

Key Points
CD44 modulates CXCL12-induced stemness features and resistance to venetoclax on AML cells.
CD44 and CXCR4 physically associate at the cell membrane upon CXCL12 induction.

Abstract
Acute myeloid leukemia (AML) has a poor prognosis under the current standard of care. In recent years, venetoclax, a BCL-2 inhibitor, was approved to treat patients who are ineligible for intensive induction chemotherapy. However, complete remission rates with venetoclax-based therapies are hampered by minimal residual disease (MRD) in a proportion of patients, leading to relapse. MRD is a result of leukemic stem cells being retained in bone marrow protective environments; activation of the CXCL12-CXCR4 pathway was shown to be relevant to this process. An important role is also played by cell adhesion molecules such as CD44, which has been shown to be crucial for the development of AML. Here we show that CD44 is involved in CXCL12 promotion of resistance to venetoclax-induced apoptosis in human AML cell lines and AML patient samples, which could be abrogated by CD44 knock down, knockout, or blocking with an anti-CD44 antibody. Split-Venus bimolecular fluorescence complementation showed that CD44 and CXCR4 physically associate at the cell membrane upon CXCL12 induction. In the venetoclax-resistant OCI-AML3 cell line, CXCL12 promoted an increase in the proportion of cells expressing high levels of embryonic stem cell core transcription factors (ESC-TFs: Sox2, Oct4, Nanog) abrogated by CD44 knockdown. This ESC-TF–expressing subpopulation which could be selected by venetoclax treatment, exhibited a basally enhanced resistance to apoptosis and expressed higher levels of CD44. Finally, we developed a novel AML xenograft model in zebrafish, which showed that CD44 knockout sensitizes OCI-AML3 cells to venetoclax treatment in vivo. Our study shows that CD44 is a potential molecular target for sensitizing AML cells to venetoclax-based therapies.
Introduction
Acute myeloid leukemia (AML) is a devastating disease. To aim for complete remission (CR), the standard approach involves intensive induction chemotherapy. The majority of patients are not eligible for this treatment because of its high toxicity. Noneligible patients receive a palliative lower-intensity therapy.1 This daunting scenario is being reshaped by the BCL-2 inhibitor venetoclax (ABT-199). In trials that used venetoclax combined with hypomethylating agents or low-dose cytarabine, the combination has achieved CR in patients who were ineligible for intensive induction therapy and those who were refractory or relapsed.2-5 Given these results, the US Food and Drug Administration granted accelerated approval to these regimens for first-line treatment of AML patients who were ineligible for intensive induction chemotherapy (https://www.fda.gov/drugs/fda-approves-venetoclax-combination-aml-adults). Nevertheless, ∼20% of AML patients treated with venetoclax and hypomethylating agent azacytidine remain refractory, and of patients who achieve a CR, a proportion of those with measurable minimal residual disease (MRD) relapse.6 MRD is sustained by a subset of resistant leukemic cells that harbor survival advantages and stemness properties.6,7 This phenotype is promoted by a protective microenvironment in the bone marrow.8 Venetoclax was shown to have a specific effect on the energy metabolism of leukemic stem cells (LSCs), enabling targeting of the LSC compartment.9 Several clinical trials currently underway are aimed at improving venetoclax-based regimens through new associations with therapeutic agents,4,5 and cytogenetic features of subclones resistant to venetoclax-based regimens are beginning to be described.10
Of the molecular pathways exploited by LSCs for their survival, the chemokine receptor CXCR4 is prominent in several types of leukemia, whereas the adhesion molecule CD44 has been shown to be relevant in AML.8 CD44 cooperates with CXCR4 for the survival of normal hematopoietic stem cells in the bone marrow niche, rich in their respective ligands, hyaluronan (HA), and CXCL12.11 CD44 also functions as a co-receptor for CXCR412 and several other receptors, modulating their signaling efficiency.13 The role of this molecular interaction in leukemic cell survival upon exposure to venetoclax remains unexplored.
Here we show that CXCL12-CXCR4 signaling, induction of resistance to venetoclax-induced apoptosis, and stemness marker expression are dependent on CD44 in vitro. In a novel zebrafish intravital imaging xenograft model, we show that the absence of CD44 in AML cells sensitizes them to venetoclax in vivo.
Methods
Patient samples
Primary AML cells were obtained from patient bone marrow at the time of diagnosis at the Third Medical Department, Paracelsus Medical University Salzburg in Salzburg, Austria (Ethics Committee approval 415-E/2009/2-2016). Mononuclear cells were isolated with Ficoll density gradient centrifugation and frozen. Patient characteristics are compiled in supplemental Table 1 (available on the Blood Web site).
Human CXCL12/SDF-1 alpha immunoassay
Enzyme-linked immunosorbent assays (ELISAs) were performed using the human CXCL12/SDF-1α Quantikine ELISA Kit (R&D Systems) according to the manufacturer’s instructions.
Flow cytometry
The OCI-AML3 and Molm13-VR (venetoclax resistant) (supplemental Methods) cell lines and the primary cells were incubated with primary monoclonal antibodies (supplemental Table 2) or corresponding isotype controls. CD44 and CXCR4 expression and cells expressing d2EGFP were identified and sorted using BD FACSAria I and FACSAria Fusion Cytometers.
Transient RNA interference and DNA transfection
OCI-AML3 cells were transfected with 5 nM small interfering RNA (siRNA) targeting CD44 (5'-CTGAAATTAGGGCCCAATT-3' and 5'-AATGGTGCATTTGGTGAAC-3' pooled) or control siRNA (5'-UAAUGUAUUGGAACGCAUAUU-3' and 5'-AGGUAGUGUAAUCGCCUUGUU-3' pooled) using the HiPerFect transfection reagent (all from QIAGEN).
Bimolecular fluorescence complementation assay
CXCR4-VN, CD44-VC, and CD44Δect-VC were transfected using ViaFect (Promega) into HEK293T cells. At 48 hours after transfection, cells were treated with HA 200 μg/mL for 1 hour or AMD3100 5 μM for 10 minutes, where indicated, or left untreated and subsequently induced with CXCL12 200 ng/mL for 10 minutes, followed by 4% paraformaldehyde fixation. Cells transfected with only CXCR4-VN were used as negative controls. Cell nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI) (Dako) for 15 minutes. Confocal images (Zeiss LSM 800) were processed using ImageJ software (National Institutes of Health, Bethesda, MD). For fusion proteins and image analysis, see supplemental Methods. Sequences of the constructs will be provided on request.
PL-SIN-EOS-S(4+)-d2EGFP-SV40-Puro vector subcloning
The pluripotency reporter vector PL-SIN-EOS-S(4+)-EGFP (gift from James Ellis; Addgene plasmid #21317; http://n2t.net/addgene:21317; RRID:Addgene_21317)14 was modified by replacing EGFP with the unstable d2EGFP (half-life 22 and 2 hours, respectively) and by adding the puromycin resistance gene expressed under SV40 promoter independently from d2EGFP (supplemental Methods).
CRISPR/Cas9 CD44 knockout
CD44 was knocked out by CRISPR/Cas9 in OCI-AML3 cells (OCI-AML3 CD44KO ) and in Molm13-VR cells (Molm13-VR CD44KO) using a lentiviral vector coding for the single-guide RNA (sgRNA) hCas9 and puromycin resistance. Control cells were generated with a CRISPR/Cas9 vector (custom cloned by Vector Builder, Inc) containing a scramble (Scr) sgRNA with no known target in the human genome. For details of the lentiviral transduction protocol and the selection strategy we followed, see supplemental Methods.
Apoptotic assay
Exponentially growing cells were seeded into wells coated with HA or control wells in technical triplicates and incubated with CXCL12 200 ng/mL. After 1 hour, venetoclax 1 µM dissolved in dimethyl sulfoxide (DMSO) or DMSO alone was added. After 4 hours of incubation, the cells were harvested and stained with annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) and analyzed by flow cytometry. The same protocol was used for AML patient samples but with a shorter venetoclax incubation time (3 hours).
For zebrafish xenografts and intravital imaging, 100 to 150 OCI-AML3 or OCI-AML3 CD44KO cells were suspended in phosphate-buffered saline and stained with CellTrace Violet (CTV; Molecular Probes, Eugene, OR) and were injected into the blastoderm of zebrafish embryos 3 hours post fertilization (hpf) and incubated at 33°C. After 2 days, the embryos were anesthetized and injected into the cardinal vein with 0.4 nL of venetoclax 2 mM mixed with CellEvent Caspase-3/7 green detection reagent 10 μM in DMSO or DMSO alone as control. Two hours post injection (hpi), 12 larvae per group were selected for live confocal imaging. For more information on intravital imaging analysis, see supplemental Methods.
Statistical analysis
Statistics were performed using GraphPad Prism 6 (GraphPad Software, San Diego, CA). After a Shapiro-Wilk test for normality, a Student t test was used to compare the means between 2 independent groups with normal distribution, and analysis of variance (ANOVA) with a Bonferroni test was used for more than 2 groups. Nonnormally distributed data sets were analyzed with the Wilcoxon rank-sum test or Kruskal-Wallis test (α = .05).
Results
Expression of CD44 and CXCR4 positively correlate in primary AML cells
In line with previous publications,15,16 the expression of the chemokine receptor CXCR4 was increased in leukemic cells from AML patients with adverse prognosis compared with those who have a favorable or intermediate prognosis (Figure 1A). CD44 and CXCR4 expression correlated positively (Figure 1B). Representative flow cytometric dot plots for 4 patients are included in supplemental Figure 1A. In sera from the same cohort of patients as in Figure 1A-B, higher concentrations of CXCL12 were detected relative to sera from healthy donors (Figure 1C). Of note, cells from patient samples express very low levels of the other CXCL12 receptor, CXCR7 (supplemental Figure 1B).
![CD44 expression correlates with CXCR4 in cells of patients with AML. (A) CXCR4 cell surface expression of primary AML cells from patients with different prognoses was determined via flow cytometry (1-way analysis of variance [ANOVA] with Bonferroni posttest). (B) Cell surface expression of CD44 and CXCR4 was correlated (n = 39; Pearson’s correlation test r = 0.3971; P = .012). (C) Serum CXCL12 level of AML patients (n = 12) as well as healthy donors (n = 15) was measured by enzyme-linked immunosorbent assay (ELISA) (Student t test P = .0003). MFI, mean fluorescent intensity. *P ≤ .05; ***P ≤ .001. The error bars indicate standard deviation.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/12/10.1182_blood.2020006343/4/m_bloodbld2020006343f1.png?Expires=1767697715&Signature=foxpJH0tATD2UJnimcXslKY8V-wwjsWbNexB1VqlY7R4wuLZUxY7wIv--BBqB26~n7TDca1tVfRT-8Sdfj67tnXb-ch3G42n5Mmfe1FcredNzAjLOq7gkrgJ4f-i~e3ViDaVF~WaEKemI0RHnom3ZT1wTb7m5MkWT0s74Co4N5wmbdGziMUfsLPGTkKPeRz-yzB1QlNaHSeJfsPxa6JbjTTYswVoLyB4nw969M0tBcFdFzHJAyuOWtVkAEXaViLEaGtAWUCOPdiC-EqH92Zexf4f88aRU8ucI6ew1HtFixfmHFHMO-JEtEOmohkhpZI57p6TZIUiYGxL6oaSTe4KHg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CD44 expression correlates with CXCR4 in cells of patients with AML. (A) CXCR4 cell surface expression of primary AML cells from patients with different prognoses was determined via flow cytometry (1-way analysis of variance [ANOVA] with Bonferroni posttest). (B) Cell surface expression of CD44 and CXCR4 was correlated (n = 39; Pearson’s correlation test r = 0.3971; P = .012). (C) Serum CXCL12 level of AML patients (n = 12) as well as healthy donors (n = 15) was measured by enzyme-linked immunosorbent assay (ELISA) (Student t test P = .0003). MFI, mean fluorescent intensity. *P ≤ .05; ***P ≤ .001. The error bars indicate standard deviation.
CD44 expression correlates with CXCR4 in cells of patients with AML. (A) CXCR4 cell surface expression of primary AML cells from patients with different prognoses was determined via flow cytometry (1-way analysis of variance [ANOVA] with Bonferroni posttest). (B) Cell surface expression of CD44 and CXCR4 was correlated (n = 39; Pearson’s correlation test r = 0.3971; P = .012). (C) Serum CXCL12 level of AML patients (n = 12) as well as healthy donors (n = 15) was measured by enzyme-linked immunosorbent assay (ELISA) (Student t test P = .0003). MFI, mean fluorescent intensity. *P ≤ .05; ***P ≤ .001. The error bars indicate standard deviation.
CXCL12 promotes resistance of AML cells to venetoclax-induced apoptosis, a process enhanced by HA
To assess whether the correlation between CXCR4 and CD44 could have functional consequences in AML cell survival, we used resistance to venetoclax-induced apoptosis as an assay. Compared with control treatment with DMSO (vehicle for venetoclax), venetoclax induced higher levels of cleaved caspase 3 (C-Cas3) in AML cell lines (Figure 2A; supplemental Figure 2A). OCI-AML3 (a venetoclax-resistant cell line17) exposed to CXCL12 before treatment with venetoclax showed lower levels of C-Cas3 compared with controls not exposed to CXCL12 (Figure 2A). Because HA is the main CD44 ligand and a component of the extracellular matrix in the bone marrow where MRD resides, we evaluated its effect in this in vitro setting. Combined with CXCL12, HA further decreased the levels of C-Cas 3 upon treatment with venetoclax (Figure 2A). Conversely, Molm13, a cell line highly sensitive to venetoclax, was less protected from apoptosis by CXCL12 and/or HA (supplemental Figure 2A). AML samples from 5 patients (3 with FLT3-internal tandem duplication [FLT3-ITD] recently shown to be associated with venetoclax resistance10) were seeded into wells coated with HA or control wells, incubated with CXCL12, and subsequently treated with venetoclax (Figure 2B). After 3 hours, the cells were stained with annexin V-FITC and PI and analyzed by flow cytometry. Venetoclax treatment significantly reduced the cell viability in all cases, except in cells seeded on HA and preincubated with CXCL12. To evaluate the contribution of cell anchorage to HA to the protective effect of CXCL12, we compared venetoclax-induced apoptosis in OCI-AML3 cells that adhered to the HA substrate with the cells that remained in suspension within the same wells. Figure 2C shows a lower number of apoptotic cells in the adherent cell fraction compared with the suspended one, an effect further enhanced by CXCL12. We also found higher MCL-1 messenger RNA (mRNA) levels (a mechanism of resistance to venetoclax17) upon CXCL12 and HA induction in primary AML cells (Figure 2D). These levels were more than one order of magnitude higher than the ones of BCL-2 in these samples (supplemental Figure 2Bi). Levels of expression of CXCR4 and CD44 were not affected by CXCL12 and/or HA induction (supplemental Figure 2Bii). Taken together, these results show that CXCL12 enhances resistance to venetoclax, an effect boosted by HA.
![CXCL12 and HA protect AML cells from venetoclax-induced apoptosis. (A) OCI-AML3 cells were preincubated with CXCL12 and/or HA where indicated, followed by venetoclax or solvent (DMSO) treatment. Lysates were probed with the antibodies indicated in the western blot. Data from 1 representative of 3 experiments are shown. The numbers above the C-Cas3 panel indicate fold changes. β-Actin was used as a loading control. Three independent experiments were quantified (Student t test). (B) 2 × 106 primary cells per mL from AML patient samples were seeded into HA-coated wells or control wells for 30 minutes in technical triplicates and incubated with CXCL12 200 ng/mL. After 1 hour, venetoclax 1 µM or DMSO (vehicle) was added to the corresponding wells. After 3 hours of incubation, the cells were harvested and stained with annexin V-fluorescein isothiocyanate (FITC) and PI and analyzed by flow cytometry. The frozen patient samples contained a population of PI+ cells, which could not be distinguished from PI+ cells at the end of the experiment, so the results were calculated using the percentage of viable cells (annexin V–/PI–). The value of each sample was expressed as a ratio between the percentage of viable control cells (incubated in the absence of HA and/or CXCL12 and treated with DMSO) and the percentage of viable cells in each of the other conditions of the same patient sample to normalize for basal differences between samples of different patients (n = 5 patient samples [3 FLT3-internal tandem duplication [FLT3-ITD]; 2 normal]) in independent experiments (1-way ANOVA). (C) OCI-AML3 cells with or without CXCL12 induction were seeded in HA-coated tissue culture plates and treated with venetoclax. Apoptotic cells (annexin V+) of the adherent and suspended populations were determined separately by flow cytometry. Three independent experiments were quantified (1-way ANOVA). (D) Primary AML cells were incubated with CXCL12 and/or HA coating for 4 hours. mRNA expression of BCL-2 and MCL-1 were determined by qPCR from 3 independent experiments with 3 different primary AML samples (Kruskal-Wallis test). *P < .05; **P < .01; ****P < .0001. The error bars indicate standard deviation. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/12/10.1182_blood.2020006343/4/m_bloodbld2020006343f2.png?Expires=1767697715&Signature=bJBM1h42cWXuuPm1oROtXg0VvEtYAqXHiJ1uNI0dsbvNbt4NrYcLi1mWol2py72TuDCiW~oGxq5D3Kk49lmPTIJARZAq-dxNTVZJXi3MXyyq2BuEm9utIiK3LWOxA7-84cPIPruYeqor42qU5XSZneEeh30g~P9F7QciggjAQRPVT2olgGUowAsvAdxisXsU9mQFwOD80fszNXhvR5owb2C-2RW89CFMx57NnQPjGbbMzR~RM9Silt77vmHtnOG0xmLqpPg0ErZUOBxtQel6TZwckk1HrRXBySFiJaSSQVnzDm4FrvFJoyHQx45GI68Nb0Q44qtPzOVd~fb~jtCTYg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CXCL12 and HA protect AML cells from venetoclax-induced apoptosis. (A) OCI-AML3 cells were preincubated with CXCL12 and/or HA where indicated, followed by venetoclax or solvent (DMSO) treatment. Lysates were probed with the antibodies indicated in the western blot. Data from 1 representative of 3 experiments are shown. The numbers above the C-Cas3 panel indicate fold changes. β-Actin was used as a loading control. Three independent experiments were quantified (Student t test). (B) 2 × 106 primary cells per mL from AML patient samples were seeded into HA-coated wells or control wells for 30 minutes in technical triplicates and incubated with CXCL12 200 ng/mL. After 1 hour, venetoclax 1 µM or DMSO (vehicle) was added to the corresponding wells. After 3 hours of incubation, the cells were harvested and stained with annexin V-fluorescein isothiocyanate (FITC) and PI and analyzed by flow cytometry. The frozen patient samples contained a population of PI+ cells, which could not be distinguished from PI+ cells at the end of the experiment, so the results were calculated using the percentage of viable cells (annexin V–/PI–). The value of each sample was expressed as a ratio between the percentage of viable control cells (incubated in the absence of HA and/or CXCL12 and treated with DMSO) and the percentage of viable cells in each of the other conditions of the same patient sample to normalize for basal differences between samples of different patients (n = 5 patient samples [3 FLT3-internal tandem duplication [FLT3-ITD]; 2 normal]) in independent experiments (1-way ANOVA). (C) OCI-AML3 cells with or without CXCL12 induction were seeded in HA-coated tissue culture plates and treated with venetoclax. Apoptotic cells (annexin V+) of the adherent and suspended populations were determined separately by flow cytometry. Three independent experiments were quantified (1-way ANOVA). (D) Primary AML cells were incubated with CXCL12 and/or HA coating for 4 hours. mRNA expression of BCL-2 and MCL-1 were determined by qPCR from 3 independent experiments with 3 different primary AML samples (Kruskal-Wallis test). *P < .05; **P < .01; ****P < .0001. The error bars indicate standard deviation. ns, not significant.
CXCL12 and HA protect AML cells from venetoclax-induced apoptosis. (A) OCI-AML3 cells were preincubated with CXCL12 and/or HA where indicated, followed by venetoclax or solvent (DMSO) treatment. Lysates were probed with the antibodies indicated in the western blot. Data from 1 representative of 3 experiments are shown. The numbers above the C-Cas3 panel indicate fold changes. β-Actin was used as a loading control. Three independent experiments were quantified (Student t test). (B) 2 × 106 primary cells per mL from AML patient samples were seeded into HA-coated wells or control wells for 30 minutes in technical triplicates and incubated with CXCL12 200 ng/mL. After 1 hour, venetoclax 1 µM or DMSO (vehicle) was added to the corresponding wells. After 3 hours of incubation, the cells were harvested and stained with annexin V-fluorescein isothiocyanate (FITC) and PI and analyzed by flow cytometry. The frozen patient samples contained a population of PI+ cells, which could not be distinguished from PI+ cells at the end of the experiment, so the results were calculated using the percentage of viable cells (annexin V–/PI–). The value of each sample was expressed as a ratio between the percentage of viable control cells (incubated in the absence of HA and/or CXCL12 and treated with DMSO) and the percentage of viable cells in each of the other conditions of the same patient sample to normalize for basal differences between samples of different patients (n = 5 patient samples [3 FLT3-internal tandem duplication [FLT3-ITD]; 2 normal]) in independent experiments (1-way ANOVA). (C) OCI-AML3 cells with or without CXCL12 induction were seeded in HA-coated tissue culture plates and treated with venetoclax. Apoptotic cells (annexin V+) of the adherent and suspended populations were determined separately by flow cytometry. Three independent experiments were quantified (1-way ANOVA). (D) Primary AML cells were incubated with CXCL12 and/or HA coating for 4 hours. mRNA expression of BCL-2 and MCL-1 were determined by qPCR from 3 independent experiments with 3 different primary AML samples (Kruskal-Wallis test). *P < .05; **P < .01; ****P < .0001. The error bars indicate standard deviation. ns, not significant.
CD44 modulates the CXCL12-induced resistance to apoptosis
To determine whether HA exerted its effect through CD44, we knocked down CD44 with siRNA in OCI-AML3 cells. This abolished the protective effect of CXCL12 against venetoclax-induced cleavage of caspase 3, independent of the presence of HA (Figure 3A-C). To confirm these results, we knocked out CD44 using CRISPR/Cas9 in OCI-AML3 cells (OCI-AML3 CD44KO; supplemental Figure 2Biv, left). Cells expressing Cas9 together with a scramble sgRNA (OCI-AML3 Scr) were used as controls. In OCI-AML3 Scr cells, CXCL12 and/or HA enhanced resistance to venetoclax; this was shown by higher cell viability measured by flow cytometry after annexin V and PI staining (Figure 3D). In contrast, OCI-AML3 CD44KO cells were sensitized to venetoclax-induced apoptosis, an effect that could not be rescued by treatment with HA or CXCL12, although those factors combined had a slight effect (Figure 3D). To complement this model, we generated Molm13 venetoclax-resistant cells (Molm13-VR) by culturing them in increasing concentrations of venetoclax for 6 weeks; we generated Molm13-VR CD44KO as well as Molm13-VR Scr by means of CRISPR/Cas9 (supplemental Figure 2Bv, left). Molm13-VR cells expressed high levels of MCL-1, but the expression of CD44, BCL-2, and BCL-xL remained unchanged (supplemental Figure 2C). Apoptosis remained inducible by relatively high concentrations of venetoclax (1 μM) in these cells but was significantly reduced by HA and the combination of HA and CXCL12 (Figure 3E). As in OCI-AML3 cells, CD44 knockout also dampened this effect. Treatment of Molm13-VR cells with the CXCR4 inhibitors AMD3100 or WZ811 mimicked this effect (Figure 3F), suggesting that the effect of CD44 could at least partially be explained by its coreceptor function on CXCR4,17 which in turn implies that this function should also be impaired by CD44 blocking antibodies. Therefore, we sought to prove this effect in an unbiased manner in AML patient samples. Upon treatment with IM7 (an antibody against all CD44 isoforms), the combination of CXCL12 and HA could not rescue AML patient bone marrow–derived cells from venetoclax-induced apoptosis, while maintaining its protective effect in the presence of an isotype control antibody (Figure 3G). This result suggests that a treatment with a CD44 inhibitor could potentially counteract venetoclax resistance induced by factors present in bone marrow niches.
![CXCL12 induction of resistance to venetoclax requires CD44. (A-C) OCI-AML3 cells transiently transfected with either CD44 siRNA or control siRNA were preincubated with CXCL12 and/or HA followed by venetoclax or DMSO treatment. Lysates were probed by western blot. A representative picture is shown. Data from 3 independent experiments were quantified. β-Actin was used as a loading control (Ctrl). The numbers above the C-Cas3 panel indicate fold changes. Three independent experiments were quantified (Kruskal-Wallis test). (D) OCI-AML3 cells and (E) venetoclax-resistant Molm13 (Molm13-VR) cells were transduced with CRISPR/Cas9 lentiviral vectors encoding either Scr or CD44 sgRNA, followed by puromycin selection and FACS sorting for cells not expressing CD44. Cells with or without CXCL12 induction were seeded in HA or solvent (phosphate-buffered saline)-coated tissue culture plates, followed by venetoclax or DMSO treatment. Viable cells were determined by flow cytometry using annexin V and PI staining, and data were analyzed using 1-way ANOVA. (F) Molm13-VR cells were preincubated with CXCR4 inhibitors (AMD3100 or WZ811) and induced with CXCL12, followed by venetoclax or DMSO treatment. Viable cells were determined by flow cytometry using annexin V and PI staining, and data were analyzed using the Student t test. (G) Primary AML cells were preincubated with an anti-CD44 antibody (IM7) or the corresponding isotype (immunoglobulin G [IgG]), seeded in HA-coated tissue culture plates, and induced with CXCL12 (200 ng/mL). After 3 hours, the cells were stained with annexin V and PI and analyzed by FACS. The results of each patient sample show the percentage of viable cells normalized as a ratio between control cells (incubated with IgG control and DMSO) and the viable cells in each of the other conditions (n = 4 patient samples [2 FLT3-ITD, 2 normal]) in independent experiments; data were analyzed using 1-way ANOVA. (H-I) OCI-AML3 cells transiently transfected with either CD44 siRNA or control siRNA were induced with or without CXCL12. mRNA expression of CD44, BCL-2, and BAX were determined by qPCR from 4 independent experiments; data were analyzed using a Wilcoxon rank-sum test. *P < .05; **P < .01; ***P < .001; ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/12/10.1182_blood.2020006343/4/m_bloodbld2020006343f3a.png?Expires=1767697715&Signature=xqc-~SFauTVeoj~3sKmtjhe2~~502VfQDw5ezbqfVIVIgjAKZh1haFwjzZdHamIP6MdYMAG830xlCw3i-NYE8MOAIsYvWkXj3LoEY6KkGGO1ARq8oOBnCJUGaQotPM3UD0pfRVHVAoWiLG-HJM21G7SJdZZD5Uyvs~O6dpERmbEizoCKz9fUQ2z0zd8RmIyUS6MQLKeGQig3t-YUlyIaQGweWxLG7FTgtYbuB09hl7blvEJyE9fpKjLtiUKfJcDbMcOFG4bYOUpaFH0dmcDuy4O9xXccMwdwSBZ04k95r4hLVfnloUKBx51U3jluPaxXUhFIyubXQQj3r~8CzVvieA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![CXCL12 induction of resistance to venetoclax requires CD44. (A-C) OCI-AML3 cells transiently transfected with either CD44 siRNA or control siRNA were preincubated with CXCL12 and/or HA followed by venetoclax or DMSO treatment. Lysates were probed by western blot. A representative picture is shown. Data from 3 independent experiments were quantified. β-Actin was used as a loading control (Ctrl). The numbers above the C-Cas3 panel indicate fold changes. Three independent experiments were quantified (Kruskal-Wallis test). (D) OCI-AML3 cells and (E) venetoclax-resistant Molm13 (Molm13-VR) cells were transduced with CRISPR/Cas9 lentiviral vectors encoding either Scr or CD44 sgRNA, followed by puromycin selection and FACS sorting for cells not expressing CD44. Cells with or without CXCL12 induction were seeded in HA or solvent (phosphate-buffered saline)-coated tissue culture plates, followed by venetoclax or DMSO treatment. Viable cells were determined by flow cytometry using annexin V and PI staining, and data were analyzed using 1-way ANOVA. (F) Molm13-VR cells were preincubated with CXCR4 inhibitors (AMD3100 or WZ811) and induced with CXCL12, followed by venetoclax or DMSO treatment. Viable cells were determined by flow cytometry using annexin V and PI staining, and data were analyzed using the Student t test. (G) Primary AML cells were preincubated with an anti-CD44 antibody (IM7) or the corresponding isotype (immunoglobulin G [IgG]), seeded in HA-coated tissue culture plates, and induced with CXCL12 (200 ng/mL). After 3 hours, the cells were stained with annexin V and PI and analyzed by FACS. The results of each patient sample show the percentage of viable cells normalized as a ratio between control cells (incubated with IgG control and DMSO) and the viable cells in each of the other conditions (n = 4 patient samples [2 FLT3-ITD, 2 normal]) in independent experiments; data were analyzed using 1-way ANOVA. (H-I) OCI-AML3 cells transiently transfected with either CD44 siRNA or control siRNA were induced with or without CXCL12. mRNA expression of CD44, BCL-2, and BAX were determined by qPCR from 4 independent experiments; data were analyzed using a Wilcoxon rank-sum test. *P < .05; **P < .01; ***P < .001; ****P < .0001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/12/10.1182_blood.2020006343/4/m_bloodbld2020006343f3b.png?Expires=1767697715&Signature=sI~l5qNNCdnPL6snjU~Wt9t0au4TD5XFC5JElh-TCjPZtsPqN7w-RK~Ac1yXN0Ljrug8GsX6bJgMPT2NgMasmtIaIUoeb7HK0yV17mOp539CBsApHVmuuHDLuAWa9lqlUpTc06FpW2Vd6Cl-9RCQX74Hs9WMI0zBUguQaaUPmvF~hUqEYH5FTcPl4gkk4PJDPUtz9AlCOpU2~uAWEC-TU7pCSqE6joDLpAuPb3U1QDTp8f6WNoLujLOK4PJzw1xiluFJMlaIgOkiXaaBvbJt9~ge0RCQU6F-H9xU6QwxAVUwnVhOOEsqVnSGlPaFV9RmuFNCY7aLT2QL2ET-FMDrjg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CXCL12 induction of resistance to venetoclax requires CD44. (A-C) OCI-AML3 cells transiently transfected with either CD44 siRNA or control siRNA were preincubated with CXCL12 and/or HA followed by venetoclax or DMSO treatment. Lysates were probed by western blot. A representative picture is shown. Data from 3 independent experiments were quantified. β-Actin was used as a loading control (Ctrl). The numbers above the C-Cas3 panel indicate fold changes. Three independent experiments were quantified (Kruskal-Wallis test). (D) OCI-AML3 cells and (E) venetoclax-resistant Molm13 (Molm13-VR) cells were transduced with CRISPR/Cas9 lentiviral vectors encoding either Scr or CD44 sgRNA, followed by puromycin selection and FACS sorting for cells not expressing CD44. Cells with or without CXCL12 induction were seeded in HA or solvent (phosphate-buffered saline)-coated tissue culture plates, followed by venetoclax or DMSO treatment. Viable cells were determined by flow cytometry using annexin V and PI staining, and data were analyzed using 1-way ANOVA. (F) Molm13-VR cells were preincubated with CXCR4 inhibitors (AMD3100 or WZ811) and induced with CXCL12, followed by venetoclax or DMSO treatment. Viable cells were determined by flow cytometry using annexin V and PI staining, and data were analyzed using the Student t test. (G) Primary AML cells were preincubated with an anti-CD44 antibody (IM7) or the corresponding isotype (immunoglobulin G [IgG]), seeded in HA-coated tissue culture plates, and induced with CXCL12 (200 ng/mL). After 3 hours, the cells were stained with annexin V and PI and analyzed by FACS. The results of each patient sample show the percentage of viable cells normalized as a ratio between control cells (incubated with IgG control and DMSO) and the viable cells in each of the other conditions (n = 4 patient samples [2 FLT3-ITD, 2 normal]) in independent experiments; data were analyzed using 1-way ANOVA. (H-I) OCI-AML3 cells transiently transfected with either CD44 siRNA or control siRNA were induced with or without CXCL12. mRNA expression of CD44, BCL-2, and BAX were determined by qPCR from 4 independent experiments; data were analyzed using a Wilcoxon rank-sum test. *P < .05; **P < .01; ***P < .001; ****P < .0001.
CXCL12 induction of resistance to venetoclax requires CD44. (A-C) OCI-AML3 cells transiently transfected with either CD44 siRNA or control siRNA were preincubated with CXCL12 and/or HA followed by venetoclax or DMSO treatment. Lysates were probed by western blot. A representative picture is shown. Data from 3 independent experiments were quantified. β-Actin was used as a loading control (Ctrl). The numbers above the C-Cas3 panel indicate fold changes. Three independent experiments were quantified (Kruskal-Wallis test). (D) OCI-AML3 cells and (E) venetoclax-resistant Molm13 (Molm13-VR) cells were transduced with CRISPR/Cas9 lentiviral vectors encoding either Scr or CD44 sgRNA, followed by puromycin selection and FACS sorting for cells not expressing CD44. Cells with or without CXCL12 induction were seeded in HA or solvent (phosphate-buffered saline)-coated tissue culture plates, followed by venetoclax or DMSO treatment. Viable cells were determined by flow cytometry using annexin V and PI staining, and data were analyzed using 1-way ANOVA. (F) Molm13-VR cells were preincubated with CXCR4 inhibitors (AMD3100 or WZ811) and induced with CXCL12, followed by venetoclax or DMSO treatment. Viable cells were determined by flow cytometry using annexin V and PI staining, and data were analyzed using the Student t test. (G) Primary AML cells were preincubated with an anti-CD44 antibody (IM7) or the corresponding isotype (immunoglobulin G [IgG]), seeded in HA-coated tissue culture plates, and induced with CXCL12 (200 ng/mL). After 3 hours, the cells were stained with annexin V and PI and analyzed by FACS. The results of each patient sample show the percentage of viable cells normalized as a ratio between control cells (incubated with IgG control and DMSO) and the viable cells in each of the other conditions (n = 4 patient samples [2 FLT3-ITD, 2 normal]) in independent experiments; data were analyzed using 1-way ANOVA. (H-I) OCI-AML3 cells transiently transfected with either CD44 siRNA or control siRNA were induced with or without CXCL12. mRNA expression of CD44, BCL-2, and BAX were determined by qPCR from 4 independent experiments; data were analyzed using a Wilcoxon rank-sum test. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Of note, neither the levels of expression of CXCR4 in OCI-AML3 cells as measured by quantitative polymerase chain reaction (qPCR) (supplemental Figure 2Biii) nor its cell surface localization as measured by flow cytometry (supplemental Figure 2Biv) were affected by CD44 knockdown (supplemental Figure 2Biii) and knockout (supplemental Figure 2Biv). The same was true for Molm13-VR cells (supplemental Figure 2Bv, right).
A disruption in the balance between pro- and antiapoptotic proteins can lead to cancer cell survival. To study the effect of the CXCL12-CXCR4 axis on this balance, we assessed the BAX:BCL-2 ratio at the mRNA level by qPCR18 in OCI-AML3 cells. This ratio was decreased upon CXCL12 induction (Figure 3H), an effect that was also abrogated when CD44 was knocked down by means of siRNA (Figure 3H-I).
CD44 has been involved in cell cycle changes in other types of leukemia.19,20 Because an increased percentage of viable cells upon a proapoptotic treatment could be the result of enhanced cell survival but also of increased cell proliferation, we assessed the cell cycle status of the AML cell lines upon CD44 knockout. Experiments that incorporated 5-ethynyl-2’-deoxyuridine in the presence or absence of HA, as well as clonogenic assays, revealed that OCI-AML3 CD44KO and Molm13-VR CD44KO cell lines did not decrease (supplemental Figure 3A) but rather had a moderate increase in proliferation rates (clonogenic assays; supplemental Figure 3B). In addition, CXCL12 did not induce proliferation of Molm13-VR cells (supplemental Figure 3C). This supports the hypothesis that the increased resistance to venetoclax in the presence of CD44 is a result of the difference in cell survival.
CD44 modulates CXCR4 signaling induced by CXCL12, being part of a molecular complex at the cell membrane
Upon CXCL12 induction of OCI-AML3 cells in vitro, addition of HA augmented ERK phosphorylation in a concentration-dependent manner (Figure 4A). Downregulation of CD44 by siRNA not only abrogated this cooperative effect of HA but also led to a decrease of CXCL12-induced signaling in the absence of HA (Figure 4B), suggesting that CD44 is required in CXCL12-induced CXCR4 signaling in AML cells, which is favored by, but does not require, CD44-HA binding. Because osteopontin is another ligand of CD4421 also present in the bone marrow,22 we tested its influence on CXCL12-induced ERK activation. We observed no significant change in ERK phosphorylation upon CXCL12 induction after treatment of OCI-AML3 cells with osteopontin (Opn) (supplemental Figure 4A).

CD44s directly interacts with CXCR4 upon CXCL12 stimulus. (A) OCI-AML3 cells were treated with CXCL12 and increasing concentrations of HA. (B) OCI-AML3 cells transiently transfected with CD44 siRNA or control siRNA were treated with CXCL12 (200 ng/mL) and/or HA (200 μg/mL). ERK phosphorylation was detected using a phospho-ERK-specific antibody by western blot. A representative picture is shown. Data from 3 independent experiments were quantified. Three independent experiments were quantified (Kruskal-Wallis test; *P < .05; **P < .01). (C) Upper rows: HEK293T cells were transfected with a plasmid encoding CXCR4 fused with the N-terminal fragment of the Venus fluorescent protein (CXCR4-VN) and with a plasmid encoding CD44 standard fused with the C-terminal domain of Venus (CD44s-VC). The transfected cells were either left uninduced or were induced with CXCL12, HA, or a combination of both as indicated. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). The bimolecular fluorescence complementation assay signal (yellow) was visualized by confocal microscopy (LSM800 Zeiss microscope 63× objective; scale bar, 20 μm). Mean corrected total cell fluorescence (CTCF) of each cell was analyzed from 4 independent experiments (n = 200 cells). Lower rows: HEK293T cells transfected as indicated above were treated with AMD3100 (5 µM for 10 minutes) or left untreated and subsequently induced with CXCL12 (200 ng/mL for 10 minutes) or left uninduced. Mean CTCF of approximately 450 cells of each condition from 2 independent experiments were analyzed by 1-way ANOVA. *P < .05; **P < .005; ***P < .0005; ****P < .0001.
CD44s directly interacts with CXCR4 upon CXCL12 stimulus. (A) OCI-AML3 cells were treated with CXCL12 and increasing concentrations of HA. (B) OCI-AML3 cells transiently transfected with CD44 siRNA or control siRNA were treated with CXCL12 (200 ng/mL) and/or HA (200 μg/mL). ERK phosphorylation was detected using a phospho-ERK-specific antibody by western blot. A representative picture is shown. Data from 3 independent experiments were quantified. Three independent experiments were quantified (Kruskal-Wallis test; *P < .05; **P < .01). (C) Upper rows: HEK293T cells were transfected with a plasmid encoding CXCR4 fused with the N-terminal fragment of the Venus fluorescent protein (CXCR4-VN) and with a plasmid encoding CD44 standard fused with the C-terminal domain of Venus (CD44s-VC). The transfected cells were either left uninduced or were induced with CXCL12, HA, or a combination of both as indicated. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). The bimolecular fluorescence complementation assay signal (yellow) was visualized by confocal microscopy (LSM800 Zeiss microscope 63× objective; scale bar, 20 μm). Mean corrected total cell fluorescence (CTCF) of each cell was analyzed from 4 independent experiments (n = 200 cells). Lower rows: HEK293T cells transfected as indicated above were treated with AMD3100 (5 µM for 10 minutes) or left untreated and subsequently induced with CXCL12 (200 ng/mL for 10 minutes) or left uninduced. Mean CTCF of approximately 450 cells of each condition from 2 independent experiments were analyzed by 1-way ANOVA. *P < .05; **P < .005; ***P < .0005; ****P < .0001.
CD44 cooperation with CXCR4 signaling in the absence of HA suggests a potential direct interaction. Using a split-Venus bimolecular fluorescence complementation assay, we sought to determine whether both receptors formed complexes upon CXCL12 stimulation. We overexpressed CXCR4 fused to an N-terminal fragment of Venus fluorescent protein (CXCR4-VN) and the standard isoform of CD44 (CD44s) fused to the complementary C-terminal fragment of Venus (CD44s-VC). Both fragments of Venus are nonfluorescent, but in close proximity (7-10 nm), they can complement each other to reconstitute a functional fluorescent protein.23,24 Because of the low transfection efficiency of our AML cell lines with these plasmids, we used HEK293T cells as a proof of principle. Upon the addition of CXCL12 to cells co-transfected with CD44s-VC and CXCR4-VN, we detected a strong fluorescent signal at the cell membrane with confocal microscopy (Figure 4C). The mean fluorescence intensity per cell was calculated in 4 independent experiments (Figure 4C, graph). HA alone did not induce a significantly higher fluorescent signal, and the combination of HA and CXCL12 did not significantly increase the signal compared with CXCL12 alone. Cells transfected with only CXCR4-VN were used as negative controls, and CD44Δect-VC (lacking CD44 extracellular domain) cotransfected with CXCR4-VN was used as a control for possible random interaction in the context of overexpressed molecules (supplemental Figure 4B). These results indicate that CXCL12 induces the recruitment of CD44 to CXCR4. Formation of the complex was decreased upon treatment with AMD3100 (Figure 4C, lower panels). We tested the presence of this complex in OCI-AML3 cells by co-immunoprecipitation and detected a CD44 band upon CXCL12 induction in CXCR4 precipitates (supplemental Figure 4C).
CD44-CXCR4 cooperation induces stemness marker expression in AML cells upon CXCL12 stimulation
Therapy-resistant cells composing the MRD deploy stem cell properties to recapitulate the disease upon relapse. Because CD44 cooperation with the CXCL12-CXCR4 axis played a role in AML cells resistance to venetoclax, we investigated its possible association to a stemness phenotype. Operationally defined LSCs can be enriched with the markers CD34+CD38–,25 although CD34– LSC populations have also been identified.26 CD34+CD38– cells usually express the core embryonic stem cells transcription factors (ESC-TFs) Sox2, Oct4, and Nanog.7,27,28 Because ESC-TFs control a stemness transcriptional program, including their own transcription in a positive feedback loop,29 we used them as a stemness feature in AML cells. We stably transduced OCI-AML3 cells with a lentiviral fluorescent Sox2/Oct4 reporter (PL-SIN-EOS-C(3+)-EiP14), which allows puromycin selection of only the cells expressing these factors; we found a significantly higher expression of BCL-2 and CD44 in the selected subpopulation compared with the unselected bulk of parental OCI-AML3 cells (supplemental Figure 5A).
We modified a similar vector to report for changes in Sox2/Oct4 expression (PL-SIN-EOS-S(4+)-d2EGFP-SV40-Puro) and transduced OCI-AML3 and Molm13-VR cells to detect cells expressing ESC-TFs within the bulk population (Figure 5Ai-iii,D). CXCL12 induction increased the proportion of the cell subpopulation expressing d2EGFP, and downregulation of CD44 impeded this effect (Figure 5Bi). CXCL12 also induced an increase in mRNA levels of ESC-TFs in OCI-AML3 cells and in primary cells derived from AML patients (Figure 5Bii-iii). Interestingly, d2EGFP+ cells also expressed approximately twofold higher mRNA levels of CD44, but not of CXCR4 (Figure 5Ci; supplemental Figure 5Aii-iii). Upon CD44 knockdown, transcript levels of ESC-TFs were drastically decreased and were unresponsive to CXCL12 (Figure 5Bii). OCI-AML3-EOS-S(+4)d2EGFP+ cells exhibited significantly higher BCL-2 mRNA levels, whereas MCL-1 and BCL-xL levels were not significantly higher (Figure 5Ci). However, MCL-1 was basally expressed at high levels in OCI-AML3 cells, whereas BCL-2 had a low basal level in this cell line (supplemental Figure 5Aii-iii). As with ESC-TF expression, primary AML cells followed a pattern of expression similar to that of antiapoptotic genes upon induction with CXCL12 (supplemental Figure 5Bi-ii).
![Expression of embryonic stem cell markers in AML cells can be enhanced by CXCL12, depends on CD44 expression, and is associated to apoptosis resistance. (A) OCI-AML3 cells express core ESC-TFs. (Ai) Modified version of the PL-SIN-EOS-S(4+)-EGFP pluripotency reporter23 (Addgene MTA [Order 35560]), expressing the unstable version of EGFP (d2EGFP: half-life, 2 hours), followed by the constitutive promoter SV40 driving the puromycin resistance gene, which allows the selection of cells carrying the reporter independently from their Oct4/Sox2 expression levels. (Aii) OCI-AML3 cells transduced with the vector described in panel Ai OCI-AML3-EOS-S(4+)d2EGFP were analyzed by FACS for the expression of d2EGFP (parental OCI-AML3 cells were used as the negative control for the gating strategy), and (Aiii) sorted for cells expressing the reporter (d2EGFP+) and cells not expressing it (d2EGFP–). (B) CXCL12 enhanced ESC-TF expression requires CD44. (Bi) OCI-AML3-EOS-S(4+)d2EGFP cells transfected with CD44 siRNA or control siRNA were treated with CXCL12 (200 ng/mL) for 1 hour or left untreated. Percentages of d2EGFP+ cells were measured by FACS (Wilcoxon rank-sum test for 3 independent experiments). (Bii) Parental OCI-AML3 cells were treated as in panel Bi, and mRNA levels of the core ESC-TFs were measured by qPCR (1-way ANOVA of 3 independent experiments). (Biii) Primary cells from 3 AML patient samples were treated with CXCL12 (200 ng/mL), and mRNA levels of the core ESC-TFs were measured by qPCR. (Ci) mRNA levels of CD44, CXCR4, and BCL-2 family members were determined by qPCR and expressed as fold change between cells sorted for d2EGFP expression as in panel Aiii. (Cii) OCI-AML3-EOS-S(4+)d2EGFP and Molm13-EOS-S(4+)d2EGFP cells were treated with venetoclax 3 μM for 16 hours, and the percentage of d2EGFP+ was measured by FACS. (D) Molm13-VR-EOS-S(4+)d2EGFP and OCI-AML3-EOS-S(4+)d2EGFP cells were sorted for the ∼20% of cells expressing the highest and the ∼20% expressing the lowest d2EGFP fluorescence intensity and were treated with venetoclax or DMSO. Apoptotic (annexin V+/PI+) cells were determined by flow cytometry; annexin V and PI staining was used. Three independent experiments were quantified with a Student t test. *P < .05; **P < .01; ***P < .001; ****P < .0001. GFP, green fluorescent protein; SSC-H, side scatter height.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/12/10.1182_blood.2020006343/4/m_bloodbld2020006343f5.png?Expires=1767697715&Signature=J4AUiqjqjwUUyXSiKK2n9LaLLrdgsX9au7Oo1YTVWK9deqHNUHFfJPBe5sv0M~41Xew03KV2DM1K61oWo4Hyjuwzed85KAeyIo7A8VdXFY1qomfjFD~3lPWXipUZTuR0Uc~BokTVEJTkywL5tZnXYMkkaSA2NxSAwid5MfoYt2JFh72zTc8dE4MIlPUS0AXHrCG6hE5Ku1A35SqkE66QtD9OiNGZMc1quf3FDLNLRnQUzrtT61XlZYcmiadRn0y-tMLeUSL4ZWn~1IIajpSrO4TS8Kvo7bvZGey0RdNteUCznSFmqNgxHT4u5Uf8mjpBSeEGP0Wg73BQdYZWMVYkHg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Expression of embryonic stem cell markers in AML cells can be enhanced by CXCL12, depends on CD44 expression, and is associated to apoptosis resistance. (A) OCI-AML3 cells express core ESC-TFs. (Ai) Modified version of the PL-SIN-EOS-S(4+)-EGFP pluripotency reporter23 (Addgene MTA [Order 35560]), expressing the unstable version of EGFP (d2EGFP: half-life, 2 hours), followed by the constitutive promoter SV40 driving the puromycin resistance gene, which allows the selection of cells carrying the reporter independently from their Oct4/Sox2 expression levels. (Aii) OCI-AML3 cells transduced with the vector described in panel Ai OCI-AML3-EOS-S(4+)d2EGFP were analyzed by FACS for the expression of d2EGFP (parental OCI-AML3 cells were used as the negative control for the gating strategy), and (Aiii) sorted for cells expressing the reporter (d2EGFP+) and cells not expressing it (d2EGFP–). (B) CXCL12 enhanced ESC-TF expression requires CD44. (Bi) OCI-AML3-EOS-S(4+)d2EGFP cells transfected with CD44 siRNA or control siRNA were treated with CXCL12 (200 ng/mL) for 1 hour or left untreated. Percentages of d2EGFP+ cells were measured by FACS (Wilcoxon rank-sum test for 3 independent experiments). (Bii) Parental OCI-AML3 cells were treated as in panel Bi, and mRNA levels of the core ESC-TFs were measured by qPCR (1-way ANOVA of 3 independent experiments). (Biii) Primary cells from 3 AML patient samples were treated with CXCL12 (200 ng/mL), and mRNA levels of the core ESC-TFs were measured by qPCR. (Ci) mRNA levels of CD44, CXCR4, and BCL-2 family members were determined by qPCR and expressed as fold change between cells sorted for d2EGFP expression as in panel Aiii. (Cii) OCI-AML3-EOS-S(4+)d2EGFP and Molm13-EOS-S(4+)d2EGFP cells were treated with venetoclax 3 μM for 16 hours, and the percentage of d2EGFP+ was measured by FACS. (D) Molm13-VR-EOS-S(4+)d2EGFP and OCI-AML3-EOS-S(4+)d2EGFP cells were sorted for the ∼20% of cells expressing the highest and the ∼20% expressing the lowest d2EGFP fluorescence intensity and were treated with venetoclax or DMSO. Apoptotic (annexin V+/PI+) cells were determined by flow cytometry; annexin V and PI staining was used. Three independent experiments were quantified with a Student t test. *P < .05; **P < .01; ***P < .001; ****P < .0001. GFP, green fluorescent protein; SSC-H, side scatter height.
Expression of embryonic stem cell markers in AML cells can be enhanced by CXCL12, depends on CD44 expression, and is associated to apoptosis resistance. (A) OCI-AML3 cells express core ESC-TFs. (Ai) Modified version of the PL-SIN-EOS-S(4+)-EGFP pluripotency reporter23 (Addgene MTA [Order 35560]), expressing the unstable version of EGFP (d2EGFP: half-life, 2 hours), followed by the constitutive promoter SV40 driving the puromycin resistance gene, which allows the selection of cells carrying the reporter independently from their Oct4/Sox2 expression levels. (Aii) OCI-AML3 cells transduced with the vector described in panel Ai OCI-AML3-EOS-S(4+)d2EGFP were analyzed by FACS for the expression of d2EGFP (parental OCI-AML3 cells were used as the negative control for the gating strategy), and (Aiii) sorted for cells expressing the reporter (d2EGFP+) and cells not expressing it (d2EGFP–). (B) CXCL12 enhanced ESC-TF expression requires CD44. (Bi) OCI-AML3-EOS-S(4+)d2EGFP cells transfected with CD44 siRNA or control siRNA were treated with CXCL12 (200 ng/mL) for 1 hour or left untreated. Percentages of d2EGFP+ cells were measured by FACS (Wilcoxon rank-sum test for 3 independent experiments). (Bii) Parental OCI-AML3 cells were treated as in panel Bi, and mRNA levels of the core ESC-TFs were measured by qPCR (1-way ANOVA of 3 independent experiments). (Biii) Primary cells from 3 AML patient samples were treated with CXCL12 (200 ng/mL), and mRNA levels of the core ESC-TFs were measured by qPCR. (Ci) mRNA levels of CD44, CXCR4, and BCL-2 family members were determined by qPCR and expressed as fold change between cells sorted for d2EGFP expression as in panel Aiii. (Cii) OCI-AML3-EOS-S(4+)d2EGFP and Molm13-EOS-S(4+)d2EGFP cells were treated with venetoclax 3 μM for 16 hours, and the percentage of d2EGFP+ was measured by FACS. (D) Molm13-VR-EOS-S(4+)d2EGFP and OCI-AML3-EOS-S(4+)d2EGFP cells were sorted for the ∼20% of cells expressing the highest and the ∼20% expressing the lowest d2EGFP fluorescence intensity and were treated with venetoclax or DMSO. Apoptotic (annexin V+/PI+) cells were determined by flow cytometry; annexin V and PI staining was used. Three independent experiments were quantified with a Student t test. *P < .05; **P < .01; ***P < .001; ****P < .0001. GFP, green fluorescent protein; SSC-H, side scatter height.
Given this antiapoptotic gene expression pattern, we hypothesized that treatment of AML cells with venetoclax would increase the proportion of cells expressing the EOS-d2EGFP reporter. Indeed, 16 hours after treatment with venetoclax, we detected a significantly higher proportion of d2EGFP+ cells in both OCI-AML3-EOS-S(+4)d2EGFP and in Molm13-VR-EOS-S(+4)d2EGFP cells (Figure 5Cii). In an inverse approach, AML cells sorted for the higher ∼20% of d2EGFP fluorescence intensity had a drastically increased resistance to venetoclax-induced apoptosis, especially in Molm13-VR cells, compared with the lowest ∼20% d2EGFP-expressing populations (Figure 5D). These experiments show that CD44 is required for the CXCL12-CXCR4 stimulation of an embryonic stem cell program, which in turn upregulates CD44 expression and is associated with an enhanced venetoclax-resistance phenotype.
CD44 has a relevant role in resistance to venetoclax-induced apoptosis of AML cells in vivo
OCI-AML3 cells engrafted in the spleen and bone marrow or detectable in peripheral blood of NSG mice 28 days post injection (dpi) into the tail vein expressed higher CD44 levels compared with their batch of origin in vitro (supplemental Figure 6A). This effect was most pronounced in cells recovered from the bone marrow (supplemental Figure 6B).
To test the role of CD44 in OCI-AML3 cell resistance to apoptosis in vivo, we injected OCI-AML3 CD44KO cells or parental cells into NSG mice. At 14 dpi, OCI-AML3 CD44KO cells exhibited a significantly lower engraftment rate (supplemental Figure 6Di-iii). Because CD44 is required for homing30 (supplemental Figure 6Div), biasing the readout of survival and/or proliferation, we established a complementary in vivo model in zebrafish that allowed similar engraftment rate of both cell lines and intravital imaging upon venetoclax treatment.
OCI-AML3 and OCI-AML3 CD44KO cells (supplemental Figure 6C) were labeled with CTV and injected into zebrafish embryos at the blastula stage (Figure 6A). Because the cells were injected before the immune system had developed, the xenografted cells were not rejected. At 48 hpf, both cell lines were found mainly in the caudal hematopoietic tissue (CHT31) and in circulation (Figure 6A). At this stage, zebrafish larvae were treated with a single dose of venetoclax injected into the cardinal vein. This rapidly decreased the number of xenografted cells in the CHT compared with vehicle (DMSO)-injected xenografted controls (supplemental Figure 7A). We observed OCI-AML3 cells migrating out of the CHT and into the circulation after engraftment, and CTV fluorescence intensity per cell decreased upon cell division. Therefore, to evaluate the survival of cells that remained engrafted in the CHT after venetoclax injection, we coinjected CellEvent Caspase-3/7 green detection reagent, a fluorescent reporter of apoptosis. At 10 hpi of venetoclax, the mean CTV fluorescence in the CHT showed a tendency toward stabilization for the parental OCI-AML3 cell line, but it continued to decrease for the OCI-AML3 CD44KO cells (Figure 6Bii).
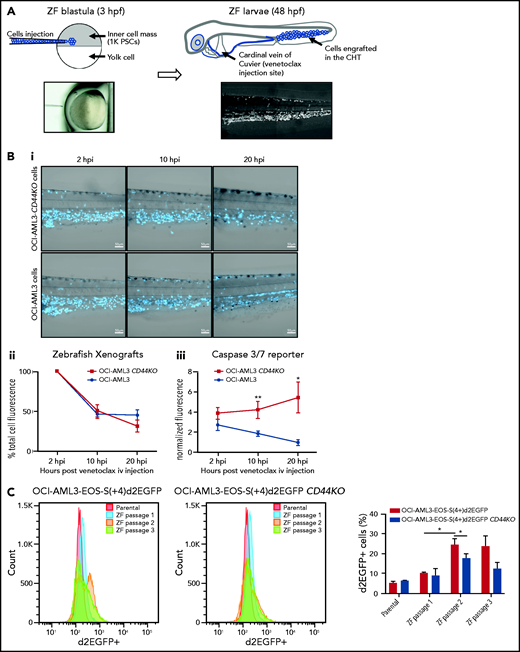
OCI-AML-3 CD44KO xenografted cells are less resistant to apoptosis upon treatment with intravenous venetoclax in an intravital imaging zebrafish model. (A) OCI-AML-3CD44KO or its parental cell line was stained with CellTraceViolet (CTV) and injected into the inner cell mass of blastula-stage zebrafish embryos. Upon development, both cell lines were tolerated and engrafted in the CHT. At 2 days after cell implantation, engrafted zebrafish larvae were treated with a single dose of 0.4 nL of venetoclax 2 mM injected into the cardinal vein in combination with a CellEventCaspase3/7 Green Detection Fluorescent Reagent. At 2, 10, and 20 hpi, intravital confocal images were taken and total CTV (blue) cell fluorescence as well as green fluorescence in the CTH was measured using ImageJ. (Bi) Representative grayscale pictures of the 3 time points in the blue channel (CTV) merged with brightfield. (Bii) Plot of the mean total CTV fluorescence in the CHT. (Biii) Plot of the mean total fluorescence of the CHT in the green channel (apoptosis reporter), normalized to the total CTV fluorescence in the same area on each xenografted zebrafish larvae (Student t test; n = 12 engrafted zebrafish larvae per group). Time lapse follow-up of individual engrafted zebrafish larvae of both cell sublines can be seen in supplemental movies. (C) OCI-AML3-EOS-S(4+)d2EGFP cells or OCI-AML3-EOS-S(4+)d2EGFP CD44KO cells were injected into zebrafish embryos as described above and incubated at 33°C. At 3 dpi, the zebrafish larvae were euthanized with ice-cold E3 medium after anesthesia with Tricaine. Larvae tails, which contain the CHT, were microdissected, dissociated by incubation in trypsin-EDTA, strained through a 40-µm mesh, and cultured in normal cell conditions. Upon proliferation, d2EGFP expression was measured by FACS, and the cells were reimplanted into zebrafish embryos initiating the next passage. Representative histograms of 1 experiment and quantification of 3 sequential in vivo passages of 3 independent experiments are shown. The error bars represent standard error. *P < .05; **P < .005. iv, intravenous; PSCs, pluripotent stem cells.
OCI-AML-3 CD44KO xenografted cells are less resistant to apoptosis upon treatment with intravenous venetoclax in an intravital imaging zebrafish model. (A) OCI-AML-3CD44KO or its parental cell line was stained with CellTraceViolet (CTV) and injected into the inner cell mass of blastula-stage zebrafish embryos. Upon development, both cell lines were tolerated and engrafted in the CHT. At 2 days after cell implantation, engrafted zebrafish larvae were treated with a single dose of 0.4 nL of venetoclax 2 mM injected into the cardinal vein in combination with a CellEventCaspase3/7 Green Detection Fluorescent Reagent. At 2, 10, and 20 hpi, intravital confocal images were taken and total CTV (blue) cell fluorescence as well as green fluorescence in the CTH was measured using ImageJ. (Bi) Representative grayscale pictures of the 3 time points in the blue channel (CTV) merged with brightfield. (Bii) Plot of the mean total CTV fluorescence in the CHT. (Biii) Plot of the mean total fluorescence of the CHT in the green channel (apoptosis reporter), normalized to the total CTV fluorescence in the same area on each xenografted zebrafish larvae (Student t test; n = 12 engrafted zebrafish larvae per group). Time lapse follow-up of individual engrafted zebrafish larvae of both cell sublines can be seen in supplemental movies. (C) OCI-AML3-EOS-S(4+)d2EGFP cells or OCI-AML3-EOS-S(4+)d2EGFP CD44KO cells were injected into zebrafish embryos as described above and incubated at 33°C. At 3 dpi, the zebrafish larvae were euthanized with ice-cold E3 medium after anesthesia with Tricaine. Larvae tails, which contain the CHT, were microdissected, dissociated by incubation in trypsin-EDTA, strained through a 40-µm mesh, and cultured in normal cell conditions. Upon proliferation, d2EGFP expression was measured by FACS, and the cells were reimplanted into zebrafish embryos initiating the next passage. Representative histograms of 1 experiment and quantification of 3 sequential in vivo passages of 3 independent experiments are shown. The error bars represent standard error. *P < .05; **P < .005. iv, intravenous; PSCs, pluripotent stem cells.
Concomitantly, the apoptosis signal showed a steady increase in OCI-AML3 CD44KO cells, while decreasing for the parental cell line. This indicates the sensitization of CD44KO cells to venetoclax treatment (Figure 6Biii; supplemental movies 1 and 2). The increase in green fluorescence intensity did not occur in controls. To evaluate whether the increased venetoclax resistance of the parental OCI-AML3 cells compared with that of their CD44KO counterparts in vivo was related to the expression of stemness markers, we used the EOS-S(4+)d2EGFP reporter and monitored its level of expression in both cell sublines upon in vivo serial passages in our zebrafish model. OCI-AML3-EOS-S(4+)d2EGFP cells had a significant increase in the d2EGFP+ population proportion between the first and the second in vivo passages and was maintained on a third passage. This increase was moderate in the OCI-AML3-EOS-S(4+)d2EGFP CD44KO cells during the same period, which generated a statistically significant higher expression of the reporter in the CD44-expressing cells (Figure 6C; supplemental Figure 7B). Taken together, these results show a role of CD44 in resistance to venetoclax-induced apoptosis in this in vivo model.
To test the relevance of the CD44-CXCR4 axis in the zebrafish in vivo model, we injected venetoclax combined with AMD3100. Although AMD3100 did not have a significant impact on cell survival when injected alone, it boosted the effect of venetoclax over the engrafted OCI-AML3 cells when both drugs were injected in combination (supplemental Figure 7Ci-ii).
Discussion
CXCL12 and the CD44 ligand HA are expressed in the bone marrow microenvironment that houses MRD after AML treatment,8 and CXCR4 on AML cells correlates with adverse prognosis.15,16 Given the groundbreaking effects of venetoclax in the treatment of AML patients, we used resistance to venetoclax-induced apoptosis to assess the impact of CXCR4-CD44 interaction in AML cell survival. Our results suggest that CXCR4 requires the coreceptor functions of CD44, positioning it as a putative molecular target in AML to enhance venetoclax-based treatment.
The increased survival capabilities of LSCs are the cause of MRD.32 In contrast to the AML cell bulk population, quiescent, functionally defined LSCs from AML in the bone marrow overexpress BCL-2.33,34 It has been demonstrated that CXCL12 stimulation of CXCR4 induces cell survival through BCL-2 among other mechanisms that influence both BCL-2 function and expression level.33 Our CD44 loss-of-function experiments indicate that the pro-survival signaling effects of CXCR4 in AML require CD44. Because HA macromers can directly bind to CXCL12,35 high-molecular-weight HA may further contribute by CXCL12 accumulation and presentation to the CXCR4-CD44 complex. It has been described that upon CXCR4 stimulation, the Ras/MAPK/ERK and the PI3K/AKT pathways are activated.36 Effectors of these pathways phosphorylate BAD,37 thus preventing its inhibitory binding to BCL-2 and BCL-xL.38,39 Specific inhibition of ERK phosphorylation was shown to induce apoptosis in OCI-AML3 cells.40 In addition, the CXCR4 inhibitor BL-8040 was described to decrease ERK and AKT phosphorylation in AML cell lines and peripheral blood samples from AML patients.34 This resulted in a significant decrease in the mRNA levels of BCL-2 in vivo. This effect was also observed in vitro in which MCL-1 levels were also significantly decreased, synergizing the effect of venetoclax.33 In parallel, AML cells in spleen and bone marrow differentiated and underwent apoptosis.33 Our results raise the possibility of regulating the CXCL12-CXCR4 pro-survival pathway through CD44.
In the venetoclax-resistant OCI-AML3 and Molm13-VR cell line models, we observed that CXCL12 increased the proportion of the cell subpopulation expressing ESC-TFs. CD44 knockdown abrogated this induction and also decreased the basal proportion of this subpopulation. In addition, cells selected for the expression of these ESC-TFs had higher levels of CD44, but not of CXCR4, thus positioning CD44 in a possible positive feedback loop to maintain this stemness feature while potentiating resistance to venetoclax-based regimens induced by CXCL12. In AML, expression of the ESC-TFs was significantly higher in the CD34+CD38– cell compartment,7,41 suggesting a role for these transcription factors in leukemic stemness.7,41 The addition of these 2 effects might contribute to MRD (Figure 7). Similarly, self-renewal of cancer stem cells in solid tumors like gliomas depends on the sonic hedgehog-GLI-Nanog axis which is disrupted by inhibition of CXCR4.42

Possible contribution of CXCR4/CD44 for MRD. Our results show that the interaction between CXCR4 and CD44 upon CXCL12 stimulation may contribute to the maintenance of a stem cell–like phenotype in AML cells by inducing a transcriptional program driven by the core ESC-TFs, which promote their own transcription as well as that of other stemness target genes.25 This upregulates CD44, which contributes to the maintenance of the phenotype in this environment. In parallel, antiapoptotic protein levels of the BCL-2 family are increased, potentiating resistance to apoptosis induced by venetoclax. Molecule cartoons were taken from ScienceSlides (VisiScience Corp) by licensed user X.Y.
Possible contribution of CXCR4/CD44 for MRD. Our results show that the interaction between CXCR4 and CD44 upon CXCL12 stimulation may contribute to the maintenance of a stem cell–like phenotype in AML cells by inducing a transcriptional program driven by the core ESC-TFs, which promote their own transcription as well as that of other stemness target genes.25 This upregulates CD44, which contributes to the maintenance of the phenotype in this environment. In parallel, antiapoptotic protein levels of the BCL-2 family are increased, potentiating resistance to apoptosis induced by venetoclax. Molecule cartoons were taken from ScienceSlides (VisiScience Corp) by licensed user X.Y.
The increased proportion of the ESC-TF–expressing OCI-AML3 subpopulation induced by the CXCL12-CXCR4-CD44 axis is unlikely to be explained by cell selection, because in the absence of venetoclax, OCI-AML3 cells expressed virtually no signs of spontaneous apoptosis. They might either stimulate an increase in the proliferation rate of cells already expressing the stemness markers or induce ESC-TF de novo expression. This last possibility would mean that this axis could be triggering the reprogramming of partially differentiated blasts to express stemness features. This phenomenon, termed cell plasticity, has profound implications for cancers that deploy cellular hierarchies.43
Although CD44 has been a longstanding putative molecular target in AML, studying it in the context of AML cell survival in vivo is hampered by inhibition of cell homing to protective niches upon injection.26 We therefore used zebrafish as a complementary in vivo AML xenograft model. The use of zebrafish to model the treatment of leukemias and other hematologic malignancies has been described before.44 However, in AML, use of a zebrafish model focused exclusively on screening compounds added to swimming water.45 The CHT is the organ to which zebrafish blood stem cells migrate and develop at the larval stage.31,46 Zebrafish Cxcl12 is expressed in this region, sharing 89% homology in the receptor-binding site with the human ortholog that binds human CXCR4.47 Coherently, the injection of a CXCR4 inhibitor in our zebrafish model decreased survival of OCI-AML3 engrafted cells upon venetoclax treatment. The CHT has been proposed to be a suitable model of cancer cells homing to the bone marrow.48 Having achieved a similar engraftment of OCI-AML3 CD44KO cells and its parental cell line in our model, allowed us to aim for readouts complementary to our mouse model.
Our data suggest that modulating the CXCL12/CXCR4 axis via CD44 inhibition in AML cells that express stem cell markers is relevant. It will be interesting to explore whether the response rates to venetoclax-based therapies correlate with CXCR4-CD44 levels in AML cells. Because MCL-1 inhibition sensitizes OCI-AML3 cells to venetoclax,31 and CD44 loss of function may affect the increase of MCL-1 expression induced by CXCL12/CXCR4, blocking CD44 may have the dual potential to sensitize cells to venetoclax while also having an impact on their stemness.
Acknowledgments
The authors thank the Institute of Biological and Chemical Systems (IBCS)-Functional Molecular Systems rodent facility and especially S. Huber, S. Müller, and S. Schrader; N. Borel from the fish facility of IBCS-Biological Information Processing; and Philipp Haitz and Lisa Geiges for their technical help.
This work is supported by German Research Foundation (DFG) grants OR124/22-1 (V.O.-R. group) and HA8151/3-1 (T.N.H.).
Authorship
Contribution: X.Y. and L.M.-S. designed experiments, performed most of the experiments, and analyzed and interpreted the data; L.M.-S. developed the zebrafish xenograft model and cowrote the article; P.M., E.B., K.S., and R.J.W. performed several experiments and analyzed the data; J.C.G. designed experiments and analyzed the data; R.G. provided the patient samples and analyzed the data; M.L.C. provided and supervised the stemness reporter vector subcloning; C.M.-T. provided expertise and access to techniques and infrastructure; T.N.H. designed experiments, analyzed the data, and was closely involved in the interpretation of data; and V.O.-R. designed the experiments, analyzed the data, interpreted the data, and wrote the article.
Conflict-of-interest disclosure: C.M.-T. is an advisory board member for Pfizer and Janssen-Cilag GmbH, has received grants and/or investigational medicinal products from Pfizer, Daiichi Sankyo, and BioLineRx, and received research funding from Bayer AG. V.O.-R. has shares in and is an advisory board member for amcure. The remaining authors declare no competing financial interests.
The peptides developed at amcure target CD44v6. CD44v6 is not the subject of this study and the peptides were not mentioned in this article.
Correspondence: Véronique Orian-Rousseau, Karlsruhe Institute of Technology, Institute of Biological and Chemical Systems-Functional Molecular Systems, Campus Nord, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany; e-mail: veronique.orian-rousseau@kit.edu.
For original data, e-mail the corresponding author.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal