

Key Points
As a result of low glutathione and glutathione peroxidase 4 levels and high 5-lipoxygenase expression, DMF induces ferroptosis in GCB DLBCL.
In ABC DLBCL, DMF induces succination of kinases IKK2 and JAK1, thus inhibiting NF-κB and JAK/STAT survival signaling.

Abstract
Despite the development of novel targeted drugs, the molecular heterogeneity of diffuse large B-cell lymphoma (DLBCL) still poses a substantial therapeutic challenge. DLBCL can be classified into at least 2 major subtypes (germinal center B cell [GCB]-like and activated B cell [ABC]-like DLBCL), each characterized by specific gene expression profiles and mutation patterns. Here we demonstrate a broad antitumor effect of dimethyl fumarate (DMF) on both DLBCL subtypes, which is mediated by the induction of ferroptosis, a form of cell death driven by the peroxidation of phospholipids. As a result of the high expression of arachidonate 5-lipoxygenase in concert with low glutathione and glutathione peroxidase 4 levels, DMF induces lipid peroxidation and thus ferroptosis, particularly in GCB DLBCL. In ABC DLBCL cells, which are addicted to NF-κB and STAT3 survival signaling, DMF treatment efficiently inhibits the activity of the IKK complex and Janus kinases. Interestingly, the BCL-2–specific BH3 mimetic ABT-199 and an inhibitor of ferroptosis suppressor protein 1 synergize with DMF in inducing cell death in DLBCL. Collectively, our findings identify the clinically approved drug DMF as a promising novel therapeutic option in the treatment of both GCB and ABC DLBCLs.
Introduction
Diffuse large B-cell lymphoma (DLBCL) represents the most frequent malignant lymphoma in adults, accounting for ∼30% to 40% of cases, and is characterized by an aggressive clinical course.1,2 With standard-of-care first-line multi-agent chemotherapy, approximately two-thirds of patients show a durable response, whereas the remaining patients are refractory to this first-line therapy or relapse after an initial response.3-5 Although DLBCL constitutes a clinically heterogeneous group, gene expression profiling has led to the identification of 2 major subtypes: germinal center B cell (GCB)-like and activated B cell (ABC)-like DLBCLs.6-9 Mutations (eg, CD79, CARD11, MYD88, or TNFAIP3) leading to constitutive NF-κB activation are enriched in the ABC DLBCL subtype; however, GCB DLBCL frequently harbors alterations in genes, such as EZH2, CREBBP, or SGK1, as well as BCL2 translocations.10-16 As a result of chronic NF-κB activity, ABC DLBCL cells constitutively produce cytokines, such as interleukin-6 (IL-6) and IL-10, which promote the activation of STAT3 in an auto- or paracrine manner.17 STAT3 activation is mediated by Janus kinases (JAKs), which phosphorylate STAT3 at tyrosine residues, resulting in its dimerization, nuclear translocation, and DNA binding and eventually in the expression of prosurvival target genes.17,18
Ferroptosis is a nonapoptotic form of cell death characterized by the accumulation of phospholipid peroxides and specific changes in cellular morphology, including mitochondrial shrinkage.19-21 As the name implies, the transition metal iron plays an essential role in the initiation and execution of ferroptosis, which involves the oxidation of polyunsaturated fatty acids. Consequently, iron-chelating agents, such as deferoxamine (DFX), can be experimentally used in certain concentrations to impair ferroptosis. In addition to iron-dependent autoxidation of lipids, lipoxygenases (LOXs), which catalyze the peroxidation of polyunsaturated fatty acids during leukotriene biosynthesis, potentially modulate ferroptosis susceptibility, although their precise role in ferroptosis is under debate.22-26 Lipid peroxides can be detoxified in a glutathione (GSH)-consuming manner by the selenoprotein glutathione peroxidase 4 (GPX4), the only enzyme capable of reducing lipid peroxides in membranes.27,28 Well-established ferroptosis inducers inhibit GPX4 activity either directly, such as RAS-selective lethal 3 (RSL3), or indirectly (eg, erastin) by blocking cystine import via the system xc− antiporter and thus decreasing GSH synthesis.19,20 Even if GPX4 is inhibited, lipophilic antioxidants, such as ferrostatin-1 (Fer-1) or α-tocopherol, can prevent lipid peroxidation and thus ferroptosis induction.21,28
Dimethyl fumarate (DMF) is a US Food and Drug Administration–approved drug used as first-line treatment of relapsing-remitting multiple sclerosis or as a systemic medication for moderate to severe psoriasis. On a molecular level, the beneficial pharmacological effects of the electrophile DMF are not well understood, but several modes of action have been proposed. It has been shown that DMF exerts a neuroprotective effect by activating the nuclear factor erythroid 2–related factor 2 (Nrf2) pathway, which promotes the expression of several antioxidant proteins and thus protects against reactive oxygen species generated during injury and inflammation.29-31 Other studies suggest that DMF reacts directly and irreversibly with accessible cysteines of a variety of proteins involved in redox regulation, glycolysis, as well as NF-κB and HIF-1α signaling.30,32-36
In this study, we demonstrate a robust antilymphoma effect of DMF on both major DLBCL subtypes. DMF inhibited NF-κB and JAK/STAT3 survival signaling in ABC DLBCL, and it efficiently induced ferroptotic cell death in GCB DLBCL cells. Combination of DMF treatment with inhibitors of the ferroptosis suppressor protein 1 (FSP1) or with BH3 mimetics synergistically induced cell death in DLBCL cells, highlighting the promising therapeutic potential of DMF in the treatment of this malignancy.
Materials and methods
Cell culture, transfection, lentiviral and retroviral transduction, and survival assays
Protocols are provided in the supplemental Data (available on the Blood Web site).
Quantification of cellular GSH levels
Levels of reduced (GSH) and oxidized (GSSG) glutathione in cellular lysates were quantified using the GSH/GSSG-Glo Assay (Promega) according to the manufacturer’s protocol.
Analysis of lipid peroxidation
To monitor ferroptosis induction, cells were stained with the lipid peroxidation sensor BODIPY 581/591 undecanoic acid (BODIPY C11; Thermo Fisher Scientific). In brief, cells were stained with 2 μM of BODIPY C11 in Hanks' balanced salt solution for 15 minutes at 37°C. After washing, dead cells were stained using SYTOX Blue dead cell stain (Thermo Fisher Scientific). The mean fluorescence intensity of oxidized BODIPY C11 was quantified by flow cytometry (BD LSRII) and normalized to the mean fluorescence intensity of the reduced probe.
Cell lysis, nuclear fractionation, immunoprecipitation, mass spectrometry, and immunoblotting
Protocols are provided in the supplemental Data.
Gene expression profiling, gene set enrichment analysis, and quantitative real-time polymerase chain reaction
Protocols are provided in the supplemental Data.
Analysis of ALOX5 promoter methylation, DNA binding of NF-κB (TransAM), and immunohistochemistry
Protocols are provided in the supplemental Data.
Xenograft mouse models, zebrafish husbandry, and yolk sac transplantation
Protocols are provided in the supplemental Data.
Results
DMF treatment impairs DLBCL growth
Because DMF has been shown to inhibit lymphocyte activation and proliferation in multiple sclerosis and psoriasis patients, we tested its potential to impair the growth of various human lymphoma cell lines. Strikingly, the survival of most DLBCL cell lines was markedly reduced by treatment with 20 μM DMF (Figure 1A-B; supplemental Figure 1A-B). DMF treatment was cytotoxic for both major DLBCL subtypes: GCB DLBCL (Figure 1A; supplemental Figure 1A,C) and ABC DLBCL (Figure 1B; supplemental Figure 1B,D). Whereas mantle cell lymphoma (MCL) cell lines exhibited a similar sensitivity toward DMF (supplemental Figure 2A), the growth of myeloid cell lines derived from lymphoma/leukemia patients and of various carcinoma/melanoma cell lines was not affected by treatment with 20 μM DMF (Figure 1C; supplemental Figure 2B). These results indicate that B-cell lymphomas, such as DLBCL and MCL, exhibit an increased DMF sensitivity.
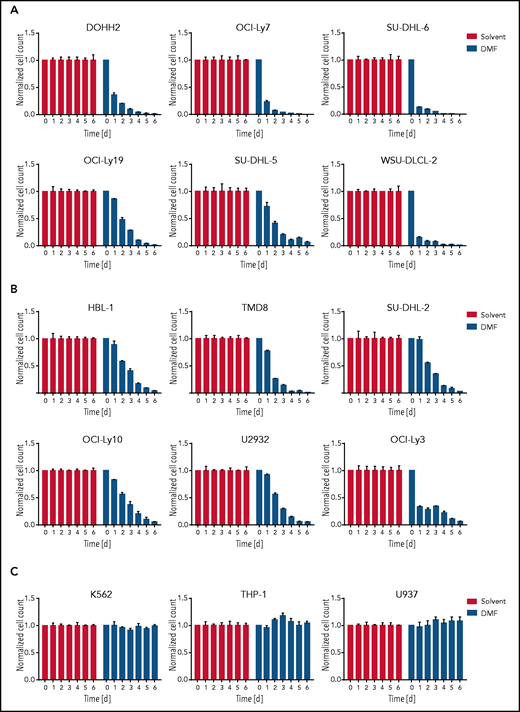
DMF treatment induces cytotoxicity in DLBCL. GCB DLBCL (A), ABC DLBCL (B), or myeloid lymphoma/leukemia (C) cell lines were treated daily with solvent or 20 μM DMF. Cell numbers were determined as indicated and normalized to the solvent control. Error bars correspond to the mean ± standard deviation. Data are representative of ≥3 independent experiments.
DMF treatment induces cytotoxicity in DLBCL. GCB DLBCL (A), ABC DLBCL (B), or myeloid lymphoma/leukemia (C) cell lines were treated daily with solvent or 20 μM DMF. Cell numbers were determined as indicated and normalized to the solvent control. Error bars correspond to the mean ± standard deviation. Data are representative of ≥3 independent experiments.
DMF induces ferroptosis in DLBCL
To gain insights into the molecular mechanism of DMF-dependent cytotoxicity in DLBCL, we investigated the importance of the electrophilic properties of DMF for its antilymphoma activity. Indeed, the structural analog dimethyl succinate, which lacks the electrophilic carbon-carbon double bond of DMF, was unable to impair the growth of DLBCL cell lines (supplemental Figure 3A-B). Similarly, the DMF metabolite monomethyl fumarate (MMF), which has been described as a potent agonist of the hydroxycarboxylic acid receptor 2, did not interfere with the survival of DLBCL cells (supplemental Figure 3A-B).30,37‐40 In contrast, the electrophile dimethyl itaconate inhibited the growth of GCB and ABC DLBCL cell lines, when applied in concentrations that induced an extent of Nrf2 stabilization similar to that induced by 20 μM DMF (supplemental Figure 3C-E). Next, we assessed the capacity of DMF to deplete reduced glutathione, which constitutes a major cellular antioxidant.31 Interestingly, we noticed that GCB DLBCL on average exhibited lower GSH levels than ABC DLBCL cell lines and that DMF efficiently diminished the GSH pool, especially in GCB DLBCL cell lines (Figure 2A-B). Similar to the well-established inhibitors of GSH synthesis γ-glutamyl-cysteine synthetase inhibitor buthionine sulfoximine (BSO) and cystine-glutamate antiporter (system xc−) inhibitor erastin, DMF treatment resulted in a rapid depletion of the GSH pool (supplemental Figure 4A). The detoxification of lipid peroxides by GPX4 critically requires reduced GSH. We therefore quantified lipid peroxidation using the oxidation-sensitive lipophilic probe BODIPY C11 in several DMF-treated DLBCL cell lines. Strikingly, low doses of DMF were sufficient to induce massive lipid peroxidation in GCB DLBCL cell lines, whereas induction of lipid peroxidation in ABC DLBCL cells required higher DMF concentrations (Figure 2C; supplemental Figure 4B). Additionally, various MCL cell lines also exhibited strong lipid peroxidation after DMF treatment; however, in myeloid cell lines, DMF only slightly reduced GSH levels and failed to induce lipid peroxidation (Figure 2B; supplemental Figure 4C-D).
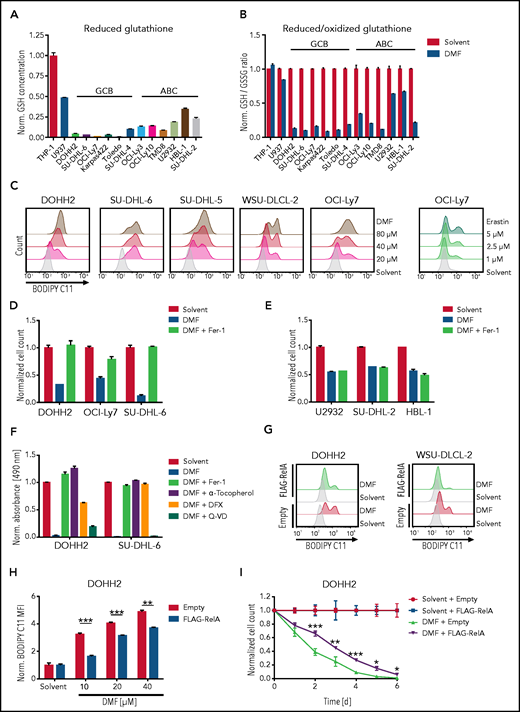
DMF treatment induces ferroptosis in DLBCL. (A) Reduced glutathione levels were quantified in various DLBCL cell lines and normalized to the myeloid cell line THP-1. (B) The indicated cell lines were treated with 20 μM of DMF for 2 hours. The ratio of reduced to oxidized glutathione was determined and normalized to the respective solvent-treated controls. (C) Lipid peroxidation was quantified by flow cytometry using the oxidation-sensitive fluorescent probe BODIPY C11. GCB DLBCL cell lines were treated with solvent or the indicated amounts of DMF and erastin for 2 and 8 hours, respectively. (D-E) GCB (D) and ABC (E) DLBCL cell lines were treated with solvent or 20 μM DMF alone, or in combination with 5 μM Fer-1 for 24 (D) and 48 (E) hours. Cell numbers were determined and normalized to the solvent control. (F) DOHH2 and SU-DHL-6 cells were treated with 20 μM DMF alone, or in combination with 5 μM Fer-1, 100 μM α-tocopherol, 100 μM DFX, or 20 μM Q-VD. Survival was quantified by MTS assay after 24 hours. (G-H) Quantification of lipid peroxidation in DMF-treated control and FLAG-RelA–overexpressing GCB DLBCL cells. The mean fluorescence intensity (MFI) of oxidized BODIPY C11 in DMF-treated cells was normalized to the MFI of the respective solvent-treated samples. (I) DOHH2 cells expressing FLAG-RelA or control vector were counted and treated daily with 20 μM DMF for 6 days. Cell counts were normalized to the solvent control. Error bars correspond to the mean ± standard deviation. Data are representative of ≥2 (A-B) or ≥3 (C-I) independent experiments. Statistical significance was calculated using the Student t test. *P < .05, **P < .01, ***P < .001.
DMF treatment induces ferroptosis in DLBCL. (A) Reduced glutathione levels were quantified in various DLBCL cell lines and normalized to the myeloid cell line THP-1. (B) The indicated cell lines were treated with 20 μM of DMF for 2 hours. The ratio of reduced to oxidized glutathione was determined and normalized to the respective solvent-treated controls. (C) Lipid peroxidation was quantified by flow cytometry using the oxidation-sensitive fluorescent probe BODIPY C11. GCB DLBCL cell lines were treated with solvent or the indicated amounts of DMF and erastin for 2 and 8 hours, respectively. (D-E) GCB (D) and ABC (E) DLBCL cell lines were treated with solvent or 20 μM DMF alone, or in combination with 5 μM Fer-1 for 24 (D) and 48 (E) hours. Cell numbers were determined and normalized to the solvent control. (F) DOHH2 and SU-DHL-6 cells were treated with 20 μM DMF alone, or in combination with 5 μM Fer-1, 100 μM α-tocopherol, 100 μM DFX, or 20 μM Q-VD. Survival was quantified by MTS assay after 24 hours. (G-H) Quantification of lipid peroxidation in DMF-treated control and FLAG-RelA–overexpressing GCB DLBCL cells. The mean fluorescence intensity (MFI) of oxidized BODIPY C11 in DMF-treated cells was normalized to the MFI of the respective solvent-treated samples. (I) DOHH2 cells expressing FLAG-RelA or control vector were counted and treated daily with 20 μM DMF for 6 days. Cell counts were normalized to the solvent control. Error bars correspond to the mean ± standard deviation. Data are representative of ≥2 (A-B) or ≥3 (C-I) independent experiments. Statistical significance was calculated using the Student t test. *P < .05, **P < .01, ***P < .001.
Because lipid peroxidation constitutes a hallmark of the nonapoptotic form of cell death termed ferroptosis, we assessed the impact of iron chelators (DFX) and lipophilic antioxidants (α-tocopherol and Fer-1) on DMF-induced lipid peroxidation and cell death. Similar to the GPX4 inhibitor RSL3, DMF induced strong lipid peroxidation, which was almost completely abolished by cotreatment with Fer-1, DFX, or α-tocopherol (supplemental Figure 5A). At a DMF dose of 20 μM, Fer-1 prevented the induction of cell death in GCB, but not in ABC DLBCL cell lines, indicating that the GCB subtype is particularly sensitive to DMF-dependent ferroptosis (Figure 2D-E; supplemental Figure 5B-C). Additionally, the ferroptosis inhibitors DFX, N-acetyl-L-cysteine, and α-tocopherol but not the broad-spectrum caspase inhibitor Q-VD protected GCB DLBCL cells from DMF-induced toxicity (Figure 2F; supplemental Figure 5D). To understand why ABC DLBCL cells are less susceptible to DMF-induced ferroptotic cell death than GCB DLBCLs, we investigated a potential involvement of the transcription factor NF-κB, which is constitutively activated in ABC DLBCL.41 Strikingly, overexpression of the NF-κB member RelA in 2 GCB DLBCL cell lines partially reduced lipid peroxidation and cytotoxicity after DMF treatment (Figure 2G-I; supplemental Figure 6A-B). This might be due to NF-κB–mediated induction of various antioxidant proteins, such as thioredoxin, NAD(P)H dehydrogenase (quinone) 1, heme oxygenase-1, manganese superoxide dismutase, and ferritin heavy chain.42 In conclusion, we demonstrated that DMF treatment efficiently depleted the already low GSH levels in GCB DLBCL cells and induced lipid peroxidation, which resulted in ferroptotic cell death.
High 5-LOX expression in GCB DLBCL correlates with ferroptosis susceptibility
To gain further insight into the molecular basis of the increased susceptibility of GCB DLBCL to DMF-induced ferroptosis, we screened for ferroptosis-associated genes that are differentially expressed in the 2 major DLBCL subtypes. While we did not detect any differences in the expression of long-chain fatty acid CoA ligase 4, FSP1, or the system xc− subunit SLC7A11, ABC DLBCL cell lines exhibited increased GPX4 levels compared with their GCB counterparts (Figure 3A; supplemental Figure 7A-C). However, the most prominent difference between the DLBCL subtypes was observed for the ALOX5 gene, which encodes the enzyme arachidonate 5-lipoxygenase; (Figure 3A-B). Immunohistochemistry of human DLBCL biopsies revealed strong perinuclear/nuclear staining for 5-LOX in ∼50% of GCB DLBCLs. In contrast, nuclear localization of 5-LOX was absent in GC B cells of reactive lymph nodes and only rarely observed in ABC DLBCL biopsies (Figure 3C-D; supplemental Figure 7D).43,44 Similar to human biopsies, nuclear 5-LOX was also detected in GCB DLBCL cell lines (supplemental Figure 7E). To shed light on the molecular basis underlying the different ALOX5 expression patterns observed in DLBCL, we investigated the DNA methylation status of the GC-rich region surrounding the transcription start site in the ALOX5 promoter, which also includes binding sites of the transcription factor Sp1.45 However, we did not detect differences in the methylation status between the DLBCL subtypes (supplemental Figure 7F). In contrast, 3 of 5 GCB DLBCL cell lines exhibited reduced 5-LOX expression in response to treatment with the Sp1 inhibitor mithramycin A (supplemental Figure 7G). Because there is so far no evidence for differences in Sp1 activity between the DLBCL subtypes, we investigated whether B-cell receptor (BCR) signaling, which is deregulated in ABC DLBCL, might influence 5-LOX expression. Indeed, inhibition of chronic active BCR signaling in ABC DLBCL with Src, BTK, SYK, and PKC inhibitors or by CD79A silencing not only reduced the expression of the NF-κB target gene TNFAIP3, but also significantly increased ALOX5 levels (Figure 3E; supplemental Figure 8A-C). In contrast, activation of GCB DLBCL cells by BCR crosslinking or stimulation with phorbol 12-myristate 13-acetate and ionomycin increased TNFAIP3 but reduced ALOX5 expression (Figure 3F; supplemental Figure 8D-E), suggesting that chronic active BCR signaling is involved in the suppression of 5-LOX expression in ABC DLBCL.
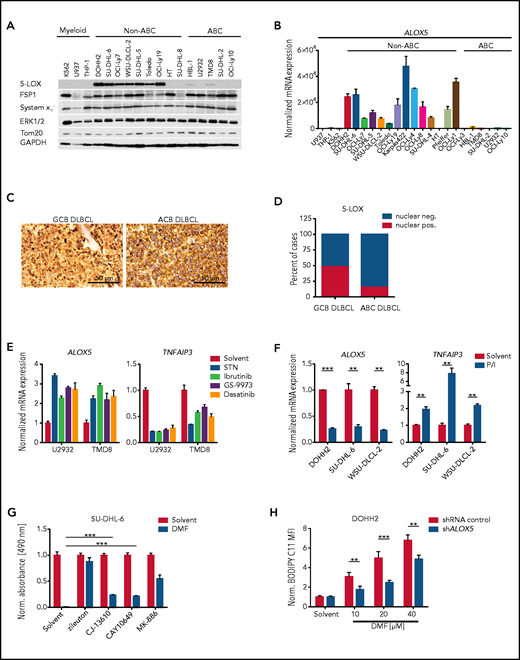
5-LOX sensitizes GCB DLBCL cells to DMF-induced ferroptosis. (A) Protein expression of 5-LOX, FSP1, and system xc− in the indicated cell lines was visualized by immunoblot analysis. ERK1/2, Tom20, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading controls. (B) ALOX5 transcript levels in DLBCL or myeloid cell lines were quantified by quantitative polymerase chain reaction and normalized to the expression in U937 cells. SDHA served as reference gene. (C) Immunohistochemical staining of 5-LOX in human GCB and ABC DLBCL biopsies. Scale bars, 50 μm. (D) Quantification of nuclear 5-LOX staining in DLBCL biopsies (GCB DLBCL, n = 89; ABC DLBCL, n = 64). (E) The indicated ABC DLBCL cell lines were treated with 5 μM sotrastaurin (STN), 0.1 μM ibrutinib, 4 μM GS-9973, or 1 μM dasatinib for 24 hours. Transcript levels of ALOX5 and TNFAIP3 were quantified and normalized to the solvent control. SDHA served as reference gene. (F) Quantification of ALOX5 and TNFAIP3 messenger RNA (mRNA) expression in the indicated GCB DLBCL cell lines activated for 8 hours with PMA/ionomycin (P/I). SDHA served as reference gene. (G) The survival of SU-DHL-6 cells treated with 20 μM DMF alone or in combination with 10 μM zileuton, 2.5 μM CJ-13610, 5 μM CAY10649, or 10 μM MK-886 was assessed after 24 hours by MTS assay. (H) Quantification of lipid peroxidation in DMF-treated control and ALOX5-silenced DOHH2 cells. The mean fluorescence intensity (MFI) of oxidized BODIPY C11 in DMF-treated cells was normalized to the MFI of the respective solvent-treated samples. Error bars correspond to the mean ± standard deviation. Data are representative of ≥2 (E) or ≥3 (A-B,F-H) independent experiments. Statistical significance was calculated using the Student t test. **P < .01, ***P < .001. sh, short hairpin.
5-LOX sensitizes GCB DLBCL cells to DMF-induced ferroptosis. (A) Protein expression of 5-LOX, FSP1, and system xc− in the indicated cell lines was visualized by immunoblot analysis. ERK1/2, Tom20, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading controls. (B) ALOX5 transcript levels in DLBCL or myeloid cell lines were quantified by quantitative polymerase chain reaction and normalized to the expression in U937 cells. SDHA served as reference gene. (C) Immunohistochemical staining of 5-LOX in human GCB and ABC DLBCL biopsies. Scale bars, 50 μm. (D) Quantification of nuclear 5-LOX staining in DLBCL biopsies (GCB DLBCL, n = 89; ABC DLBCL, n = 64). (E) The indicated ABC DLBCL cell lines were treated with 5 μM sotrastaurin (STN), 0.1 μM ibrutinib, 4 μM GS-9973, or 1 μM dasatinib for 24 hours. Transcript levels of ALOX5 and TNFAIP3 were quantified and normalized to the solvent control. SDHA served as reference gene. (F) Quantification of ALOX5 and TNFAIP3 messenger RNA (mRNA) expression in the indicated GCB DLBCL cell lines activated for 8 hours with PMA/ionomycin (P/I). SDHA served as reference gene. (G) The survival of SU-DHL-6 cells treated with 20 μM DMF alone or in combination with 10 μM zileuton, 2.5 μM CJ-13610, 5 μM CAY10649, or 10 μM MK-886 was assessed after 24 hours by MTS assay. (H) Quantification of lipid peroxidation in DMF-treated control and ALOX5-silenced DOHH2 cells. The mean fluorescence intensity (MFI) of oxidized BODIPY C11 in DMF-treated cells was normalized to the MFI of the respective solvent-treated samples. Error bars correspond to the mean ± standard deviation. Data are representative of ≥2 (E) or ≥3 (A-B,F-H) independent experiments. Statistical significance was calculated using the Student t test. **P < .01, ***P < .001. sh, short hairpin.
5-LOX promotes ferroptosis induction by DMF
To investigate the importance of 5-LOX in DMF-induced ferroptosis, we inhibited its activity with the LOX inhibitor nordihydroguaiaretic acid (NDGA) or the 5-LOX–specific inhibitor zileuton. Both NDGA and zileuton efficiently protected GCB DLBCL cell lines from DMF-induced lipid peroxidation and cytotoxicity (supplemental Figure 9A-D). Unexpectedly, short hairpin RNA–mediated silencing of 5-LOX expression only slightly alleviated DMF-induced toxicity, indicating that the antioxidant properties of NDGA and zileuton rather than LOX inhibition mediate their protective role against DMF (supplemental Figure 9E).26 Cotreatment with 5-LOX inhibitors, which lack this radical-trapping activity (ie, CJ-13610 and CAY10649), or with an inhibitor targeting the 5-LOX–activating protein (ie, MK-886) resulted in partial but not complete rescue from DMF-mediated cytotoxicity (Figure 3G; supplemental Figure 9F). Interestingly, ALOX5 silencing increased the relative amount of GSH in DLBCL cells and reduced DMF-induced lipid peroxidation (Figure 3H; supplemental Figure 9G-H). Moreover, cells surviving repetitive treatments with DMF exhibited reduced 5-LOX protein expression, suggesting that even though 5-LOX is not essential for DMF-induced ferroptosis, it might contribute to the general oxidative milieu within the cell and thus to ferroptosis sensitivity (supplemental Figure 9I). The cell line HT, one of the few GCB DLBCL cell lines that lacks 5-LOX expression, exhibited rather high GSH levels and was resistant to both DMF-induced lipid peroxidation and cytotoxicity (Figure 3A-B; supplemental Figures 1A and 10A-C). Forced expression of FLAG-tagged 5-LOX in HT cells promoted DMF-induced lipid peroxidation, indicating that 5-LOX expression in GCB DLBCL sensitizes to DMF-mediated ferroptosis induction (supplemental Figure 10D-E). Taken together, high 5-LOX expression in GCB DLBCL was correlated with an increased sensitivity to DMF-induced ferroptosis.
DMF reduces NF-κB activity by inhibition of the IKK complex
Unlike in GCB DLBCL, low doses of DMF were unable to induce ferroptosis in ABC DLBCL cell lines (Figure 2D-E). To elucidate the molecular mechanism of its antilymphoma effect in ABC DLBCL, we quantified DMF-induced alterations in the global gene expression profile of HBL-1 cells by RNA sequencing (Figure 4A). Gene set enrichment analysis identified several NF-κB gene signatures downregulated upon DMF treatment, comprising classical NF-κB target genes, such as NFKBIA, NFKBIZ, TNFAIP3, BIRC3, and CFLAR (Figure 4A-C; supplemental Figure 11A-C).
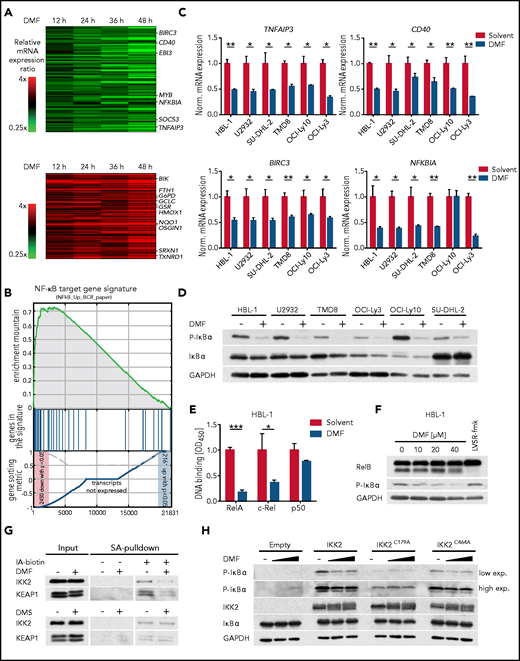
DMF potently inhibits IKK activity. (A) Heatmaps of differentially expressed genes in HBL-1 cells treated with DMF for 12, 24, 36, and 48 hours compared with the solvent control. Gene expression changes are depicted according to the color scale. (B) Gene set enrichment analysis identified a gene set describing NF-κB targets to be downregulated by DMF treatment. (C) The indicated ABC DLBCL cell lines were treated with solvent or 20 μM DMF for 6 hours. Transcript levels of TNFAIP3, CD40, BIRC3, and NFKBIA were quantified by quantitative polymerase chain reaction. Expression of DMF-treated cells was normalized to the respective solvent control. SDHA served as reference gene. (D) ABC DLBCL cell lines were treated with 40 μM DMF for 4 hours. S32/36 phosphorylation of IκBα was visualized by immunoblot analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control. (E) After treatment with either solvent or DMF for 16 hours, nuclear fractions were isolated and binding of the NF-κB members RelA, c-Rel, and p50 to their consensus nucleotide sequence was quantified by TransAM assay. (F) HBL-1 cells were treated with the indicated DMF concentrations or with the MALT1 inhibitor LVSR-fmk (2 μM) for 4 hours in combination with the proteasome inhibitor MG-132 (5 μM) 90 minutes before cell lysis. MALT1-mediated RelB cleavage was visualized by immunoblot analysis. GAPDH served as loading control. (G) HBL-1 cells were treated with solvent, 40 μM DMF, or 40 μM DMS for 4 hours. After lysis, proteins containing reactive cysteine residues were labeled with biotin-coupled iodoacetamide (IA-biotin) and subsequently pulled down with streptavidin (SA) agarose. The accessibility of cysteine residues in IKK2 and KEAP1 was analyzed by immunoblotting. (H) HEK293T cells were transfected with the indicated IKK2 constructs, treated for 1 hour with solvent or DMF, and analyzed for IκBα phosphorylation by immunoblotting. GAPDH served as loading control. Data are representative of ≥2 (E,G) or ≥3 (C-D,F,H) independent experiments. Statistical significance was calculated using the Student t test. *P < .05, **P < .01, ***P < .001. mRNA, messenger RNA.
DMF potently inhibits IKK activity. (A) Heatmaps of differentially expressed genes in HBL-1 cells treated with DMF for 12, 24, 36, and 48 hours compared with the solvent control. Gene expression changes are depicted according to the color scale. (B) Gene set enrichment analysis identified a gene set describing NF-κB targets to be downregulated by DMF treatment. (C) The indicated ABC DLBCL cell lines were treated with solvent or 20 μM DMF for 6 hours. Transcript levels of TNFAIP3, CD40, BIRC3, and NFKBIA were quantified by quantitative polymerase chain reaction. Expression of DMF-treated cells was normalized to the respective solvent control. SDHA served as reference gene. (D) ABC DLBCL cell lines were treated with 40 μM DMF for 4 hours. S32/36 phosphorylation of IκBα was visualized by immunoblot analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control. (E) After treatment with either solvent or DMF for 16 hours, nuclear fractions were isolated and binding of the NF-κB members RelA, c-Rel, and p50 to their consensus nucleotide sequence was quantified by TransAM assay. (F) HBL-1 cells were treated with the indicated DMF concentrations or with the MALT1 inhibitor LVSR-fmk (2 μM) for 4 hours in combination with the proteasome inhibitor MG-132 (5 μM) 90 minutes before cell lysis. MALT1-mediated RelB cleavage was visualized by immunoblot analysis. GAPDH served as loading control. (G) HBL-1 cells were treated with solvent, 40 μM DMF, or 40 μM DMS for 4 hours. After lysis, proteins containing reactive cysteine residues were labeled with biotin-coupled iodoacetamide (IA-biotin) and subsequently pulled down with streptavidin (SA) agarose. The accessibility of cysteine residues in IKK2 and KEAP1 was analyzed by immunoblotting. (H) HEK293T cells were transfected with the indicated IKK2 constructs, treated for 1 hour with solvent or DMF, and analyzed for IκBα phosphorylation by immunoblotting. GAPDH served as loading control. Data are representative of ≥2 (E,G) or ≥3 (C-D,F,H) independent experiments. Statistical significance was calculated using the Student t test. *P < .05, **P < .01, ***P < .001. mRNA, messenger RNA.
To unravel the molecular basis of the DMF-dependent NF-κB inhibition in ABC DLBCL, we assessed the functionality of the IKK complex, an essential player in canonical NF-κB signaling. DMF treatment impaired the phosphorylation of IκBα at S32/36, indicating that DMF disrupted the chronic activation of the IKK complex in ABC DLBCL (Figure 4D; supplemental Figure 12A). Consistent with the established inhibitory role of IκBα, DMF treatment resulted in markedly decreased RelA translocation to the nucleus as well as in impaired binding of RelA and c-Rel to their consensus DNA sequence (Figure 4E; supplemental Figure 12B).
Because many genes that were upregulated in response to DMF treatment represented classical Nrf2 target genes (eg, GCLM, TXNRD1, NQO1, SLC7A11, and FTH1), we explored a potential role of Nrf2 in DMF-dependent NF-κB inhibition (Figure 4A; supplemental Figure 11C). However, neither inhibition of IKK activity nor DMF-induced cytotoxicity was affected by Nrf2 silencing, demonstrating that DMF-mediated NF-κB inhibition and cell death were independent of Nrf2 (supplemental Figure 12C-E). Accordingly, although treatment of ABC DLBCL cell lines with the DMF metabolite MMF provoked an Nrf2 response, it failed to impair IκBα phosphorylation (supplemental Figure 12F). Because the GPX4 inhibitor RSL3, as well as BSO and erastin, which deplete the GSH pool and induce a prooxidative state in the cell, were also unable to inhibit IKK activity in ABC DLBCL, we concluded that protein or lipid oxidation was not involved in the DMF-mediated inhibition of NF-κB signaling (supplemental Figure 12G).
In addition, DMF treatment did not impair upstream BCR and MAPK signaling, because DMF treatment did not reduce the phosphorylation of BLNK, PLCγ2, ERK1/2, AKT, JNK1/2, or p38 (supplemental Figure 13A). BCR-mediated NF-κB activation critically relies on the assembly of the CARD11-BCL10-MALT1 (CBM) complex, which upon polyubiquitination not only recruits and activates the IKK complex but also promotes MALT1 protease activation, which in turn supports NF-κB signaling by cleavage and inactivation of negative regulators, such as A20 or RelB.10,46 Neither K63-linked polyubiquitination of BCL10 nor MALT1-mediated RelB cleavage was affected by DMF treatment (Figure 4F; supplemental Figure 13B-C).
To investigate whether IKK2 was directly modified by DMF, proteins with reactive cysteines were labeled with biotin-coupled iodoacetamide and isolated using streptavidin beads. In solvent-treated cells, IKK2 was precipitated because of its accessible reactive cysteine residues. DMF treatment before labeling, however, significantly reduced the reaction of the iodoacetamide probe with IKK2 and KEAP1, the latter a key sensor of oxidative and electrophilic stress that harbors several reactive cysteine residues (Figure 4G).47 Mass spectrometric analysis of endogenous IKK complex in DMF-treated ABC DLBCL cells identified succination of several cysteine residues (eg, C179 in the kinase domain and C464 in the leucine zipper domain of IKK2 as well as C347 in the ubiquitin-binding domain of NEMO; supplemental Figure 14A-B).48-50 Interestingly, the cysteines found to be succinated by DMF are highly conserved across various species (supplemental Figure 15A-B). To evaluate the importance of C179 and C464 in DMF-dependent IKK2 inhibition, we assessed the capacity of the respective cysteine-to-alanine mutants of IKK2 to catalyze the phosphorylation of IκBα. As previously reported, the IKK2C179A mutant exhibited impaired kinase activity, whereas the activity of IKK2C464A was similar to that of the wild-type protein.50 Strikingly, whereas the kinase activity of wild-type IKK2 and the IKK2C464A variant was reduced by DMF treatment, the residual activity of the IKK2C179A mutant was resistant to DMF, indicating that succination of this cysteine residue is key for the observed DMF-mediated IKK2 inhibition (Figure 4H). Collectively, DMF did not alter upstream BCR signaling or CBM complex formation in ABC DLBCL, but impaired NF-κB signaling by directly inhibiting IKK activity.
JAKs are inhibited directly by DMF
In addition to IκBα phosphorylation, DMF treatment simultaneously decreased the constitutive STAT3 phosphorylation observed in ABC DLBCL cell lines, indicating a direct role of DMF in STAT3 inhibition (Figure 5A; supplemental Figure 16A). DMF treatment also strongly impaired STAT3 phosphorylation in ABC DLBCL cell lines that were activated with exogenous IL-6 or IL-10 (Figure 5B; supplemental Figure 16B-C). Similar to IKK inhibition, neither MMF, dimethyl succinate, BSO, erastin, nor RSL3 affected the STAT3 phosphorylation status (supplemental Figure 16D-E). Because we noticed that both the steady-state and the interferon-α–induced phosphorylation of STAT1 were also impaired by DMF treatment, we hypothesized that DMF might directly inhibit the activity of JAKs (Figure 5A,C). Indeed, DMF treatment decreased the autophosphorylation of JAK1 and TYK2 in untreated or interferon-α–stimulated ABC DLBCL cell lines (Figure 5D). Strikingly, all JAKs investigated showed reduced labeling with biotin-coupled iodoacetamide upon DMF treatment, indicating a DMF-mediated succination of these kinases (Figure 5E). To identify which cysteine residues were modified by DMF, we immunoprecipitated JAK1 from solvent- or DMF-treated ABC DLBCL cells and analyzed potential succination events by mass spectrometry. Succination of 2 cysteine residues of JAK1 (ie, C257 and C731) was detected in the DMF-treated samples only (Figure 5F; supplemental Figure 17A). Whereas C731 is located in the catalytically inactive protein kinase 1 domain, we hypothesized that succination of the strongly conserved C257 within the FERM domain, which mediates the interaction with cytokine receptors, may be responsible for the observed inhibitory effect of DMF on JAK1 activity (Figure 5F).51 Structural analysis confirmed the accessibility of C257 and thus corroborated the idea of a direct modification of JAK1 by DMF (Figure 5G). Strikingly, DMF treatment indeed weakened the interaction of the α subunit of the IL-10 receptor with wild-type JAK1 but not with the JAK1C257A mutant (Figure 5H; supplemental Figure 17B). Because the activity of the JAK1C257A mutant was still partially impaired by DMF treatment, further modification of JAK1 might contribute to the DMF-mediated inhibition of the JAK/STAT pathway (supplemental Figure 17C). HBL-1 cells that were transduced to express a hyperactive STAT3 mutant (STAT3C) and treated with exogenous IL-6/IL-10 exhibited an increased resistance to DMF-mediated cytotoxicity, confirming the importance of JAK inhibition for DMF-mediated toxicity (Figure 5I). Collectively, our data identify the direct modification and inhibition of JAKs by DMF as important mechanisms for its toxicity in ABC DLBCL.
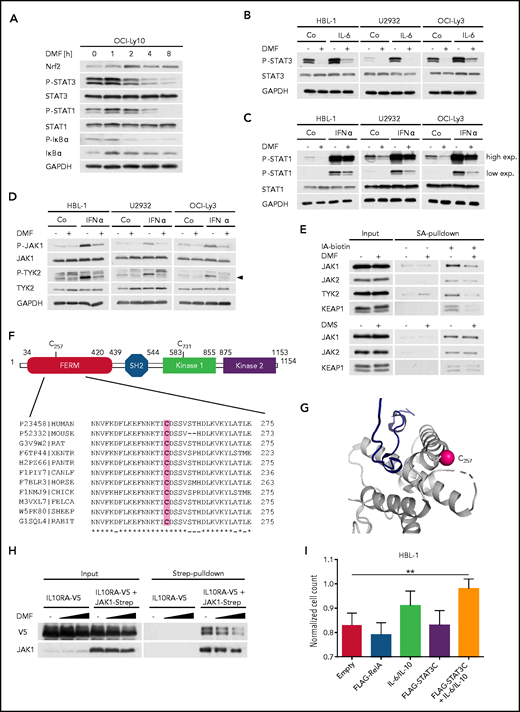
DMF impairs the activity of JAKs. (A) The ABC DLBCL cell line OCI-Ly10 was treated with 40 μM DMF for the specified times and the phosphorylation of STAT3, STAT1, and IκBα was assessed by immunoblot analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control. (B-C) ABC DLBCL cell lines were treated with solvent, 40 μM DMF, and/or 3 ng/mL recombinant human IL-6 (B) or 1 ng/mL interferon-α (IFN-α) (C), as indicated. Phosphorylation of STAT1/3 was visualized by immunoblot analysis. GAPDH served as loading control. (D) Analysis of JAK1 and TYK2 autophosphorylation in solvent-, IFN-α–, and/or DMF-treated ABC DLBCL cell lines. Arrowhead indicates phosphorylated TYK2 (P-TYK2). GAPDH served as loading control. (E) HBL-1 cells treated with solvent, 40 μM DMF, or 40 μM DMS were lysed, and reactive cysteine-containing proteins were labeled with biotin-coupled iodoacetamide (IA-biotin), isolated using streptavidin (SA) agarose, and analyzed by immunoblotting. (F) Schematic representation of JAK1 domain structure including FERM domain, Src homology 2 (SH2) domain, and kinase domains. Alignment of JAK1 sequences surrounding the conserved C257 from various species. (G) Using PDB entry 5IXI, human JAK1 was pictured around the modified cysteine residue (C257) to emphasize its solvent accessibility. The sulfur atom of the cysteine side chain is marked by a pink sphere. The protein chain (gray) is shown in cartoon representation. The close proximity of C257 to the receptor (dark blue) binding site is shown. (H) HEK293T cells were transiently transfected with V5-tagged IL-10 receptor (α subunit; IL10RA-V5) alone or in combination with Strep-tagged JAK1. Twenty-four hours after transfection, cells were treated with solvent or DMF for 1 hour and lysed; JAK1 was pulled down using Strep-Tactin beads. The interaction between IL10RA-V5 and JAK1-Strep was visualized by immunoblot analysis. (I) HBL-1 cells expressing control vector, FLAG-RelA, or FLAG-STAT3C were treated daily with solvent or 20 μM DMF in combination with recombinant human IL-6 and IL-10 (3 ng/mL each), as indicated. Cell numbers were determined after 48 hours and normalized to the respective controls lacking DMF. Data are representative of ≥2 (E,I) or 3 (A-D,H) independent experiments. Statistical significance was calculated using the Student t test. **P < .01.
DMF impairs the activity of JAKs. (A) The ABC DLBCL cell line OCI-Ly10 was treated with 40 μM DMF for the specified times and the phosphorylation of STAT3, STAT1, and IκBα was assessed by immunoblot analysis. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as loading control. (B-C) ABC DLBCL cell lines were treated with solvent, 40 μM DMF, and/or 3 ng/mL recombinant human IL-6 (B) or 1 ng/mL interferon-α (IFN-α) (C), as indicated. Phosphorylation of STAT1/3 was visualized by immunoblot analysis. GAPDH served as loading control. (D) Analysis of JAK1 and TYK2 autophosphorylation in solvent-, IFN-α–, and/or DMF-treated ABC DLBCL cell lines. Arrowhead indicates phosphorylated TYK2 (P-TYK2). GAPDH served as loading control. (E) HBL-1 cells treated with solvent, 40 μM DMF, or 40 μM DMS were lysed, and reactive cysteine-containing proteins were labeled with biotin-coupled iodoacetamide (IA-biotin), isolated using streptavidin (SA) agarose, and analyzed by immunoblotting. (F) Schematic representation of JAK1 domain structure including FERM domain, Src homology 2 (SH2) domain, and kinase domains. Alignment of JAK1 sequences surrounding the conserved C257 from various species. (G) Using PDB entry 5IXI, human JAK1 was pictured around the modified cysteine residue (C257) to emphasize its solvent accessibility. The sulfur atom of the cysteine side chain is marked by a pink sphere. The protein chain (gray) is shown in cartoon representation. The close proximity of C257 to the receptor (dark blue) binding site is shown. (H) HEK293T cells were transiently transfected with V5-tagged IL-10 receptor (α subunit; IL10RA-V5) alone or in combination with Strep-tagged JAK1. Twenty-four hours after transfection, cells were treated with solvent or DMF for 1 hour and lysed; JAK1 was pulled down using Strep-Tactin beads. The interaction between IL10RA-V5 and JAK1-Strep was visualized by immunoblot analysis. (I) HBL-1 cells expressing control vector, FLAG-RelA, or FLAG-STAT3C were treated daily with solvent or 20 μM DMF in combination with recombinant human IL-6 and IL-10 (3 ng/mL each), as indicated. Cell numbers were determined after 48 hours and normalized to the respective controls lacking DMF. Data are representative of ≥2 (E,I) or 3 (A-D,H) independent experiments. Statistical significance was calculated using the Student t test. **P < .01.
Inhibition of FSP1 and BCL-2 synergizes with DMF treatment
To investigate the therapeutic potential of DMF in vivo, we transplanted human DLBCL cell lines into zebrafish embryos that were subsequently incubated in solvent- or DMF-supplemented medium. A dose of 5 μM DMF was sufficient to prevent tumor formation in >50% (SU-DHL-6) or 70% (DOHH2) of the animals (Figure 6A-B).
To further sensitize DLBCL cells to ferroptotic cell death, we pharmacologically targeted the GSH-independent FSP1.52 Strikingly, combined treatment with DMF and FSP1 inhibitor synergistically induced cytotoxicity in GCB DLBCL cell lines (Figure 6C; supplemental Figure 18A). Moreover, the BCL-2 inhibitor ABT-199, but neither the PI3K inhibitor AZD8835 nor the tubulin polymerization inhibitor vincristine, showed promising combinatorial effects with DMF treatment in ABC DLBCL cells (Figure 6D; supplemental Figures 18B and 19A-B). To validate the efficacy of the combinatorial treatment with DMF and ABT-199 in vivo, we used HBL-1 and patient-derived (VFN-D1) xenograft mouse models. Although DMF treatment alone was sufficient to significantly reduce tumor growth, combination with ABT-199 strongly enhanced its antilymphoma effect (Figure 6E-F). Collectively, our data reveal potent antitumor activity of DMF both in vitro and in vivo and suggest that combined treatment of DMF with either FSP1 inhibitors or BH3 mimetics might represent a promising novel strategy in DLBCL therapy.
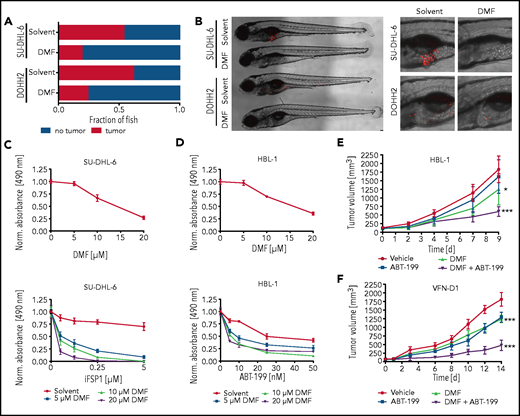
Inhibition of FSP1 and BCL-2 synergizes with DMF treatment. (A) SU-DHL-6 and DOHH2 cells were transplanted into zebrafish embryos. Tumor formation in animals treated with either solvent or 5 μM DMF was quantified by microscopy after 3 days (n ≥ 21). DMF treatment significantly reduced the frequency of tumor-bearing animals (P = .001 for SU-DHL-6; P = .043 for DOHH2). (B) Representative images of transplanted zebrafish embryos after solvent or DMF treatment. The respective tumor cells were fluorescently labeled and visualized by microscopy. The pictures on the right represent magnified sections of the zebrafish embryos. Scale bar, 100 μm. (C-D) SU-DHL-6 or HBL-1 cells were treated with DMF alone (top panels) or in combination with either FSP1 inhibitor (iFSP1) (C) or ABT-199 (D) (bottom panels). Cell survival was quantified by MTS assay after 24 (C) or 72 (D) hours . The combination index (CI) for DMF and iFSP1 in SU-DHL-6 is ≤0.55, and for DMF and ABT-199 in HBL-1 is ≤0.75. (E-F) HBL-1 (E) or VFN-D1 patient-derived (F) xenograft mice were treated either with vehicle, ABT-199, DMF, or the combination of ABT-199 and DMF, as indicated. Tumor volume was quantified by caliper measurements up to 14 days after start of the treatment. Each group consists of ≥7 animals. Statistical significance was calculated by the comparison of each treatment group with the vehicle control. Data are representative of ≥3 independent experiments (A-D). *P < .05, ***P < .001.
Inhibition of FSP1 and BCL-2 synergizes with DMF treatment. (A) SU-DHL-6 and DOHH2 cells were transplanted into zebrafish embryos. Tumor formation in animals treated with either solvent or 5 μM DMF was quantified by microscopy after 3 days (n ≥ 21). DMF treatment significantly reduced the frequency of tumor-bearing animals (P = .001 for SU-DHL-6; P = .043 for DOHH2). (B) Representative images of transplanted zebrafish embryos after solvent or DMF treatment. The respective tumor cells were fluorescently labeled and visualized by microscopy. The pictures on the right represent magnified sections of the zebrafish embryos. Scale bar, 100 μm. (C-D) SU-DHL-6 or HBL-1 cells were treated with DMF alone (top panels) or in combination with either FSP1 inhibitor (iFSP1) (C) or ABT-199 (D) (bottom panels). Cell survival was quantified by MTS assay after 24 (C) or 72 (D) hours . The combination index (CI) for DMF and iFSP1 in SU-DHL-6 is ≤0.55, and for DMF and ABT-199 in HBL-1 is ≤0.75. (E-F) HBL-1 (E) or VFN-D1 patient-derived (F) xenograft mice were treated either with vehicle, ABT-199, DMF, or the combination of ABT-199 and DMF, as indicated. Tumor volume was quantified by caliper measurements up to 14 days after start of the treatment. Each group consists of ≥7 animals. Statistical significance was calculated by the comparison of each treatment group with the vehicle control. Data are representative of ≥3 independent experiments (A-D). *P < .05, ***P < .001.
Discussion
Here we demonstrate a broad antilymphoma effect of DMF, an established, approved, and even during long-term application well-tolerated drug, which is currently used for the treatment of psoriasis and relapsing-remitting multiple sclerosis.30,53 Although DMF treatment was found to diminish lymphocyte numbers in the blood of patients, clinical trials failed to provide any evidence for an increased susceptibility to infections, even though single cases of progressive multifocal leukoencephalopathy were reported. Despite its profound cytotoxicity in both major DLBCL subtypes, we uncovered different modes of action underlying the broad cytotoxic effect of DMF; whereas DMF-induced cell death in GCB DLBCL exhibited all typical hallmarks of ferroptosis (ie, lipid peroxidation, iron dependency, and protection by lipophilic radical-trapping antioxidants), DMF-induced cytotoxicity in ABC DLBCL was mainly characterized by inhibition of NF-κB and JAK/STAT survival signaling.19-21
In contrast to BSO and erastin, which directly or indirectly inhibit GSH synthesis, DMF potently and rapidly depletes GSH by succination.54 Because of its distinct mode of action, DMF cannot be easily incorporated into the currently defined classes of ferroptosis inducers, which include cystine import inhibitors, GPX4 inhibitors, GPX4/CoQ10 depleters, and lipid peroxidation inducers. We therefore propose a new group of ferroptosis inducers comprising electrophilic substances that potently deplete the cellular GSH pool.55
Our data suggest that GCB DLBCL cell lines are highly susceptible to DMF-induced ferroptosis because of their inherently low GSH levels in concert with low GPX4 and high 5-LOX expression. The nuclear localization of 5-LOX observed in GCB DLBCL cell lines and in ∼50% of GCB DLBCL biopsies indicates that 5-LOX may be enzymatically active, which requires its recruitment to the 5-LOX–activating protein residing at the nuclear membrane.43,44 Although the transcription factor Sp1 contributes to 5-LOX expression, it remains unclear why a subset of GCB DLBCLs shows high 5-LOX levels. Interestingly, B-cell activation seems to play an important role in the regulation of 5-LOX expression, because PKC activity inversely correlates with 5-LOX levels. In B cells, PKC controls among others the activation of the transcription factors NF-κB and AP-1, but it is still unknown whether these are involved in 5-LOX regulation.10 The contribution of Sp1 to the ferroptosis susceptibility of DLBCL also requires further clarification, particularly because it has been associated with 5-LOX and GPX4 expression.45,56,57 We propose a model in which 5-LOX expression is driven by constitutively active transcription factors, but is simultaneously suppressed by chronic B-cell activation in ABC DLBCL cells.
Our data suggest that inhibition of classical NF-κB and STAT3 activity, both essential for ABC DLBCL survival, contributes to the DMF-induced cytotoxicity observed in this subtype.10,17,41 Inhibition of NF-κB signaling by DMF has been previously reported in various cellular systems, but the proposed modes of action have differed substantially and included HO-1–mediated NF-κB inhibition, reduction of K63-linked polyubiquitination, and direct modification of RelA or upstream regulators of NF-κB, such as PKC.32,58‐61 In ABC DLBCL cell lines, DMF does not affect upstream BCR signaling, CBM complex formation, MALT1 protease activity, or K63-linked polyubiquitination of BCL10. Instead, DMF directly succinates a reactive cysteine residue, which resides in the activation loop of the IKK2 kinase domain and thus inhibits IKK activity in DLBCL cell lines.50
In addition to its inhibitory effect on NF-κB, we were able to demonstrate that DMF directly modifies JAKs. We propose that succination of the highly conserved C257 located in the FERM domain of JAK1 weakens the interaction between JAK1 and the IL-10 receptor and possibly other cytokine receptors. Expression of STAT3C, a STAT3 mutant with an increased DNA-binding affinity, and its activation with exogenous IL-6/IL-10 partially protected ABC DLBCL cell lines from DMF-mediated cytotoxicity. Therefore, we propose a model in which the DMF-dependent cytotoxicity observed in ABC DLBCL relies on the inhibition of both NF-κB and JAK/STAT survival signaling.
Because 40% to 80% of primary DLBCL samples exhibit high levels of BCL-2 expression, the combination of BH3 mimetics, such as ABT-199, with DMF represents an attractive novel therapeutic strategy.62-66 This seems especially promising in ABC DLBCL, because BCL-2 is an established regulator of apoptosis but not of ferroptosis. In GCB DLBCL, the simultaneous pharmacological inhibition of the GSH-GPX4 axis and the FSP1-CoQ10-NAD(P)H system, which cooperate in the prevention of excessive lipid peroxidation, synergistically induced cell death.52,67 Therefore, combining DMF with other drugs impairing the cellular capacity to detoxify lipid peroxides might provide an attractive option for the future treatment of DLBCL patients.
Acknowledgments
The authors thank Margot Thome, Dirk Schwarzer, Georg Zocher, Franziska Klose, Corinna Kosnopfel, and Marcus Conrad for helpful discussion and technical advice.
This work was supported by grants from the Collaborative Research Center Transregio (SFB/TR 156 [S.H.] and SFB/TR 209 [K.S.-O., S.H.]), the Emmy-Noether program of the Deutsche Forschungsgemeinschaft (DFG) and the DFG under Germany’s Excellence Strategy EXC2180 (S.H.), the Deutsche Krebshilfe (G.L., S.H.), grants from the Ministry of Health of the Czech Republic (NV19-08-00144) and Grant Agency of the Czech Republic (GA20-25308S) (P.K.), and a grant from the Swiss National Science Foundation (310030 179239) (C.L.).
Authorship
Contribution: A.S., P. Bucher, M. Grimm, P. Berning, M.B., W.X., M.K., M.Z., M.-A.J., C.S., H.H., P.V., and P.K. performed experiments; A.S., M.K., M.Z., M. Grau, P.K., K.B., A.R., G.O., C.L., K.S.-O., G.L., and S.H. analyzed data; A.S., G.L., and S.H. wrote the manuscript, on which all other authors commented; and A.S., G.L., and S.H. conceived and coordinated the study.
Conflict-of-interest disclosure: G.L. declares the following potential conflict of interests with respect to the current study: honoraria from Bayer, AstraZeneca, NanoString, Celgene, Gilead, Janssen, and Roche; consulting or advisory role for AstraZeneca, Bayer, Celgene, Gilead, Janssen, NanoString, and Roche; speaker’s bureau for Bayer, Celgene, Gilead, Janssen, and Roche; research funding from AQUINOX, AstraZeneca, Bayer, Celgene, Gilead, Janssen, and Roche; and travel, accommodation, and expenses reimbursed by Bayer, Celgene, Gilead, Janssen, Roche, and Verastem. The remaining authors declare no competing financial interests.
Correspondence: Stephan Hailfinger, Interfaculty Institute of Biochemistry, University of Tübingen, Auf der Morgenstelle 34, 72076 Tübingen, Germany; e-mail: stephan.hailfinger@uni-tuebingen.de.
Data can be found under accession #GSE164267.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal