881 | 59 (15-390) |
| ALT, U/L | 45 (28-76) | 46 (29-68) | 34 (19-88) | .455 | NT |
| LDH, U/L | 415 (375-498) | 426 (382-504) | 394 (312-497) | .502 | 219 (199-410) |
| Medication at admission to hospital, n (%) | |||||
| Oral anticoagulation | 1 (4.8) | 1 (7.1) | 0 (0.0) | .424 | 0 (0.0) |
| ACEi/ARB | 9 (42.9) | 5 (35.7) | 4 (57.1) | .515 | 2 (40) |
| Aldosterone inhibitors | 2 (9.5) | 0 (0.0) | 2 (28.6) | .051 | 0 (0.0) |
| Diuretics | 2 (9.5) | 1 (7.1) | 1 (14.3) | .696 | 3 (5) |
| Calcium channel blockers | 3 (14.3) | 2 (14.3) | 1 (14.3) | .869 | 0 (0.0) |
| Beta blockers | 7 (33.3) | 3 (21.4) | 4 (57.1) | .152 | 2 (40) |
| Statins | 5 (23.8) | 2 (14.3) | 3 (42.9) | .210 | 1 (20) |
| ASA | 5 (23.8) | 2 (14.3) | 3 (42.9) | .210 | 2 (40) |
| P2Y12 blockers | 0 (0.0) | 0 (0.0) | 0 (0.0) | NA | 0 (0.0) |
Bold indicates significant P value.
ACEi, angiotensin-converting enzyme inhibitors; ALT, alanine aminotransferase; ARB, angiotensin II receptor blockers; ASA, acetylsalicylic acid; AST, aspartate aminotransferase; CAD, cardiac artery disease; CK, creatine kinase; GFR, glomerular filtration rate; INR, international normalized ratio; LDH, lactate dehydrogenase; NA, not applicable; NT, not tested; NT pro BNP, N-terminal pro b-type natriuretic peptide; PLT, platelet; RV, right ventricular; SD, standard deviation.
Obesity (body mass index >30 kg/m2).
In the ICU, all patients received controlled respiratory support, and 6 had additional venous-venous extracorporeal membrane oxygenation. Anticoagulation with heparin was administrated at prophylactic doses (400 IE/h) in 12 of 21 (57%) patients or at therapeutic doses (two- to threefold aPTT) in 9 of 21 (43%) patients, depending on patient’s risk factors for thrombosis. Eight of 21 (38%) patients had additional antiplatelet therapy with aspirin. At the day of material submission, patients had a median SOFA score of 8 (range, 2-17), median platelet counts of 151 × 109/L (range, 35-500 × 109/L), median aPTT of 39 seconds (range, 20-98 seconds), median international normalized ratio of 1.0 (range, 0.9-1.5), median D-dimer of 4 µg/mL (range, 1-42 µg/mL), and median fibrinogen of 551 mg/dL (range, 273-961 mg/dL; supplemental Table 1).
During the 30-day follow-up, 7 of 21 (33%) died, 13 of 21 (62%) patients developed thrombocytopenia (range platelet count, 9-149 × 109/L), and 12 of 21 (57%) patients had at least 1 thromboembolic complication including kidney, spleen, and liver infarction (n = 7), catheter-associated thrombosis (n = 2), clotting of the extracorporeal circulation system (n = 1), pulmonary embolism (n = 1), cerebral infarction (n = 1), and myocardial infarction (n = 1; supplemental Table 1).
Severe COVID-19 infection is associated with platelet apoptosis
To evaluate the mitochondrial function in platelets from COVID-19 patients, the mitochondrial ΔΨm, was assessed. As shown in Figure 1A, significantly increased ΔΨm depolarization was found in platelets isolated from COVID-19 patients in the ICU compared with healthy donors (1.385 ± 0.071 vs 0.997 ± 0.050, P = .0001), as well as to non-ICU COVID-19 patients (1.385 ± 0.071 vs 0.851 ± 0.096, P = .0046; Figure 1A; supplemental Figure 1A). Of note, a comparable ΔΨm depolarization was observed between ICU COVID-19 and ICU non–COVID-19 patients (1.385 ± 0.071 vs1.369 ± 0.112, P = .921; Figure 1A; supplemental Figure 1A).
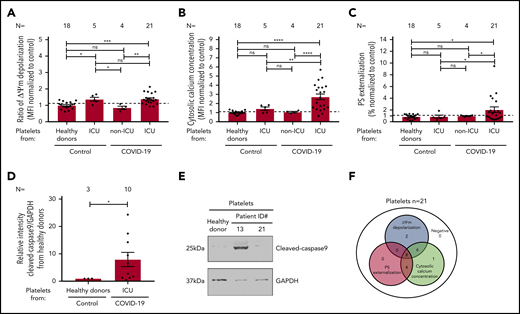
Platelet apoptosis in COVID-19 patients. (A-C) Changes in apoptosis pathways were analyzed by assessing the depolarization of the ΔΨm (A), cytosolic calcium concentration (B), and PS externalization (C) in platelets from COVID-19 patients in the ICU or COVID-19 patients not in the ICU, as well as ICU non–COVID-19 patients (control group) and healthy donors, respectively. (D) Quantification of cleaved-caspase 9 level in platelets from COVID-19 patients in the ICU normalized to healthy donors. (E) Representative western blot showing GAPDH and cleaved-caspase 9 proteins in platelets from COVID-19 patients in the ICU. Protein bands were detected with the infrared imaging system (Odyssey, LI.COR, Lincoln, NE). (F) Diagram indicating the number of COVID-19 patients in the ICU positive for each apoptotic parameter: ΔΨm depolarization, cytosolic calcium concentration, and PS externalization. Data are presented as mean ± standard error mean (SEM) of the measured fold increase compared with control. Not significant, *P < .05, **P < .01, ***P < .001, ****P < .0001. The number of patients and healthy donors tested is reported in each graphic. Dashed lines represent the cutoffs determined from healthy donors as mean of fold increase (FI) + 2× SEM.
Platelet apoptosis in COVID-19 patients. (A-C) Changes in apoptosis pathways were analyzed by assessing the depolarization of the ΔΨm (A), cytosolic calcium concentration (B), and PS externalization (C) in platelets from COVID-19 patients in the ICU or COVID-19 patients not in the ICU, as well as ICU non–COVID-19 patients (control group) and healthy donors, respectively. (D) Quantification of cleaved-caspase 9 level in platelets from COVID-19 patients in the ICU normalized to healthy donors. (E) Representative western blot showing GAPDH and cleaved-caspase 9 proteins in platelets from COVID-19 patients in the ICU. Protein bands were detected with the infrared imaging system (Odyssey, LI.COR, Lincoln, NE). (F) Diagram indicating the number of COVID-19 patients in the ICU positive for each apoptotic parameter: ΔΨm depolarization, cytosolic calcium concentration, and PS externalization. Data are presented as mean ± standard error mean (SEM) of the measured fold increase compared with control. Not significant, *P < .05, **P < .01, ***P < .001, ****P < .0001. The number of patients and healthy donors tested is reported in each graphic. Dashed lines represent the cutoffs determined from healthy donors as mean of fold increase (FI) + 2× SEM.
Next, we determined the cytosolic Ca2+ concentration in platelets from COVID-19 patients using the intracellular dye Fluo-3. Platelets from COVID-19 patients in the ICU showed a significant increase in the cytosolic Ca2+ concentration compared with healthy donors (2.729 ± 0.314 vs 1.007 ± 0.043, P < .0001), non-ICU COVID-19 patients (2.729 ± 0.314 vs 1.060 ± 0.069, P < .0001), and ICU non–COVID-19 patients (2.729 ± 0.314 vs 1.432 ± 0.190, P = .0018; Figure 1B; supplemental Figure 1B).
To further confirm the potential involvement of platelet apoptosis in COVID-19 patients in the ICU, we evaluated surface PS externalization on platelets. This was significantly higher in platelets from COVID-19 patients in the ICU compared with healthy volunteers (2.051 ± 0.476 vs 0.865 ± 0.088, P = .0290) and non-ICU COVID-19 patients (2.051 ± 0.476 vs 0.993 ± 0.064, P = .0392; Figure 1C; supplemental Figure 1C), as well as ICU non–COVID-19 patients (2.051 ± 0.476 vs 0.869 ± 0.263, P = .0404, Figure 1C; supplemental Figure 1C). Moreover, western blot analyses showed increased level of cleaved caspase 9 levels in platelets from COVID-19 patients in the ICU compared with healthy donors (7.94 ± 2.61 vs 1.00 ± 0.00, P = .0260; Figure 1D-E). In association with mitochondrial dysfunction and increased cytosolic Ca2+ concentration, these data indicate an activation of the intrinsic pathway of apoptosis in platelets during COVID-19 infection. Overall, as shown in Figure 1F, platelets from 16 of 21 (76%) patients showed ΔΨm depolarization, 19 of 21 (90%) had increased cytosolic Ca2+ concentration, and 12 of 21 (57%) patients revealed enhanced PS externalization (Figure 1F).
Association of apoptosis markers with laboratory parameters and clinical outcomes
To draw a conclusion on the potential clinical impact of our findings, we limited the analysis of the correlations for apoptosis markers to platelet count, SOFA score, D-dimer, and COVID-19 antibody levels at the days of blood sampling. We found that platelet count is negatively correlated with PS externalization (r = −0.6177, P = .0028; Figure 2A) and with cytosolic Ca2+ concentration (r = −0.5666; P = .0299; Figure 2B). A strong positive correlation was also observed for PS externalization with D-dimer (r = 0.4473; P = .0420; Figure 2C), SOFA score (r = 0.5635; P = .0078; Figure 2D), and SARS-CoV2-antibodies (r = 0.5745; P = .0064; supplemental Figure 3).
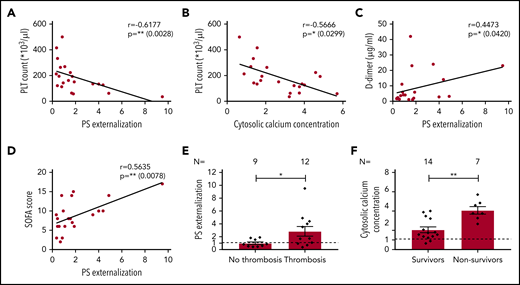
Association among platelet apoptosis and clinical biomarkers, thromboembolic complications, and mortality. The correlations between platelet apoptosis parameters and PLT count, as well as D-dimer and SOFA score measured at the same day of platelet testing, were assessed. (A-B) An association was observed between PLT count and PS externalization (A) and cytosolic calcium concentration (B). (C) Moreover, a significant correlation was detected for D-dimer and PS externalization. (D) The clinical relevance of PS externalization was assessed using the SOFA score and revealed a significant positive correlation. Pearson’s correlation coefficients were calculated and are shown in the panels. (E-F) The PS externalization (E) and the cytosolic calcium concentration (F) were determined and compared between COVID-19 patients in the ICU depending on the incidence of thromboembolic complications and mortality, respectively. Data are presented as mean of the measured FI compared with control. Not significant, *P < .05, **P < .01, ***P < .001, ****P < .0001. The number of patients and healthy donors tested is reported in each graphic. Dashed lines represent the cutoffs determined from healthy donors as mean of FI + 2× SEM.
Association among platelet apoptosis and clinical biomarkers, thromboembolic complications, and mortality. The correlations between platelet apoptosis parameters and PLT count, as well as D-dimer and SOFA score measured at the same day of platelet testing, were assessed. (A-B) An association was observed between PLT count and PS externalization (A) and cytosolic calcium concentration (B). (C) Moreover, a significant correlation was detected for D-dimer and PS externalization. (D) The clinical relevance of PS externalization was assessed using the SOFA score and revealed a significant positive correlation. Pearson’s correlation coefficients were calculated and are shown in the panels. (E-F) The PS externalization (E) and the cytosolic calcium concentration (F) were determined and compared between COVID-19 patients in the ICU depending on the incidence of thromboembolic complications and mortality, respectively. Data are presented as mean of the measured FI compared with control. Not significant, *P < .05, **P < .01, ***P < .001, ****P < .0001. The number of patients and healthy donors tested is reported in each graphic. Dashed lines represent the cutoffs determined from healthy donors as mean of FI + 2× SEM.
Next, we analyzed the markers of apoptosis in relation to the incidence of thromboembolic complications and mortality during the 30-day follow-up. We found that COVID-19 patients in the ICU with thromboembolic complications had significantly higher levels of PS externalization compared with those without thrombosis (2.85 ± 0.75 vs 0.99 ± 0.20, P = .0340; Figure 2E). In addition, cytosolic Ca2+ concentration was significantly higher in nonsurvivors compared with survivors (4.07 ± 0.39 vs 2.05 ± 0.30, P = .0012; Figure 2F).
Sera from COVID-19 patients in the ICU cause IgG-mediated platelet apoptosis via cross-linking Fcγ receptor IIA
To explore the mechanism of platelet apoptosis in COVID-19, patient sera were incubated with washed platelets from healthy donors. As shown in Figure 3A, COVID-19 sera induced a significantly higher ΔΨm depolarization compared with sera from healthy donors (1.52 ± 0.117 vs 0.958 ± 0.082, P = .0086) and sera from non–COVID-19 patients in the ICU (1.52 ± 0.117 vs 1.099 ± 0.057, P = .0036; Figure 3A; supplemental Figure 2A). Cytosolic Ca2+ concentration was also significantly increased compared with healthy volunteers (1.372 ± 0.074 vs 0.984 ± 0.055, P = .0036; Figure 3B; supplemental Figure 2B) and non–COVID-19 patients in the ICU (1.372 ± 0.074 vs 1.086 ± 0.066, P = .0113; Figure 3B; supplemental Figure 2B). Similarly, enhanced PS externalization was found in platelets on incubation with COVID-19 sera from patients in the ICU compared with healthy donors (1.624 ± 0.126 vs 0.969 ± 0.100, P = .0051; Figure 3C; supplemental Figure 2C) and non–COVID-19 sera from patients in the ICU (1.624 ± 0.126 vs 0.953 ± 0.169, P = .0214; Figure 3C; supplemental Figure 2C). In addition, significantly higher levels of cleaved caspase 9 were found after incubation with sera from COVID-19 patients compared with healthy controls (3.40 ± 0.65 vs 1.00 ± 0.00, P = .0030; Figure 3D-E). In the whole cohort, sera from 13 of 21 (62%) patients induced enhanced ΔΨm depolarization, 8 of 19 (42%) patients had increased cytosolic Ca2+ concentration, and 13 of 21 (62%) patient sera mediated increased PS externalization on platelets of healthy donors (Figure 3F).
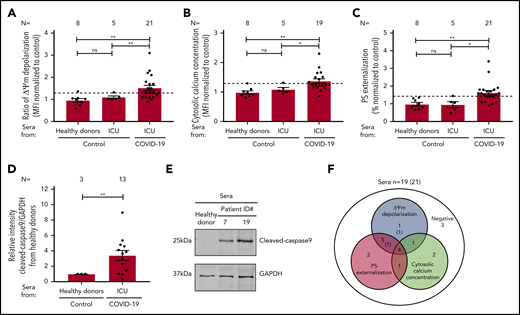
Impact of sera from COVID-19 patients on platelet apoptosis. (A-C) Changes in apoptosis pathways induced by sera from COVID-19 patients in the ICU, non–COVID-19 patients in the ICU (control group), and healthy donors were investigated by assessing the depolarization of the ΔΨm (A), cytosolic calcium concentration (B) (n = 19 because of the lack of biomaterial for 2 patients), and PS externalization (C). (D) Quantification of cleaved caspase 9 level in platelets from healthy donors after incubation with patient sera, normalized to sera from healthy donors. (E) Representative western blot of GAPDH and cleaved caspase 9 proteins. Protein bands were detected with the infrared imaging system (Odyssey, LI.COR). (F) Diagram indicating the number of COVID-19 patients in the ICU positive for each apoptotic parameter: ΔΨm depolarization, cytosolic calcium concentration, and PS externalization. The 2 sera tested only for ΔΨm depolarization and PS externalization are in parentheses. Data are presented as mean ± SEM of the measured FI compared with control. Not significant, *P < .05, **P < .01, ***P < .001, ****P < .0001. The number of sera tested is reported in each graphic. Dashed lines represent the cutoffs determined testing sera from healthy donors as mean of FI + 2× SEM.
Impact of sera from COVID-19 patients on platelet apoptosis. (A-C) Changes in apoptosis pathways induced by sera from COVID-19 patients in the ICU, non–COVID-19 patients in the ICU (control group), and healthy donors were investigated by assessing the depolarization of the ΔΨm (A), cytosolic calcium concentration (B) (n = 19 because of the lack of biomaterial for 2 patients), and PS externalization (C). (D) Quantification of cleaved caspase 9 level in platelets from healthy donors after incubation with patient sera, normalized to sera from healthy donors. (E) Representative western blot of GAPDH and cleaved caspase 9 proteins. Protein bands were detected with the infrared imaging system (Odyssey, LI.COR). (F) Diagram indicating the number of COVID-19 patients in the ICU positive for each apoptotic parameter: ΔΨm depolarization, cytosolic calcium concentration, and PS externalization. The 2 sera tested only for ΔΨm depolarization and PS externalization are in parentheses. Data are presented as mean ± SEM of the measured FI compared with control. Not significant, *P < .05, **P < .01, ***P < .001, ****P < .0001. The number of sera tested is reported in each graphic. Dashed lines represent the cutoffs determined testing sera from healthy donors as mean of FI + 2× SEM.
To further dissect the pathways of the antibody-mediated platelet apoptosis, IgG fractions were isolated from apoptosis-inducing sera tested in the absence or presence of an Fcγ receptor IIA (FcγΡΙΙΑ)-blocking monoclonal antibody (mAb; IV.3). Interestingly, IgG fractions from COVID-19 sera induced significant upregulation in all 3 apoptosis markers. ΔΨm depolarization significantly increased in platelets isolated from COVID-19 patients in the ICU compared with healthy donors (1.54 ± 0.14 vs 1.00 ± 0.00, P = .0051; Figure 4A) and non-ICU COVID-19 patients (1.54 ± 0.14 vs 1.16 ± 0.08, P = .0249; Figure 4A). Cytosolic Ca2+ concentration was increased when comparing healthy donors (1.36 ± 0.05 vs 1.00 ± 0.00, P < .0001) with non-ICU COVID-19 patients (1.36 ± 0.05 vs 1.10 ± 0.02, P < .0005), and a similar cytosolic Ca2+ concentration was found compared with non–COVID-19 patients in the ICU (1.36 ± 0.05 vs 1.23 ± 0.06, P = .651; Figure 4B). PS externalization in platelets was significantly higher in platelets from COVID-19 patients in the ICU compared with healthy volunteers (1.70 ± 0.27 vs 0.70 ± 0.27, P = .0330; Figure 4C) and was nonsignificant compared with non-ICU COVID-19 patients (1.70 ± 0.27 vs 1.04 ± 0.27, P = .7093; Figure 4C) and non–COVID-19 patients in the ICU (1.70 ± 0.27 vs 1.08 ± 0.08, P = .0547; Figure 4C).
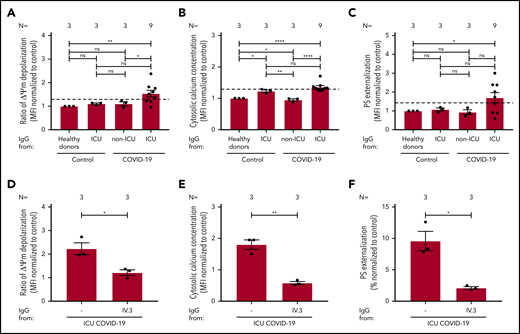
IgG fractions from COVID-19 patients in the ICU induce platelet apoptosis via crosslinking FcγRIIA. (A-C) Changes in apoptosis pathways induced by IgG fraction from COVID-19 patients in the ICU or COVID-19 patients not in the ICU, as well as non–COVID-19 patients in the ICU (control group) and healthy donors, were analyzed by assessing the depolarization of the ΔΨm (A), cytosolic calcium concentration (B), and PS externalization (C) in platelets from 3 different healthy donors. (D-F) The same assays were performed in the presence of the IV.3 monoclonal antibody (mAb) to block FcγRIIA signaling. Each dot represents 2 experiments with platelets from 2 different donors. Data are presented as mean ± SEM of the measured FI compared with control. Not significant, *P < .05, **P < .01, ***P < .001, ****P < .0001. The number of sera tested is reported in each graphic.
IgG fractions from COVID-19 patients in the ICU induce platelet apoptosis via crosslinking FcγRIIA. (A-C) Changes in apoptosis pathways induced by IgG fraction from COVID-19 patients in the ICU or COVID-19 patients not in the ICU, as well as non–COVID-19 patients in the ICU (control group) and healthy donors, were analyzed by assessing the depolarization of the ΔΨm (A), cytosolic calcium concentration (B), and PS externalization (C) in platelets from 3 different healthy donors. (D-F) The same assays were performed in the presence of the IV.3 monoclonal antibody (mAb) to block FcγRIIA signaling. Each dot represents 2 experiments with platelets from 2 different donors. Data are presented as mean ± SEM of the measured FI compared with control. Not significant, *P < .05, **P < .01, ***P < .001, ****P < .0001. The number of sera tested is reported in each graphic.
In contrast, a significant reduction of platelet apoptosis was observed after blocking platelet FcγRIIA: ΔΨm depolarization (2.23 ± 0.25 vs 1.22 ± 0.12, P = .0216; Figure 4D), cytosolic Ca2+ concentration (1.80 ± 0.15 vs 0.58 ± 0.04, P = .0015; Figure 4E), and PS externalization in platelets (9.59 ± 1.52 vs 2.12 ± 0.20, P = .0371; Figure 4F). Of note, protein staining of the isolated IgG showed no hint of IgG aggregates or immune complexes (data not shown).
To elucidate whether apoptosis is driven by loss of mitochondrial integrity, we investigated the impact of cyclosporine A (CsA) and the pan caspase-inhibitor Q-VD-OPh on IgG-mediated platelet apoptosis. CsA efficiently blocked ΔΨm depolarization (2.55 ± 0.27 vs 1.16 ± 0.15, P = .0313; Figure 5A) and PS externalization (5.91 ± 0.84 vs 1.13 ± 0.15, P = .0313; Figure 5B) compared with nontreated platelets. Q-VD-OPh showed no significant inhibition of ΔΨm depolarization (3.70 ± 0.67 vs 3.08 ± 0.75, P = .2188; Figure 5C) but significantly reduced PS externalization (5.25 ± 1.38 vs 3.54 ± 1.06, P = .0313; Figure 5D) compared with nontreated platelets. Taken together, with the observed cleavage of caspase 9, these data indicate that IgG fractions from severe COVID-19 patients induce procoagulant/apoptotic platelets, leading to the expression of PS by different pathways.
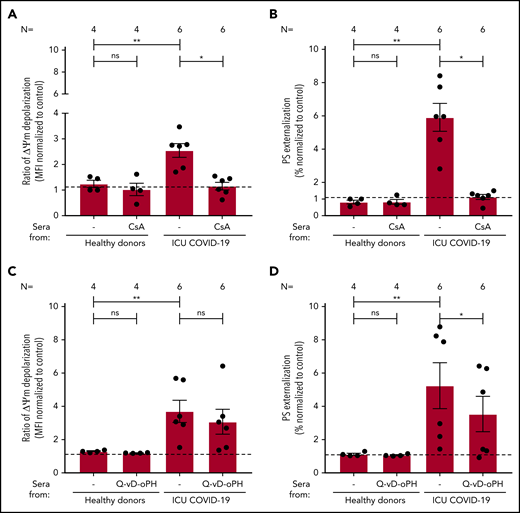
The impact of CsA and Q-VD-OPh on antibody-mediated procoagulant platelets. (A-B) CsA and Q-VD-OPh inhibition of COVID-19–induced ΔΨ depolarization (A) and PS externalization (B). (C-D) CsA significantly inhibits the depolarization of ΔΨm and PS expression in COVID-19–positive patients. Q-VD-OPh was not able to inhibit ΔΨm depolarization (C) but significantly reduced PS externalization (D). Data are presented as mean ± SEM of the measured FI compared with control. Not significant, *P < .05, **P < .01, ***P < .001, ****P < .0001. The number of sera tested is reported in each graphic.
The impact of CsA and Q-VD-OPh on antibody-mediated procoagulant platelets. (A-B) CsA and Q-VD-OPh inhibition of COVID-19–induced ΔΨ depolarization (A) and PS externalization (B). (C-D) CsA significantly inhibits the depolarization of ΔΨm and PS expression in COVID-19–positive patients. Q-VD-OPh was not able to inhibit ΔΨm depolarization (C) but significantly reduced PS externalization (D). Data are presented as mean ± SEM of the measured FI compared with control. Not significant, *P < .05, **P < .01, ***P < .001, ****P < .0001. The number of sera tested is reported in each graphic.
Discussion
COVID-19–infected individuals have a heightened risk for developing thromboembolic complications, and a link has been suggested between low platelet count and severity of disease and mortality.27-29 Here, we show that severe COVID-19 is associated with antibody-mediated upregulation of platelet apoptosis. In addition, we found a correlation between platelet apoptosis markers and SOFA score, plasma levels of D-dimer, and the incidence of thromboembolic complications in severe COVID-19 patients. These data indicate that platelet apoptosis may contribute to sustained inflammation and increased thromboembolic risk in COVID-19 patients and could potentially present a potential therapeutic target.
A subpopulation with severe COVID-19 in our study had an increase in platelet apoptosis markers. The exact mechanism of COVID-19–induced platelet apoptosis has not been studied thus far. Our data show depolarization of the mitochondrial inner transmembrane potential in a subgroup and enhanced cytosolic Ca2+ concentration in most of the patients with severe COVID-19 in the ICU. Together with the increased PS externalization and cleavage of caspase 9, these results suggest that platelet apoptosis in severe COVID-19 is activated via the intrinsic pathway. Apoptosis is also described for septic patients.30 However, apoptosis seems to be different from apoptosis in septic patients. Notably, we could find increased ΔΨm depolarization in platelets of septic patients, but we could not show any increase in cytosolic Ca2+ concentration or in PS externalization patient platelets in the non–COVID-19 control group in the ICU. After incubation of serum from septic patients with platelets of healthy donors, we observed no effect hinting toward an additional serum factor for apoptosis in COVID-19 patients.
No enhanced apoptosis was observed in patient platelets or in healthy platelets after incubation of sera isolated from non–COVID-19 control patients in the ICU, suggesting that the activation of the apoptotic pathway may be a specific effect induced by the COVID-19 infection through a serum component. However, it remains unclear whether apoptosis alone is sufficient to support thromboembolic status in these patients or whether it is linked to platelet activation. Cytosolic Ca2+ is a marker for platelet apoptosis and agonist-induced activation. Therefore, our data indicate that IgGs from severe COVID-19 patients induce procoagulant platelets that might contribute to the increased risk for thromboembolic complications.31,32 In apoptotic platelets, lifetime is shortened, and platelet function is also reduced. In our cohort, severe thrombocytopenia was not common among COVID-19 patients, which was also described for the SARS-CoV-1 in 2008.33 In fact, SARS-CoV-1, which caused the epidemic outbreak in 2003, has also been previously shown to induce apoptosis in Vero cells in a virus replication-dependent manner,34 despite missing thrombocytopenia. Downregulation of Bcl-2, the activation of effector caspase 3, and the upregulation of the proapoptotic protein Bax were detected, suggesting the involvement of the caspase family and the activation of the mitochondrial signaling pathway. In fact, our data also showed that platelets from severe COVID-19 patients have higher levels of cleaved caspase 9.
Prolonged exposure to higher concentrations of platelet agonists has been shown to induce the collapse of mitochondrial membrane potential and subsequent platelet apoptosis.35 Enhanced platelet activation leading to platelet deposition in damaged pulmonary blood vessels has been demonstrated recently for COVID-19.36 To exclude potential contribution of platelet hyperactive status in our cohort, we assessed the impact of patients’ sera on platelet apoptosis.
Recent data showed that autoantibodies from patients with autoimmune thrombocytopenia are able to induce platelet apoptosis.37 The incubation of sera and IgG fractions from severe COVID-19 patients with platelets from healthy donors induced significant changes in apoptosis markers, including ΔΨm depolarization, increased cytosolic Ca2+ concentration, caspase 9 cleavage, and finally PS externalization. More importantly, the antibody-mediated platelet apoptosis was inhibited by blocking the FcγRIIA receptors using a specifc mAb. These data indicate that IgG antibodies contribute to the increased PS expression on platelets of patients with severe COVID-19 infection. However, we cannot exclude other cofactor(s) that could also induce procoagulant platelets in vivo. Interestingly, we found an association between the IgG binding to the Spike protein of SARS-CoV2 and the PS externalization after incubation with patient sera. However, it still remains unclear whether it is a direct IgG–virus complex interaction such as previously described for the influenza virus H1N1 infection38 and dengue fever15 or an indirect effect after binding to platelet nonspecific targets.
Nevertheless, our data indicate that severe COVID-19 is associated with antibody-mediated activation of the intrinsic pathway via crosslinking FcγRIIA receptors by IgG antibodies against to-be-identified target antigen(s). These findings might offer new therapeutic options such as blockade of the FcγRIIA receptor signaling by tyrosine kinase inhibitors, which have been suggested to have a potential use to prevent platelet activation in heparin-induced thrombocytopenia.39
The clinical relevance of platelet apoptosis is supported by the significant negative correlation between PS levels on platelet surface with platelet count, the positive correlation with D-dimer plasma levels, and most importantly with the SOFA score. Meanwhile, it is well established that COVID-19 is associated with increased risk for thromboembolic complications. During the 30-day follow-up, 57% of our patients had thromboembolic complications. Platelets from COVID-19 patients in the ICU with thromboembolic events showed significantly increased externalization of the apoptosis marker PS compared with those with no thrombosis. This finding suggests a link between thromboembolism and platelet apoptosis in COVID-19 as has been suggested for acute pulmonary embolism.40
Our study has limitations. First, as an observational study, we cannot conclude that the reported associations between platelet markers and laboratory parameters, as well as clinical outcomes, are causal or specific for infection with SARS-CoV-2. Second, we cannot exclude the possibility of remaining residual confounding or unmeasured potential confounders. Third, the low number of patients does not enable a final and robust multivariate statistical analysis. In addition, we observed a small difference in age between COVID-19 patients in the ICU and controls. However, age of the patient is not classically associated, and little is known about the impact of age on platelet apoptosis.
It should also be emphasized that it is not clear whether the platelet apoptosis described here is a driver of disease severity or a mere consequence of hypercoagulation in severe (ICU) COVID-19 patients. Indeed, the definitive pathophysiology of COVID-19 and answering questions of causality will likely await the development of model systems for the disease. Our data showed that severe COVID-19 infection induces procoagulant platelets (loss of the mitochondrial potential associated with PS exposure and caspase stimulation). Despite the small number of PS+ platelets that was observed in our study, these cells have a remarkable potential to accelerate the thrombus formation as described in the literature.5 In addition, in ongoing studies, IgG fractions from COVID-19 patients that induced PS externalization showed a marked ability to induce rheologic changes in whole blood, leading to increased thrombus formation. We believe that our findings present another piece of the puzzle and will motivate further research into the role of platelet apoptosis in COVID-19–associated thromboembolic complications.
Data presented in this study may build a basis for future studies to dissect platelet-mediated pathologic mechanisms involved in the progression of COVID-19, and a larger multicenter study is warranted to investigate the predictive power of platelet apoptosis markers in well-phenotyped longitudinal cohorts. To summarize, sera from severe COIVD-19 patients increase PS exposure on platelets. This exposure is triggered by elevated cytosolic calcium, which induces procoagulant and apoptotic cells, and inhibited via caspase inhibition, indicating a significant contribution of the apoptosis pathways.41,42
These data may indicate procoagulant platelet and platelet apoptosis as a potential target of COVID-19 treatment as has been suggested for ARDS43 and other platelet disorders.32,37,44
Data may be requested for academic collaboration from the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank their students Umut Tasdelen, Andreas Witzemann, and Jonas Funk for great contributions, and Wissam Abou Khalel, Karoline Weich, and Flavianna Rigoni for excellent technical support.
This work was supported by grants from the German Research Foundation and from the Herzstiftung (Bakchoul 5158/4 and Tübinger Studie für Gerinnungsstörung bei COVID-19 [Tubinger Study for Coagulopathy in COVID-19]) (T.B.), Deutsche Forschungsgemeinschaft Collaborative Research Centres 240/TR 240 “Platelets—molecular, cellular, and systemic functions in health and disease” (project 374031971) Teilprojekt (TP; subproject) A03 (H.S.), TP B01 (M.G.), TP B07 (P.R. and B.N.), Tübinger Programm zur Frauenförderung (Tubinger Program for Female Scientists)-Gleichstellungsförderung to K.A. (2563-0-0), and “Ministerium für Wissenschaft, Forschung und Kunst Baden-Württemberg” (T.B. and M.G.).
Authorship
Contribution: K.A., T.B., and P.R. designed the study; H.H., M.M., M.B., N.M., M.G., and P.R. were responsible for the treatment of the patients; K.A., S.H., D.R., and M.M. collected and analyzed the clinical data; K.A., I.M., J.Z., L.P., and A.S. performed the experiments; I.M., K.A., T.B., and P.R. collected the data; H.B. provided pathologic findings on thrombosis; K.A., I.M., J.Z., L.P., H.S., B.N., T.B., and P.R. analyzed the data, interpreted the results, and wrote the manuscript; and all authors read and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Tamam Bakchoul, Center for Clinical Transfusion Medicine, University Hospital of Tuebingen, Otfried-Mueller-Strß 4/1, 72076 Tuebingen, Germany; e-mail: tamam.bakchoul@med.uni-tuebingen.de.
