

Key Points
In myeloma patients with a median 4 prior therapy lines, adding dexamethasone to isatuximab increased response rates from 23.9% to 43.6%.
Dexamethasone improved isatuximab efficacy with no detrimental effect on safety, supporting the use of this combination regimen.

Abstract
This phase 2 study evaluated isatuximab as monotherapy or combined with dexamethasone in relapsed/refractory multiple myeloma (RRMM). Patients had RRMM refractory to an immunomodulatory drug (IMiD) and a proteasome inhibitor (PI) or had received ≥3 prior lines of therapy incorporating an IMiD and PI. Patients received isatuximab either as monotherapy (20 mg/kg on days 1, 8, 15, and 22 [once weekly] of cycle 1 followed by 20 mg/kg on days 1 and 15 of subsequent cycles; Isa group) or in combination with dexamethasone (40 mg/d [20 mg/d in patients aged ≥75 years] once weekly; Isa-dex group). Treated patients (N = 164) had received a median of 4 (range, 2-10) prior treatment lines. Patients received a median of 5 (1-24) and 7 (1-22) treatment cycles; at data cutoff, 13 (11.9%) of 109 and 15 (27.3%) of 55 patients remained on treatment in the Isa and Isa-dex arms, respectively. Overall response rate (primary efficacy end point) was 23.9% in the Isa arm and 43.6% in the Isa-dex arm (odds ratio, 0.405; 95% confidence interval, 0.192-0.859; P = .008). Median progression-free survival and overall survival were 4.9 and 18.9 months for Isa, and 10.2 and 17.3 months for Isa-dex. Infusion reactions (mostly grade 1/2) and hematologic abnormalities were the most common adverse events. There was a similar incidence of grade 3 or higher infections in both groups (22.0% and 21.8%). In conclusion, addition of dexamethasone to isatuximab increased response rates and survival outcomes with no detrimental effect on safety. This trial was registered at www.clinicaltrials.gov as #NCT01084252.
Introduction
CD38 is a cell surface receptor, ectoenzyme, and an attractive target for the treatment of multiple myeloma (MM) as it is reliably expressed on malignant plasma cells.1,2 Isatuximab is a CD38-targeting immunoglobulin G1 monoclonal antibody approved in the United States and the European Union in combination with pomalidomide and dexamethasone for the treatment of relapsed/refractory MM (RRMM).3,4 Isatuximab eliminates MM cells via antibody-dependent cellular cytotoxicity, antibody-dependent cellular phagocytosis, complement-dependent cytotoxicity, and direct apoptosis; it may also affect the tumor immunosuppressive environment via inhibition of CD38 adenosinergic activity.5-7
Dexamethasone is a commonly used backbone treatment of multiagent regimens.8-10 Steroids are also used for infusion reaction (IR) prophylaxis with monoclonal antibodies.11,12 In addition to a direct antitumor effect, steroids have immunomodulatory properties,13 the effects of which when given with immunomodulatory monoclonal antibodies have not been characterized.
In the dose-finding stage 1 of an international phase 1/2 study to evaluate the safety and efficacy of single-agent isatuximab in patients with RRMM, patients received doses ranging from 3 to 20 mg/kg.14 In patients with a median of 5 prior lines of therapy, isatuximab was generally well tolerated and active at doses ≥10 mg/kg, with an overall response rate (ORR; primary end point) of 20% to 29%. There was no clear dose–response relationship observed between 10 and 20 mg/kg, and pharmacokinetic (PK) analysis supported a choice of 20 mg/kg once weekly/once every 2 weeks for stage 2 of the study. The antitumor activity of this single-agent isatuximab dose in patients with RRMM was confirmed in the Japanese phase 1/2 ISLANDS (Isatuximab Single Agent Study in Japanese Relapsed AND Refractory Multiple Myeloma Patients; ORR, 36.4% [12 of 33]) study.15
We report results from stage 2 of the international phase 1/2 study, evaluating the efficacy and safety of isatuximab 20 mg/kg once weekly/once every 2 weeks as monotherapy or in combination with dexamethasone.
Methods
Study design
The dose-finding stage of this phase 1/2 randomized, international, multicenter study has been published previously (clinicaltrials.gov identifier #NCT01084252).14 The objective of stage 2 of the study was to evaluate the activity of isatuximab at the selected dose/schedule from the dose-finding stage, as monotherapy and combined with dexamethasone in patients with RRMM.
The study was conducted according to the principles of the Declaration of Helsinki and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Good Clinical Practice. The protocol was approved by the appropriate institutional review boards/ethics committees. All patients provided written informed consent.
Study population
Eligible patients had MM refractory to both an immunomodulatory agent (IMiD) and a proteasome inhibitor (PI), or had been treated with ≥3 prior lines of therapy including an IMiD and a PI. Patients had to have received an alkylating agent, achieved at least a minimal response to a prior line of therapy, and could have received prior stem cell transplant. Patients were aged ≥18 years, with an Eastern Cooperative Oncology Group performance status ≤2 or Karnofsky performance status ≥60, serum creatinine level ≤2 times the upper limit of normal and/or estimated glomerular filtration rate (per Modification of Diet in Renal Disease equation) ≥15 mL/min/1.73 m2, and measurable disease. No other anticancer medications were permitted for the study duration except palliative radiotherapy.
Treatment
Patients were randomized in a 2:1 ratio using an interactive voice/Web response system to receive IV isatuximab 20 mg/kg once weekly (days 1, 8, 15, and 22) for 4 infusions followed by 20 mg/kg every 2 weeks (days 1 and 15), either as monotherapy (Isa arm) or in combination with dexamethasone (Isa-dex arm). Dexamethasone was administered IV or orally at a dose of 40 mg/d (or 20 mg/d for patients aged ≥75 years) on days 1, 8, 15, and 22 of each 28-day cycle.
The isatuximab infusion rate was initially 175 mg/h, increasing to a maximum of 400 mg/h in the absence of IRs. All patients received IR prophylaxis 15 to 30 minutes before isatuximab infusion consisting of diphenhydramine 25 to 50 mg IV (or equivalent), methylprednisolone 100 mg IV (or equivalent; Isa arm only), ranitidine 50 mg IV (or equivalents for each), and acetaminophen (paracetamol) 650 to 1000 mg taken orally. Dexamethasone was administered instead of methylprednisolone in the Isa-dex arm as pre-medication and as part of the combination treatment.
No isatuximab dose reductions were permitted. No more than 2 dose reductions of dexamethasone were allowed (for toxicity); after a decrease, re-escalation to the previous dose level was not permitted. Cycle delay up to 14 days, and dose delay up to 3 days within a cycle, was allowed. In cycle 1 only, if dose delay beyond 3 days was required, the dose could be omitted, and the cycle extended from 28 to 35 days to allow for 4 infusions of isatuximab to be given. No more than 2 consecutive omissions of isatuximab infusion were permitted.
In the Isa-dex arm, if one study drug was prematurely discontinued, patients could continue receiving the other. Patients continued treatment until disease progression, an unacceptable adverse event (AE) occurred, or any other reason for discontinuation.
Outcomes
The primary efficacy end point was ORR, defined as the proportion of patients with a partial response (PR) or better, assessed by the Independent Adjudication Committee (IAC) using International Myeloma Working Group criteria.16 Efficacy was also assessed by investigators using local laboratory results. Disease assessment was made on day 1 of cycle 2, and every 4 weeks, whenever disease progression was suspected, to confirm PR or complete response (CR), and at the end-of-treatment visit.
Secondary efficacy end points included duration of response, clinical benefit response rate (minimal response or better), progression-free survival (PFS), and overall survival (OS).
Patients with documented disease progression were followed up 60 days after the last dose of study treatment (isatuximab or dexamethasone, whichever was stopped last) and then every 3 months until death or the cutoff date. Patients without disease progression at the end of treatment and who had not started another anticancer therapy were followed up for efficacy and safety every 4 weeks until disease progression, death, or the cutoff date.
Safety was evaluated based on physical examination, laboratory test results, and reports of AEs graded per National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03. Antidrug antibodies were assessed on days 1 and 15 of the first cycle, at the first dose of each subsequent cycle, the end of treatment, and 60 days’ posttreatment.
Population PK analysis by using nonlinear mixed effects modeling was conducted based on isatuximab concentrations in blood samples collected before each isatuximab dose. Exploratory analyses investigated change in paraprotein, analysis of Fc γ receptor (FCGR3A) polymorphism from baseline blood samples, the relationship between adaptive immune responses (T-cell receptor [TCR] clonality), and immune phenotyping with parameters of clinical response. TCR DNA sequencing for repertoire profiling was conducted on whole blood samples collected before isatuximab administration on day 1 of cycles 1, 3, and 5 to quantify expansion of individual T-cell clones.
Specific cell protein marker quantification for immunophenotyping was performed on whole blood samples collected before isatuximab administration on day 1 of cycles 1 and 3. Blood samples were lysed with fluorescence-activated cell sorter lysing solution and stained with 6 multi-fluorochrome (8-color) antibody panels and a CD38-fluorescein isothiocyanate multi-epitope antibody that binds to epitopes distinct from the epitope bound by isatuximab (CYT-38F2, Cytognos S.L.). The analysis was performed by using fluorescence-activated cell sorter Navios or LSRII flow cytometers, and data were analyzed by using Infinicyt flow cytometry software (Cytognos S.L.). Immune cell subpopulations were represented as a percentage of total nucleated cells unless indicated otherwise.
DNA from patient blood samples was assessed by using an immunoSEQ Assay (Adaptive Biotechnologies) for TCR rearrangements. Analysis was performed by using prequalified multiplex polymerase chain reaction assays (TR2015CRO-V-019) that included primers targeting the variable (V) and joining (J) gene families. Priming pairs of V and J were used to amplify somatically recombined TCRs. Each initially amplified region was amplified a second time with forward and reverse primers containing a universal sequence and an adaptor sequence needed for DNA sequencing by Illumina.17
The T-cell clonality metric quantitates the extent of clonal expansion by measuring the shape of clone frequency distribution after deep sequencing of the TCRβ gene on genomic DNA extracted from patient blood samples. Values range from 0 to 1; values approaching 1 indicate a nearly monoclonal population.
Statistical analyses
Efficacy and safety analyses were performed on the safety population comprising patients who received ≥1 full or partial dose of isatuximab for the Isa arm and patients who received dexamethasone in addition to isatuximab for the Isa-dex arm. ORR difference between arms was tested for significance by using Fisher’s exact test with a one-sided significance level of 0.025. Estimates of survival (PFS and OS) were calculated by using the Kaplan-Meier method and log-rank test at a one-sided significance level of 0.025. FCGR3A polymorphism analysis was summarized with descriptive statistics. Immunophenotyping analyses were summarized with descriptive statistics and presented according to treatment arm and ORR; statistical tests were not performed due to a limited sample size for some subgroups. P values for the analysis of median changes from baseline of TCR clonality were derived by using a Wilcoxon signed rank test. The other analysis for TCR clonality was summarized with descriptive statistics.
Results
Patients and treatment
The final analysis cutoff date for stage 2 of this phase 2 study was January 21, 2019, twelve months after the last enrolled patient completed the first cycle of treatment. Of the 165 patients randomized to treatment, 109 in the Isa arm and 55 in the Isa-dex arm were treated and included in the safety population. At data cutoff, 13 (11.9%) of 109 patients in the Isa arm and 15 (27.3%) of 55 patients in the Isa-dex arm remained on treatment. The most common reason for treatment discontinuation was disease progression (Isa, 64.2%; Isa-dex, 60.0%) (Figure 1).
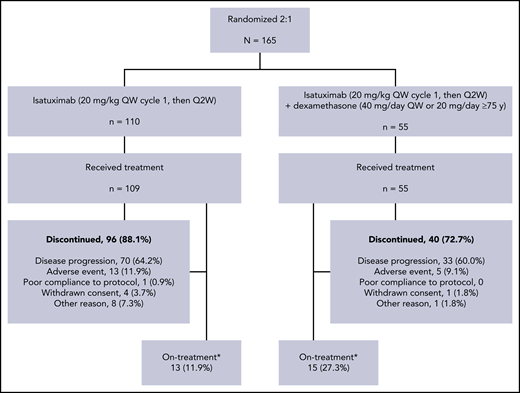
Study design and treatment disposition. Q2W, every 2 weeks; QW, once weekly. *At study cutoff January 21, 2019 (12 months after the last patient who enrolled completed the first cycle of treatment).
Study design and treatment disposition. Q2W, every 2 weeks; QW, once weekly. *At study cutoff January 21, 2019 (12 months after the last patient who enrolled completed the first cycle of treatment).
Demographic data and patient medical history are presented in Table 1. There were no notable imbalances in patient characteristics. In the Isa and Isa-dex arms, 21% and 22% of patients, respectively, had high-risk cytogenetics, defined as del(17p), t(4:14), and/or t(14;16) according to central or local (if central was not available) laboratory data. Four patients in each arm with a creatinine clearance level of 15 to 30 mL/min/1.73 m2 were included.
Patient demographic data, disease characteristics, and treatment history (randomized population)
| Variable | Isatuximab monotherapy (n = 109) | Isatuximab + dexamethasone (n = 55) |
|---|---|---|
| Age, median (range), y | 68 (37–84) | 66 (42–85) |
| ≥75 y | 21 (19.3) | 12 (21.8) |
| Female, n (%) | 58 (53.2) | 26 (47.3) |
| Race, n (%) | ||
| White | 91 (83.5) | 45 (81.8) |
| Black or African American | 5 (4.6) | 3 (5.5) |
| Asian | 0 | 1 (1.8) |
| Other | 13 (11.9) | 6 (10.9) |
| Ethnicity, n (%) | ||
| Hispanic or Latino | 19 (17.4) | 9 (16.4) |
| Not Hispanic or Latino | 90 (82.6) | 46 (83.6) |
| Geographic region, n (%) | ||
| Europe | 62 (56.9) | 31 (56.4) |
| North America | 23 (21.1) | 10 (18.2) |
| Other | 24 (22.0) | 14 (25.5) |
| ECOG PS score (Karnofsky PS), n (%) | ||
| 0 (100%) | 48 (44.0) | 27 (49.1) |
| 1 (80–90%) | 54 (49.5) | 22 (40.0) |
| 2 (60–70%) | 7 (6.4) | 6 (10.9) |
| Time since initial diagnosis, median (range), y | 5.3 (0.7–21.1) | 5.6 (1.2–23.0) |
| Type of myeloma at diagnosis, n (%) | ||
| Immunoglobulin A | 20 (18.3) | 12 (21.8) |
| Immunoglobulin G | 45 (41.3) | 29 (52.7) |
| Light chain (κ and λ) | 33 (30.3) | 10 (18.2) |
| Measurable paraprotein at baseline,*n (%) | ||
| Serum M-protein | 75 (68.8) | 40 (72.7) |
| Urine M-protein | 12 (11.0) | 10 (18.2) |
| κ light chain | 11 (10.1) | 1 (1.8) |
| λ light chain | 10 (9.2) | 3 (5.5) |
| ISS stage at baseline,†n (%) | ||
| I | 27 (24.8) | 15 (27.3) |
| II | 37 (33.9) | 20 (36.4) |
| III | 45 (41.3) | 20 (36.4) |
| Bone marrow plasma cells, median (range), % | 23.5 (0.0-100) | 29.0 (1.0-100) |
| Plasmacytoma at baseline,† n (%) | 21 (19.3) | 17 (30.9) |
| High-risk cytogenetics,†‡ n (%) | 23 (21.1) | 12 (21.8) |
| CrCl 15-60 mL/min/1.73 m2,§ n (%) | 38 (34.9) | 18 (32.7) |
| Prior lines of therapy, median (range) | 4 (2-10) | 4 (2-10) |
| Prior regimens, median (range) | 5 (2-16) | 5 (2-14) |
| ≥1 prior stem cell transplant, n (%) | 59 (54.1) | 28 (50.9) |
| Refractory to an IMiD,ǁ n (%) | 93 (85.3) | 50 (90.9) |
| Refractory to lenalidomide | 77 (70.6) | 34 (61.8) |
| Refractory to pomalidomide | 41 (37.6) | 23 (41.8) |
| Refractory to a PI,ǁ n (%) | 88 (80.7) | 46 (83.6) |
| Refractory to bortezomib | 71 (65.1) | 37 (67.3) |
| Refractory to carfilzomib | 30 (27.5) | 11 (20.0) |
| Refractory to alkylating agent, n (%)ǁ | 75 (68.8) | 34 (61.8) |
| Double refractory, n (%)ǁ,¶ | 76 (69.7) | 42 (76.4) |
| Quadruple refractory, n (%)ǁ,# | 7 (6.4) | 4 (7.3) |
| Refractory to last regimen, n (%)ǁ | 99 (90.8) | 49 (89.1) |
| Variable | Isatuximab monotherapy (n = 109) | Isatuximab + dexamethasone (n = 55) |
|---|---|---|
| Age, median (range), y | 68 (37–84) | 66 (42–85) |
| ≥75 y | 21 (19.3) | 12 (21.8) |
| Female, n (%) | 58 (53.2) | 26 (47.3) |
| Race, n (%) | ||
| White | 91 (83.5) | 45 (81.8) |
| Black or African American | 5 (4.6) | 3 (5.5) |
| Asian | 0 | 1 (1.8) |
| Other | 13 (11.9) | 6 (10.9) |
| Ethnicity, n (%) | ||
| Hispanic or Latino | 19 (17.4) | 9 (16.4) |
| Not Hispanic or Latino | 90 (82.6) | 46 (83.6) |
| Geographic region, n (%) | ||
| Europe | 62 (56.9) | 31 (56.4) |
| North America | 23 (21.1) | 10 (18.2) |
| Other | 24 (22.0) | 14 (25.5) |
| ECOG PS score (Karnofsky PS), n (%) | ||
| 0 (100%) | 48 (44.0) | 27 (49.1) |
| 1 (80–90%) | 54 (49.5) | 22 (40.0) |
| 2 (60–70%) | 7 (6.4) | 6 (10.9) |
| Time since initial diagnosis, median (range), y | 5.3 (0.7–21.1) | 5.6 (1.2–23.0) |
| Type of myeloma at diagnosis, n (%) | ||
| Immunoglobulin A | 20 (18.3) | 12 (21.8) |
| Immunoglobulin G | 45 (41.3) | 29 (52.7) |
| Light chain (κ and λ) | 33 (30.3) | 10 (18.2) |
| Measurable paraprotein at baseline,*n (%) | ||
| Serum M-protein | 75 (68.8) | 40 (72.7) |
| Urine M-protein | 12 (11.0) | 10 (18.2) |
| κ light chain | 11 (10.1) | 1 (1.8) |
| λ light chain | 10 (9.2) | 3 (5.5) |
| ISS stage at baseline,†n (%) | ||
| I | 27 (24.8) | 15 (27.3) |
| II | 37 (33.9) | 20 (36.4) |
| III | 45 (41.3) | 20 (36.4) |
| Bone marrow plasma cells, median (range), % | 23.5 (0.0-100) | 29.0 (1.0-100) |
| Plasmacytoma at baseline,† n (%) | 21 (19.3) | 17 (30.9) |
| High-risk cytogenetics,†‡ n (%) | 23 (21.1) | 12 (21.8) |
| CrCl 15-60 mL/min/1.73 m2,§ n (%) | 38 (34.9) | 18 (32.7) |
| Prior lines of therapy, median (range) | 4 (2-10) | 4 (2-10) |
| Prior regimens, median (range) | 5 (2-16) | 5 (2-14) |
| ≥1 prior stem cell transplant, n (%) | 59 (54.1) | 28 (50.9) |
| Refractory to an IMiD,ǁ n (%) | 93 (85.3) | 50 (90.9) |
| Refractory to lenalidomide | 77 (70.6) | 34 (61.8) |
| Refractory to pomalidomide | 41 (37.6) | 23 (41.8) |
| Refractory to a PI,ǁ n (%) | 88 (80.7) | 46 (83.6) |
| Refractory to bortezomib | 71 (65.1) | 37 (67.3) |
| Refractory to carfilzomib | 30 (27.5) | 11 (20.0) |
| Refractory to alkylating agent, n (%)ǁ | 75 (68.8) | 34 (61.8) |
| Double refractory, n (%)ǁ,¶ | 76 (69.7) | 42 (76.4) |
| Quadruple refractory, n (%)ǁ,# | 7 (6.4) | 4 (7.3) |
| Refractory to last regimen, n (%)ǁ | 99 (90.8) | 49 (89.1) |
CrCl, creatinine clearance; ECOG, Eastern Cooperative Oncology Group; ISS, International Staging System; PS, performance status.
One patient in each arm had data missing.
Based on central laboratory data.
High-risk cytogenetics status, defined as presence of t(4;14) and/or del(17p), was unknown in 48 patients (29 patients in Isa arm and 19 patients in Isa-dex arm), based on central laboratory data (n = 100) and local laboratory data (n = 16).
Eight patients (4 in each arm) had CrCl 15 to 30 mL/min/1.73 m2.
ǁRefractory disease defined according to International Myeloma Working Group criteria.
Refractory to IMiD and PI.
Refractory to lenalidomide, bortezomib, pomalidomide, and carfilzomib.
Patients in both arms had a median of 4 (range, 2-10) prior lines of therapy. In the Isa and Isa-dex arms, 69.7% and 76.4% of patients, respectively, were refractory to both an IMiD and PI (double refractory), and >89% of patients in each arm were refractory to their last regimen.
Patients received a median (range) of 5 (1-24) and 7 (1-22) cycles, and the duration of exposure to treatment was 18.9 (1.0-97.0) and 30.0 (1.0-91.9) weeks in the Isa and Isa-dex arms, respectively. The median relative dose intensity was >97% in both treatment arms.
The duration of the first infusion of isatuximab 20 mg/kg was a median of 5.0 hours in the Isa arm and 4.8 hours in the Isa-dex arm. This was reduced to a median of 4.5 hours in both arms for subsequent infusions.
Efficacy
Best responses per IAC assessment are given in Figure 2. The ORR was 23.9% (26 of 109 patients; 95% confidence interval [CI], 0.162-0.330) in the Isa arm and 43.6% (24 of 55 patients; 95% CI, 0.303-0.577) in the Isa-dex arm (odds ratio [OR], 0.405; 95% CI, 0.192-0.859; P = .008). Responses were either PRs (Isa, 14.7%; Isa-dex, 23.6%) or very good partial responses [VGPRs; Isa, 9.2%; Isa-dex, 20.0%). The overall clinical benefit response rate was 43.1% (47 of 109 patients) and 54.5% (30 of 55 patients) with Isa and Isa-dex, respectively.

Confirmed responses (safety population). Confirmed responses are those assessed by IAC and defined according to International Myeloma Working Group criteria for all treated patients. Serum immunofixation electrophoresis interference has not been investigated and may underestimate depth of response.
Confirmed responses (safety population). Confirmed responses are those assessed by IAC and defined according to International Myeloma Working Group criteria for all treated patients. Serum immunofixation electrophoresis interference has not been investigated and may underestimate depth of response.
Per investigator assessment, the ORR was 27.5% (30 of 109 patients) with Isa and 45.5% (25 of 55 patients) with Isa-dex (OR, 0.456; 95% CI, 0.220-0.951; P < .02). These findings are in accordance with the IAC assessment of disease response.
When disease responses in the Isa and Isa-dex arms were analyzed according to a prespecified subgroup, the ORR was 23.7% (18 of 76) and 38.1% (16 of 42) in those who were double refractory to IMiD and PI; 22.8% (23 of 101) and 41.2% (21 of 51) for those with ≥3 prior lines of therapy, including ≥1 PI and ≥1 IMiD; and 19.6% (10 of 51) and 39.3% (11 of 28) for patients with ≥4 prior lines of therapy whose disease was refractory to ≥2 PIs and ≥2 IMiDs. Based on cytogenetics, the ORR was 4.3% (1 of 23) and 16.7% (2 of 12) for high-risk patients, and 31.6% (18 of 57) and 58.3% (14 of 24) for standard-risk patients in the Isa and Isa-dex arms, respectively.
When responses were examined according to cumulative dexamethasone exposure, ORR in patients with a total cumulative exposure greater than the median (840 mg) for all patients was 70.4% (19 of 27 patients). In patients with a cumulative dexamethasone exposure lower than the median for all patients, ORR was 17.9% (5 of 28 patients).
Median (range) time to first response was 1.0 (1-9) months and 1.0 (1-11) months in the Isa and Isa-dex arms, respectively (supplemental Figure 1, available on the Blood Web site). Median (range) time to best response was 2.0 (2-10) and 2.1 (2-12) months in the Isa and Isa-dex arms (supplemental Figure 1). Median (range) duration of response (PR or better) was 9.3 (5.6-not reached) months with Isa and 14.1 (11.3-19.2) months with Isa-dex. Duration of response was similar when based on investigator assessment (Isa, 7.6 months; Isa-dex, 13.3 months).
PFS per IAC assessment was estimated based on 65 events occurring in 59.6% of patients in the Isa arm and 31 events occurring in 56.4% of patients in the Isa-dex arm. Median PFS was 4.9 months (95% CI, 3.9-7.7) in the Isa arm and 10.2 months (95% CI, 4.9-17.3) in the Isa-dex arm (hazard ratio [HR], 0.677; 95% CI, 0.440-1.043; P < .04) (Figure 3A). PFS was similar when based on investigator assessment of response (Isa, 4.8 months; Isa-dex, 7.6 months).

Survival outcomes according to treatment arm, per IAC assessment (safety population). (A) PFS. (B) OS.
Survival outcomes according to treatment arm, per IAC assessment (safety population). (A) PFS. (B) OS.
The median (range) duration of follow-up for OS was 12.9 (1-23) months with Isa and 13.4 (1-24) months with Isa-dex. OS was estimated based on 50 deaths (45.9% of patients) in the Isa arm and 22 deaths (40.0% of patients) in the Isa-dex arm. Median OS was 18.9 months (95% CI, 13.6-23.1) with Isa and 17.3 months (95% CI, 15.4-not reached) with Isa-dex (HR, 0.799; 95% CI, 0.484-1.321; P = .19) (Figure 3B). OS at 12 months was 63.5% (95% CI, 54.4-72.7) and 73.8% (62.0-85.6), with Isa and Isa-dex, respectively.
In the Isa and Isa-dex arms, 72 (66.1%) of 109 patients and 26 (47.3%) of 55 patients went on to receive further anticancer treatment, with a median time to next treatment of 5.8 months (95% CI, 4.9-8.9) and 15.2 months (95% CI, 6.7-18.4), respectively. These patients most commonly received regimens containing dexamethasone (86.1% and 80.8%) followed by PIs (58.3% and 61.5%), IMiDs (51.4% and 34.6%), alkylating agents (37.5% and 46.2%), or daratumumab (19.4% and 11.5%) in the Isa and Isa-dex arms.
Change in paraprotein
Of the patients with ≥1 postbaseline paraprotein assessment, 36 (37.1%) of 97 patients and 29 (58.0%) of 50 patients had a ≥50% reduction in the Isa and Isa-dex arms, respectively (Figure 4). Two patients in the Isa arm and 5 patients in the Isa-dex arm achieved a 100% reduction in M-protein.

Waterfall plot of best percent change in paraprotein in individual patients (safety population).
Waterfall plot of best percent change in paraprotein in individual patients (safety population).
Safety
More than 91% of patients had ≥1 treatment-emergent AE (TEAE). Any-grade TEAEs and grade 3 or higher TEAEs occurring in ≥5% of all patients along with hematologic abnormalities are presented in Table 2. Grade 3 or higher TEAEs considered to be possibly drug-related occurred in 13.8% and 18.2% of patients in the Isa and Isa-dex arms, respectively.
TEAEs and grade 3 or higher TEAEs (occurring in ≥5% of all patients by preferred term) and hematologic abnormalities (safety population)
| System organ class | Isatuximab (n = 109) | Isatuximab + dexamethasone (n = 55) | ||
|---|---|---|---|---|
| Preferred term | All grades | Grade 3 or higher | All grades | Grade 3 or higher |
| TEAEs, n (%) | 100 (91.7) | 53 (48.6) | 51 (92.7) | 33 (60.0) |
| Injury, poisoning, and procedural complications | 46 (42.2) | 7 (6.4) | 26 (47.3) | 3 (5.5) |
| IRs* | 44 (40.4) | 5 (4.6) | 22 (40.0) | 2 (3.6) |
| Infections and infestations | 65 (59.6) | 24 (22.0) | 33 (60.0) | 12 (21.8) |
| URTI | 14 (12.8) | 1 (0.9) | 8 (14.5) | 1 (1.8) |
| Pneumonia | 10 (9.2) | 7 (6.4) | 6 (10.9) | 3 (5.5) |
| Bronchitis | 7 (6.4) | 1 (0.9) | 4 (7.3) | 3 (5.5) |
| Nasopharyngitis | 4 (3.7) | 0 | 7 (12.7) | 0 |
| Respiratory tract infection | 5 (4.6) | 3 (2.8) | 4 (7.3) | 1 (1.8) |
| Urinary tract infection | 8 (7.3) | 1 (0.9) | 1 (1.8) | 0 |
| Respiratory, thoracic, and mediastinal disorders | 50 (45.9) | 11 (10.1) | 23 (41.8) | 2 (3.6) |
| Cough | 19 (17.4) | 0 | 9 (16.4) | 0 |
| Dyspnea | 19 (17.4) | 2 (1.8) | 8 (14.5) | 0 |
| Nasal congestion | 9 (8.3) | 0 | 2 (3.6) | 0 |
| Metabolism and nutrition disorders | 20 (18.3) | 3 (2.8) | 7 (12.7) | 0 |
| Decreased appetite | 12 (11.0) | 1 (0.9) | 4 (7.3) | 0 |
| Psychiatric disorders | 13 (11.9) | 0 | 17 (30.9) | 3 (5.5) |
| Insomnia | 2 (1.8) | 0 | 14 (25.5) | 1 (1.8) |
| Nervous system disorders | 37 (33.9) | 7 (6.4) | 21 (38.2) | 5 (9.1) |
| Headache | 14 (12.8) | 0 | 8 (14.5) | 0 |
| Gastrointestinal disorders | 41 (37.6) | 3 (2.8) | 27 (49.1) | 4 (7.3) |
| Diarrhea | 21 (19.3) | 0 | 11 (20.0) | 2 (3.6) |
| Nausea | 16 (14.7) | 1 (0.9) | 8 (14.5) | 0 |
| Vomiting | 14 (12.8) | 1 (0.9) | 3 (5.5) | 0 |
| Constipation | 10 (9.2) | 1 (0.9) | 3 (5.5) | 0 |
| Dyspepsia | 2 (1.8) | 0 | 4 (7.3) | 0 |
| Musculoskeletal and connective tissue disorders | 58 (53.2) | 7 (6.4) | 27 (49.1) | 4 (7.3) |
| Back pain | 22 (20.2) | 2 (1.8) | 9 (16.4) | 0 |
| Pain in extremity | 9 (8.3) | 1 (0.9) | 9 (16.4) | 2 (3.6) |
| Arthralgia | 9 (8.3) | 0 | 4 (7.3) | 0 |
| Musculoskeletal chest pain | 9 (8.3) | 0 | 4 (7.3) | 0 |
| Bone pain | 10 (9.2) | 0 | 1 (1.8) | 0 |
| Myalgia | 8 (7.3) | 0 | 3 (5.5) | 1 (1.8) |
| Musculoskeletal pain | 7 (6.4) | 2 (1.8) | 3 (5.5) | 0 |
| General disorders and administration site conditions | 45 (41.3) | 11 (10.1) | 26 (47.3) | 4 (7.3) |
| Fatigue | 19 (17.4) | 3 (2.8) | 10 (18.2) | 0 |
| Asthenia | 8 (7.3) | 1 (0.9) | 6 (10.9) | 1 (1.8) |
| Chills | 9 (8.3) | 0 | 5 (9.1) | 0 |
| Pyrexia | 5 (4.6) | 0 | 7 (12.7) | 1 (1.8) |
| Peripheral edema | 5 (4.6) | 0 | 5 (9.1) | 0 |
| Disease progression | 7 (6.4) | 7 (6.4) | 2 (3.6) | 2 (3.6) |
| Pain | 6 (5.5) | 1 (0.9) | 3 (5.5) | 0 |
| Hematologic abnormalities, n/N (%)† | ||||
| Anemia | 104/109 (95.4) | 25/109 (22.9) | 52/54 (96.3) | 8/54 (14.8) |
| Platelet count decreased | 73/109 (67.0) | 20/109 (18.3) | 32/54 (59.3) | 8/54 (14.8) |
| Neutrophil count decreased | 72/109 (66.1) | 20/109 (18.3) | 20/54 (37.0) | 7/54 (13.0) |
| Lymphocyte count decreased | 91/109 (83.5) | 30/109 (27.5) | 47/54 (87.0) | 26/54 (48.1) |
| System organ class | Isatuximab (n = 109) | Isatuximab + dexamethasone (n = 55) | ||
|---|---|---|---|---|
| Preferred term | All grades | Grade 3 or higher | All grades | Grade 3 or higher |
| TEAEs, n (%) | 100 (91.7) | 53 (48.6) | 51 (92.7) | 33 (60.0) |
| Injury, poisoning, and procedural complications | 46 (42.2) | 7 (6.4) | 26 (47.3) | 3 (5.5) |
| IRs* | 44 (40.4) | 5 (4.6) | 22 (40.0) | 2 (3.6) |
| Infections and infestations | 65 (59.6) | 24 (22.0) | 33 (60.0) | 12 (21.8) |
| URTI | 14 (12.8) | 1 (0.9) | 8 (14.5) | 1 (1.8) |
| Pneumonia | 10 (9.2) | 7 (6.4) | 6 (10.9) | 3 (5.5) |
| Bronchitis | 7 (6.4) | 1 (0.9) | 4 (7.3) | 3 (5.5) |
| Nasopharyngitis | 4 (3.7) | 0 | 7 (12.7) | 0 |
| Respiratory tract infection | 5 (4.6) | 3 (2.8) | 4 (7.3) | 1 (1.8) |
| Urinary tract infection | 8 (7.3) | 1 (0.9) | 1 (1.8) | 0 |
| Respiratory, thoracic, and mediastinal disorders | 50 (45.9) | 11 (10.1) | 23 (41.8) | 2 (3.6) |
| Cough | 19 (17.4) | 0 | 9 (16.4) | 0 |
| Dyspnea | 19 (17.4) | 2 (1.8) | 8 (14.5) | 0 |
| Nasal congestion | 9 (8.3) | 0 | 2 (3.6) | 0 |
| Metabolism and nutrition disorders | 20 (18.3) | 3 (2.8) | 7 (12.7) | 0 |
| Decreased appetite | 12 (11.0) | 1 (0.9) | 4 (7.3) | 0 |
| Psychiatric disorders | 13 (11.9) | 0 | 17 (30.9) | 3 (5.5) |
| Insomnia | 2 (1.8) | 0 | 14 (25.5) | 1 (1.8) |
| Nervous system disorders | 37 (33.9) | 7 (6.4) | 21 (38.2) | 5 (9.1) |
| Headache | 14 (12.8) | 0 | 8 (14.5) | 0 |
| Gastrointestinal disorders | 41 (37.6) | 3 (2.8) | 27 (49.1) | 4 (7.3) |
| Diarrhea | 21 (19.3) | 0 | 11 (20.0) | 2 (3.6) |
| Nausea | 16 (14.7) | 1 (0.9) | 8 (14.5) | 0 |
| Vomiting | 14 (12.8) | 1 (0.9) | 3 (5.5) | 0 |
| Constipation | 10 (9.2) | 1 (0.9) | 3 (5.5) | 0 |
| Dyspepsia | 2 (1.8) | 0 | 4 (7.3) | 0 |
| Musculoskeletal and connective tissue disorders | 58 (53.2) | 7 (6.4) | 27 (49.1) | 4 (7.3) |
| Back pain | 22 (20.2) | 2 (1.8) | 9 (16.4) | 0 |
| Pain in extremity | 9 (8.3) | 1 (0.9) | 9 (16.4) | 2 (3.6) |
| Arthralgia | 9 (8.3) | 0 | 4 (7.3) | 0 |
| Musculoskeletal chest pain | 9 (8.3) | 0 | 4 (7.3) | 0 |
| Bone pain | 10 (9.2) | 0 | 1 (1.8) | 0 |
| Myalgia | 8 (7.3) | 0 | 3 (5.5) | 1 (1.8) |
| Musculoskeletal pain | 7 (6.4) | 2 (1.8) | 3 (5.5) | 0 |
| General disorders and administration site conditions | 45 (41.3) | 11 (10.1) | 26 (47.3) | 4 (7.3) |
| Fatigue | 19 (17.4) | 3 (2.8) | 10 (18.2) | 0 |
| Asthenia | 8 (7.3) | 1 (0.9) | 6 (10.9) | 1 (1.8) |
| Chills | 9 (8.3) | 0 | 5 (9.1) | 0 |
| Pyrexia | 5 (4.6) | 0 | 7 (12.7) | 1 (1.8) |
| Peripheral edema | 5 (4.6) | 0 | 5 (9.1) | 0 |
| Disease progression | 7 (6.4) | 7 (6.4) | 2 (3.6) | 2 (3.6) |
| Pain | 6 (5.5) | 1 (0.9) | 3 (5.5) | 0 |
| Hematologic abnormalities, n/N (%)† | ||||
| Anemia | 104/109 (95.4) | 25/109 (22.9) | 52/54 (96.3) | 8/54 (14.8) |
| Platelet count decreased | 73/109 (67.0) | 20/109 (18.3) | 32/54 (59.3) | 8/54 (14.8) |
| Neutrophil count decreased | 72/109 (66.1) | 20/109 (18.3) | 20/54 (37.0) | 7/54 (13.0) |
| Lymphocyte count decreased | 91/109 (83.5) | 30/109 (27.5) | 47/54 (87.0) | 26/54 (48.1) |
URTI, upper respiratory tract infection.
IRs of grade 3 or higher were prespecified as an AE of special interest.
Based on laboratory values measured during the on-treatment period.
IRs and hematologic abnormalities were the most common TEAEs (Table 2). IRs were reported in 44 (40.4%) of 109 patients and 22 (40.0%) of 55 patients in the Isa and Isa-dex arms, respectively. Most reactions were grade 1 or 2; five (4.6%) patients in the Isa arm and two (3.6%) patients in the Isa-dex arm had a grade 3 or higher IR. Overall, 6 (3.7%) of 164 patients discontinued the study due to IRs. Most IRs (98.6%) occurred with the first infusion, and no IRs occurred from the third infusion onward. There was a similar incidence of laboratory-measured grade 3 or higher neutropenia (18.3% and 13.0%) and grade 3 or higher infections (22.0% and 21.8%) in the Isa and Isa-dex arms, respectively.
The addition of dexamethasone increased the incidence of any-grade TEAEs in the System Organ Class psychiatric disorders (from 11.9% to 30.9%, driven by insomnia [Isa, 1.8%; Isa-dex, 25.5%]), and the System Organ Class gastrointestinal disorders (Table 2). The gastrointestinal TEAEs most frequently reported in the Isa-dex arm were diarrhea (11 patients [20.0%], nausea (8 patients [14.5%]), and dyspepsia (4 patients [7.3%]); of these, only dyspepsia appeared elevated compared with the Isa arm (2 patients [1.8%]), and there was an accompanying decrease in vomiting and constipation.
Serious TEAEs occurred in 51 (46.8%) of 109 patients and 25 (45.5%) of 55 patients in the Isa and Isa-dex arms, respectively. The most commonly occurring serious TEAEs, which were predominantly respiratory infections, disease progression, and infusion-related reactions, are presented in supplemental Table 1. These were considered possibly drug related in 14 (12.8%) of 109 patients in the Isa arm and 6 (10.9%) of 55 patients in the Isa-dex arm.
Treatment was discontinued as a result of a TEAE in 13 (11.9%) of 109 patients in the Isa arm and 5 (9.1%) of 55 patients in the Isa-dex arm. There were 19 fatal TEAEs (14 [12.8%] in the Isa arm and 5 [9.1%] in the Isa-dex arm). Two deaths were considered related to treatment with isatuximab (1 case of respiratory tract infection and 1 case of sepsis); no deaths were considered related to dexamethasone.
One of the 160 evaluable patients tested positive for anti-isatuximab antibodies during the on-treatment period (Isa arm; cycle 1 day 15) but was negative when next tested (cycle 2 day 1) and remained so for the remainder of treatment (4 cycles started).
PK variables
At the dosing regimen used in this study (20 mg/kg once weekly in cycle 1, then every 2 weeks), the estimated median (50th percentile) and 90th percentile time to reach steady state was 43 days and 89 days, respectively, in the overall isatuximab PK population. There was a 3.02 and 2.85 ratio in the geometric mean of the trough concentration between cycle 2 day 1 and cycle 1 day 8 in the Isa and Isa-dex arms, respectively. There was a 1.68 and 1.92 ratio in the geometric means of the concentration at the end of infusion between cycle 4 day 1 and cycle 1 day 1 in the Isa and Isa-dex arms, respectively. The PK analysis showed that the addition of dexamethasone did not modify the PK parameters of isatuximab.
Fc γ receptor polymorphism
ORRs seemed similar between the FCGR3A F158V gene variants, including those with high affinity for NK cell–immunoglobulin G binding (V/V) and low binding affinity (V/F or F/F).18,19 In the Isa group, the ORR was 22.2% (2 of 9) with V/V; 22.5% (9 of 40) with F/F; and 24.1% (14 of 58) with F/V. In the Isa-dex group, the ORR was 53.8% (7 of 13) with V/V; 42.9% (6 of 14) with F/F; and 40.0% (10 of 25) with F/F.
Immune monitoring and TCR repertoire
Peripheral blood immune monitoring showed an overall decrease in total NK cells from cycle 1 day 1 to cycle 3 day 1 that was unaffected by the addition of dexamethasone and was similar in responders and nonresponders (supplemental Figure 2A). However, a detailed analysis of the NK cell compartment revealed a trend for median values of the CD38+ NK cells, particularly the CD38+ CD56+dim, CD16+bright cytotoxic subset, to remain stable in responding patients (supplemental Figure 2B-C). Although we may have expected the CD38+ NK cells to be depleted by the anti-CD38 regimen, as shown for daratumumab,20 this did not seem to be the case among responders to isatuximab. The reasons for this are unclear; however, we may speculate that these cells continue to contribute to the anti-myeloma response through ADCC.
CD3+ T cells and the cytotoxic CD8+ T-cell subsets showed a modest increment from cycle 1 day 1 to cycle 3 day 1 in all patients except responders to Isa-dex (supplemental Figure 2D-E), whereas CD4+ T cells remained stable (data not shown). A trend for reduced median values of regulatory T cells was observed, particularly in responders to Isa-dex (supplemental Figure 2F).
Isatuximab monotherapy induced expansion of clonotypic T cells, as shown by the median increase in TCR clonality from cycle 1 to cycle 3 (24.4%; P < .0001) and cycle 5 (48.1%; P < .0001) in peripheral blood, suggesting a T-cell immune response. In the Isa-dex arm, there was a similar expansion of clonotypic T cells from cycle 1 to cycle 3 (39.3%; P < .0001) and cycle 5 (51.6%; P < .0001) (Figure 5), indicating that TCR clonality was not modified by the addition of dexamethasone.

The addition of dexamethasone, with its increase in ORR, was not associated with a further increase in TCR clonality. Also, the median changes in T-cell clonality from baseline to cycle 3 day 1 among responders were similar in both the Isa (31.8%) and Isa-dex (39.8%) arms. However, higher depth of response seemed to be associated with increased TCR clonality; median changes from baseline were higher in patients achieving VGPR or better (Isa, 61.3%; Isa-dex, 61.5%) compared with PR (Isa, 23.8%; Isa-dex, 24.7%) and PR or worse (Isa, 23.7%; Isa-dex, 23.5%) and was independent of treatment arm.
Discussion
In phase 2 stage 2 of this randomized phase 1/2 study in heavily pretreated patients with RRMM, responses rates were clinically significant with isatuximab 20 mg/kg once weekly (cycle 1)/every 2 weeks as monotherapy (ORR of 23.9% by IAC, 27.5% by investigator assessment) and in combination with dexamethasone (ORR of 43.6% by IAC, 45.5% by investigator assessment).
The study was not powered for comparison between treatment arms. However, the addition of dexamethasone to isatuximab monotherapy resulted in a significant increase in ORR (OR, 0.405; P = .008) with improved depth of response and evidence of increased PFS (HR, 0.677 [P < .04]; median PFS of 4.9 months with Isa and 10.2 months with Isa-dex). No patients in this study achieved CR; however, the true CR rate may be underestimated because of isatuximab interference with M-protein measurement.21 The HR for OS favored Isa-dex compared with Isa but did not reach statistical significance (HR, 0.799; P = .19); after 12 months, 64% and 74% of patients in the Isa and Isa-dex arms were still alive, respectively. OS is affected by subsequent therapy, and there was a longer time to next treatment in the Isa-dex arm, with more patients in the Isa arm receiving subsequent IMiDs, PIs, and daratumumab than in the Isa-dex arm. It was interesting to observe that median PFS with Isa-dex in this study was longer (10.2 months) than that reported with daratumumab monotherapy (3.7 months), although the difference in 12 month OS was not as wide (74% vs 65%, respectively). However, such direct comparisons must be interpreted with caution, as we note that differences in participating countries for our study (North and South America, Europe, Russia, and Israel) compared with the SIRIUS (Daratumumab Monotherapy in Patients With Treatment-Refractory Multiple Myeloma) study (United States, Canada, and Spain) could have influenced the type and availability of subsequent rescue therapies.
Isatuximab was generally well tolerated at a dosage of 20 mg/kg once weekly (cycle 1)/every 2 weeks. As seen previously,14,22,23 IRs were the most common TEAE, occurring in ∼40% of patients in both arms. All patients received infusion management prophylaxis, and IRs were mostly mild to moderate in nature, with <5% of patients experiencing grade 3 or higher IRs. Discontinuations due to a TEAE were low, and thus the therapeutic potential of isatuximab does not seem to be limited by its safety profile.
The addition of dexamethasone to isatuximab resulted in moderate steroid-related toxicity that was manageable, did not change the incidence of IRs or high-grade infections, and resulted in a similar number of serious AEs and discontinuations due to AEs or fatal AEs. There was no increase in hematologic toxicity or infections with Isa-dex compared with Isa.
There were no meaningful differences in ORR with isatuximab in patients with the NK cell high-affinity FCGR3A 158V/V variant compared with patients with the low-affinity 158F/F or 158F/V variants. Patients may benefit from isatuximab regardless of FCGR3A genotypes.
Immune monitoring of regulatory T-cell and NK cell populations showed similar kinetics with both Isa and Isa-dex, and confirmed that dexamethasone had no detrimental effect in the number of CD8+ cytotoxic T cells. Interestingly, there was a lower decrease in the cytotoxic subset of NK CD38+ cells in responders compared with nonresponders. Abnormal T-cell function is associated with immunosenescence in the MM tumor microenvironment.24 Induction of clonal T-cell expansion has previously been observed as part of the immunomodulatory action of anti-CD38 antibodies.25 We confirm that T-cell clonal expansion also occurs with isatuximab treatment, suggesting progress toward reconstitution of T-cell adaptive immunity. This seems to coincide with an immune response to myeloma-associated antigens (including CD38) in patients treated with isatuximab monotherapy.26 TCR clonality was not further modified by the addition of dexamethasone, despite a known inhibitory effect of corticosteroids on naive T-cell differentiation.27 Thus, the increased response rate in the Isa-dex arm did not seem to be associated with increased T-cell clonality; however, T-cell clonality did seem greater among patients with deeper responses (VGPR or better vs PR or worse) when measured on day 1 of cycle 3.
The ORR of 44% with Isa-dex in this study compares favorably to the clinical response rate observed for pomalidomide plus dexamethasone (33% ORR),28 carfilzomib plus dexamethasone (55%),29 selinexor plus dexamethasone (22%), and the novel alkylating agent melflufen plus dexamethasone (40%)30 in other populations of heavily pretreated patients with RRMM. Clinical trials of daratumumab monotherapy in patients with RRMM, which included corticosteroids as part of the premedication and postmedication IR prophylaxis, observed ORRs in the range of 29% to 36%.31,32 Uniquely, in the Isa-dex arm, there was a median PFS benefit of 10.2 months, which is longer than has been shown with IMiDs33 or with daratumumab monotherapy (median PFS range, 3.7-5.6 months).31,32
The efficacy and safety of isatuximab 10 mg/kg in combination with pomalidomide plus dexamethasone for the treatment of RRMM were confirmed in the phase 3 ICARIA-MM (Isatuximab 10 mg/kg QW/Q2W in Combination with pomalidomide And low-dose dexamethasone veRsus pomalidomide and low-dose dexamethasone In patients with relapsed And refractory Multiple Myeloma) study (#NCT02990338).34 In ICARIA-MM, PFS was significantly improved with isatuximab in combination with pomalidomide plus dexamethasone vs pomalidomide and dexamethasone (median, 11.53 vs 6.47 months; HR, 0.596; 95% CI, 0.436-0.814; P = .001).
Despite 2 patients in the Isa arm and 5 patients in the Isa-dex arm achieving a 100% reduction in M-protein, there were no CRs recorded. However, interference of therapeutic antibodies with immunofixation and serum protein electrophoresis assays used in response assessments may therefore lead to underestimation of CR and VGPR.35
The addition of dexamethasone to isatuximab resulted in a clear increase in response, depth of response, and a trend toward improved long-term outcomes with no detrimental effect on safety. This study provides the rationale for including dexamethasone in effective combination regimens with isatuximab.
Qualified researchers can request access to patient-level data and related study documents, including the clinical study report, study protocol with any amendments, blank case report forms, statistical analysis plan, and data set specifications. Patient-level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi’s data-sharing criteria, eligible studies, and process for requesting access are available at: https://www.clinicalstudydatarequest.com.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank all patients and investigators involved in the study. They are grateful to Kathryn Corzo and Ai-Min Hui for contributing to the development of the study design. Medical writing support (including development of a draft outline and subsequent drafts in consultation with the authors, assembling tables and figures, collating author comments, copyediting, fact checking, and referencing) was provided by Julianna Solomons at Aspire Scientific, and funded by Sanofi Genzyme.
This study was funded by Sanofi Genzyme.
Authorship
Contribution: J.Y.Y., S.M., and R.V. were involved in designing the study; M.D., S.B., P.A., M. Capra, M. Cavo, C.C., C.G., V.H., M.J., V.V., E.Y.R., and R.V. were study investigators; B.P. performed the immunophenotyping analysis; J.Y.Y, R.S., M.H., and S.M. analyzed the data; and all authors critically reviewed and approved the submitted manuscript.
Conflict-of-interest disclosure: M.D. has received honoraria from Amgen, Bristol Myers Squibb, Celgene, Janssen, and Takeda. S.B. reports consultancy for Janssen-Cilag and Takeda; has received honoraria from Amgen, Bristol Myers Squibb, Celgene, and Janssen; and an advisory role with Amgen, Celgene, Janssen, and Karyopharm. P.A. reports consultancy for Takeda and an advisory role with Amgen, Janssen, and Takeda. M. Capra reports speakers bureau for Amgen, Janssen, and Roche. M. Cavo reports consultancy, honoraria, and speakers bureau for Amgen, Bristol Myers Squibb, Celgene, Janssen, and Takeda; and other from Janssen and Celgene. C.C. has received travel/accommodation expenses from Amgen. C.G. reports consultancy for Celgene, Millennium, and Onyx; has received honoraria from Amgen, Bristol Myers Squibb, Celgene, and Millennium; reports speakers bureau and travel/accommodation expenses from Celgene and Millennium; and research funding from Celgene. V.H. reports consultancy/advisory board for AbbVie, Amgen, Celgene, Janssen, Sanofi, and Takeda; and speakers bureau for Amgen, Celgene, Janssen, Sanofi, and Takeda. M.J. reports consultancy/honoraria/speakers bureau for AbbVie, Celgene, Janssen, Novartis, and Takeda. V.V. reports consultancy for Biocad, Janssen, and Roche; and speakers bureau for Astellas, Bristol Myers Squibb, Janssen, Roche, and Takeda. J.Y.Y., R.S., and S.M. are employees of Sanofi. M.H. is an employee of Ividata, working for Sanofi. B.P. has received honoraria for lectures from and membership on advisory boards with Amgen, Bristol Myers Squibb, Celgene, Janssen, Merck, Novartis, Roche, and Sanofi; unrestricted grants from Celgene, EngMab, Sanofi, and Takeda; and consultancy for Celgene, Janssen, Sanofi, and Takeda. R.V. has received honoraria and travel expenses from Bristol Myers Squibb, Celgene, Janssen, Merck, Novartis, Onyx, and Takeda; and research funding from Onyx and Takeda. E.Y.R. declares no competing financial interests.
Correspondence: Meletios Dimopoulos, National and Kapodistrian University of Athens, School of Medicine, Department of Clinical Therapeutics, Alexandra Hospital, 80 Vas. Sofias, 11528 Athens, Greece; e-mail: mdimop@med.uoa.gr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal