


Abstract
Diffuse large B-cell lymphoma (DLBCL) is a heterogeneous diagnostic category comprising distinct molecular subtypes characterized by diverse genetic aberrations that dictate patient outcome. As roughly one-third of patients with DLBCL are not cured by current standard chemoimmunotherapy, a better understanding of the molecular pathogenesis is warranted to improve outcome. B-cell receptor (BCR) signaling is crucial for the development, growth, and survival of normal B cells and a substantial fraction of malignant B cells. Various analyses revealed genetic alterations of central components of the BCR or its downstream signaling effectors in some subtypes of DLBCL. Thus, BCR signaling and the downstream NF-κB and phosphatidylinositol 3-kinase (PI3K) cascades have been proposed as potential targets for the treatment of patients with DLBCL. As one of the main effectors of BCR activation, PI3K-mediated signals play a crucial role in the pathogenesis and survival of DLBCL. In this review, we summarize our current understanding of BCR signaling with a special focus on the PI3K pathway in DLBCL and how to use this knowledge therapeutically.
Introduction
Diffuse large B-cell lymphoma (DLBCL) represents the most common lymphoma subtype in adults accounting for roughly 40% of all newly diagnosed lymphoma cases.1,2 DLBCL is a heterogeneous diagnostic category in terms of genetic aberrations, oncogenic addictions, clinical presentation, and response to therapy.3-5 Gene expression profiling identified 2 major molecular subtypes termed activated B-cell–like (ABC) and germinal center B-cell–like (GCB) DLBCL.6 Both molecular subtypes are characterized by distinct gene expression profiles, divergent genetic aberrations, and different outcomes.3,6,7 Utilization of next generation sequencing techniques has recently led to the refinement of the ABC/GCB DLBCL distinction by identifying novel clusters and subtypes within ABC and GCB DLBCL.4,5
The addition of the anti-CD20 antibody rituximab to the CHOP regimen (R-CHOP; cyclophosphamide, doxorubicin, vincristine, and prednisone+rituximab) has significantly improved outcome of patients with DLBCL.8-12 However, still a significant fraction of patients are not cured by R-CHOP and subsequent therapies.13,14 Especially for these patients, a better understanding of the molecular pathogenesis is warranted to use novel targeted agents in an optimal manner.
A significant proportion of DLBCLs are addicted to B-cell receptor (BCR) signaling that activates downstream oncogenic pathways such as nuclear factor–κB (NF-κB) or phosphatidylinositol 3-kinase (PI3K).15 Therefore, various specific inhibitors targeting these signaling cascades have been evaluated in clinical trials, predominantly in patients with relapsed and/or refractory (r/r) disease. In this review, we provide an overview of the current understanding of BCR signaling with a special focus on the PI3K pathway and its role in the pathogenesis of DLBCL. In addition, we discuss in detail the available clinical data and the potential mechanisms of resistance to PI3K inhibitors.
Physiologic B-cell receptor signaling
The BCR contains a cell surface antibody composed of 2 pairs of membrane-bound immunoglobulin heavy and light chains.16 The coreceptor heterodimer comprising CD79A (Igα) and CD79B (Igβ) forms a complex with the BCR that is essential for intracellular signal transduction.16 BCR signaling is centrally involved in the activation, survival, and proliferation of both normal and malignant B cells (Figure 1).17 An individual B cell expresses 1 BCR of a single specificity. Upon antigen stimulation, the BCR aggregates and the immunoreceptor tyrosine-based activation motifs (ITAMs) on the cytoplasmic tails of CD79A and CD79B are phosphorylated by members of the SRC tyrosine kinase family including LYN.17,18 Dual-phosphorylated ITAMs recruit and, in concert with SRC family kinase members, activate the spleen tyrosine kinase (SYK).19,20 One of the SYK substrates is the adaptor protein B-cell linker protein, which itself promotes SYK kinase activity, forming a positive feedback loop.21 Other substrates of SYK include VAV, BTK, and phospholipase C-γ2 (PLCγ2), all of which are essential for various aspects of B-cell activation.22-25 In addition, activated SYK phosphorylates the cytoplasmic tail of the B-cell coreceptor CD19 and B-cell adaptor for PI3K, which provide binding sites for the p85 subunit of PI3K.26 Phosphorylated by BTK, PLCγ2 becomes active and hydrolyzes phosphatidylinositol 4,5-bisphosphate to generate diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3), which regulates intracellular calcium levels. DAG and calcium flux activate protein kinase Cβ at the plasma membrane, which in turn phosphorylates the caspase recruitment domain-containing protein 11 (CARD11).27,28 The scaffolding protein CARD11, together with BCL10, binds to the mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1), thus forming the CARD11-BCL10-MALT1 signalosome complex that mediates downstream activation of the NF-κB pathway (Figure 1).29,30 In addition to the antigen-dependent signaling, the BCR also transmits tonic signals via PI3K that are essential for the survival of resting B cells.31,32 This tonic BCR signaling is antigen-independent and largely relies on PI3K signaling.33
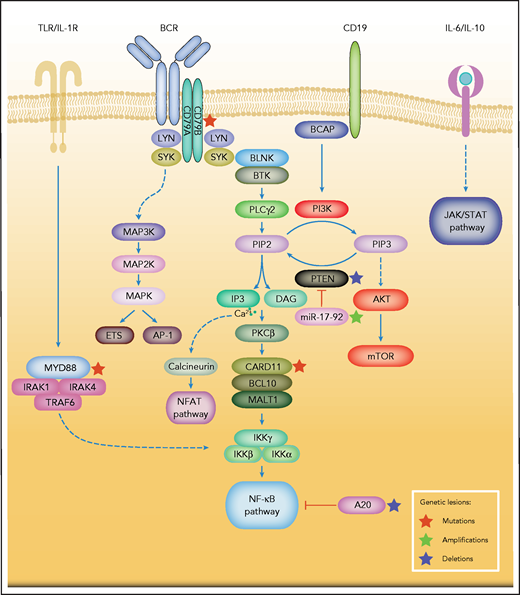
Simplified depiction of BCR, PI3K, and NF-κB signaling in DLBCL. Activation of the BCR induces a signaling cascade that culminates in the activation of the NF-κB, PI3K/mTOR, MAPK, and NFAT pathways. Other key survival pathways in DLBCL are depicted. Constitutive MYD88 signaling also induces activation of the NF-κB pathway. Genetic aberrations are indicated by colored asterisks.
Simplified depiction of BCR, PI3K, and NF-κB signaling in DLBCL. Activation of the BCR induces a signaling cascade that culminates in the activation of the NF-κB, PI3K/mTOR, MAPK, and NFAT pathways. Other key survival pathways in DLBCL are depicted. Constitutive MYD88 signaling also induces activation of the NF-κB pathway. Genetic aberrations are indicated by colored asterisks.
A tight regulation of BCR signaling is crucial for normal B-cell survival and is achieved by the activation of several inhibitory phosphatases when BCR signaling strength exceeds the maximum threshold. For example, SHP1 dephosphorylates ITAMs and SYK, whereas SHIP1 and PTEN hydrolyze phosphatidylinositol 3,4,5-trisphosphate (PIP3), and thus attenuate downstream PI3K and AKT signaling.34-36 As a member of the SRC family of tyrosine kinases, LYN is involved in the initiation and amplification of BCR signals by phosphorylation of tyrosine residues in the cytoplasmic domain of CD19 and ITAMs of CD79A/B (Figure 1).37 The SH3 domain of LYN associates with the p85 subunit of PI3K, increasing its activity.38 LYN also negatively regulates BCR signals through the phosphorylation of the immunoreceptor tyrosine-based inhibition motifs (ITIMs) of several inhibitory receptors, including CD22, FcγRIIb1, and CD5.37
During B-cell development, PI3K as the downstream signaling cascade of the BCR and its precursor receptor, the pre–B-cell receptor (pre-BCR), plays an essential role.39,40 In contrast to the mature BCR, the pre-BCR consists of 2 µ heavy chains and 2 surrogate light chains and it is expressed on late pro-B cells during the transition to the large pre-B stage.39 To date, several lines of evidence have suggested that the PI3K pathway is critical in the transmission of pre-BCR signals that drive the transition to pro-B cells: (1) autonomous pre-BCR signaling activates the PI3K/AKT pathway in pre-B cells41; (2) deficiency of CD19, which amplifies pre-BCR-derived signals, impairs PI3K activity and reduces the number of pre-B cells.40 Reconstitution of CD19-deficient B cells with wild-type CD19, but not a CD19 mutant that does not recruit PI3K, restored this phenotype42; (3) combined inactivation of both the ubiquitously expressed PI3K p110α and the leukocyte-specific p110δ catalytic subunits leads to a near-complete developmental block at the pre–B-cell stage.43 However, PI3K activity plays different roles in early/advanced B-cell stages. A deficiency of downstream AKT1/AKT2 results in increased numbers of pro-, pre-, and immature B cells, whereas the survival of mature B cells is significantly compromised, suggesting that the survival of early-stage B cells might be independent of PI3K signaling.44 Conversely, mature resting B cells rely on PI3K signaling, as BCR-deficient mature B cells can be rescued by constitutively active PI3K signaling alone.33
Once activated by a foreign antigen, B cells either differentiate directly into antibody-secreting cells or enter the germinal center in secondary lymphoid tissues, where they undergo activation-induced cytidine deaminase–driven iterative cycles of somatic hypermutation and immunoglobulin class-switch recombination.45,46 PI3K activity plays an important role in the balance between IgM-expressing antibody-secreting cells and class-switched B cells. On the one hand, ablation of PTEN expression in B cells promotes PI3K activity and thus results in increased IgM, but reduced IgGs and IgA serum levels.47 Inhibition of PI3K in wild-type B cells, on the other hand, enhances secretion of IgGs and IgA, demonstrating a central role of PI3K in the regulation of class-switch recombination and somatic hypermutation.48
B-cell receptor signaling in DLBCL
Various genetic aberrations that have been detected in primary DLBCL samples result in constitutive BCR signaling and downstream NF-κB and PI3K pathway activation.15 A hallmark of ABC DLBCLs is chronic active BCR signaling and BCR clustering on the cell surface (Figure 1).15,49 ABC DLBCLs harbor CD79B and CD79A aberrations that result in an increase in cell surface BCR expression and decreased activity of the negative BCR regulator LYN.15 In addition, in ABC DLBCL, roughly 10% of samples harbor mutations of CARD11 that induce constitutive NF-κB signaling.50 Nevertheless, the majority of ABC DLBCLs exhibit a wild-type status for CD79A/B and CARD11, although these ABC DLBCLs are also frequently dependent on chronic active BCR signaling. Thus, a role of self-antigens has been proposed in these lymphomas.51
Survival of ABC DLBCL models that are characterized by high expression of known NF-κB target genes and by constitutive IκB kinase (IKK) activity52 depends on constitutive NF-κB signaling. This has been shown by the introduction of a “super repressor” form of IκBα that cannot be phosphorylated by IKK, as well as the expression of dominant negative forms of IKKβ are toxic to ABC DLBCL models.52 In addition, selective pharmacologic IKK inhibition induces toxicity in ABC DLBCL models.53
In addition to chronic active BCR signaling, NF-κB is activated via Toll-like receptors (TLRs) and the adaptor molecule myeloid differentiation primary response 88 (MyD88).54MYD88 mutations were detectable in ∼40% of ABC DLBCLs, and ∼29% of all ABC DLBCL samples harbored the hotspot L265P mutant in the MyD88 Toll/IL-1 receptor domain.54 The synergistic toxicity of MyD88 and CARD11/CD79A knockdown indicated that MyD88 and BCR signaling provide nonredundant survival signals to ABC DLBCL cells.54 Recently, a MyD88-TLR9-BCR multiprotein supercomplex (My-T-BCR) was identified.55 Inhibitors of BCR signaling decreased the formation and function of the My-T-BCR supercomplex in ABC DLBCL models.55 In a retrospective analysis of lymphoma samples derived from patients with r/r DLBCL who were treated with ibrutinib in a phase 2 study,56 the presence of the My-T-BCR correlated with response to ibrutinib.55,56 In this analysis, the percentage of My-T-BCR+ cells was significantly higher in lymphomas that responded to ibrutinib, compared with those that progressed while on treatment.55 Despite the low number of analyzed biopsy specimens (n = 8), these results potentially suggest that the presence of My-T-BCR can predict response to ibrutinib.55
PI3K signaling in DLBCL
In response to different stimuli such as growth factors, cytokines, or insulin, the PI3Ks are activated and transduce intracellular signaling to activate pathways that regulate various critical cellular processes, including cell cycle, cell survival, metabolism, and cell motility.57 PI3Ks constitute a family of lipid kinases that catalyze the phosphorylation of the 3'-hydroxyl group of phosphatidylinositides, leading to the accumulation of the second messenger PIP3, which recruits multiple oncogenic proteins to the plasma membrane.58 Consequently, AKT promotes the activity of the mammalian target of rapamycin (mTOR) pathway (Figure 2).59 This PI3K-AKT-mTOR signaling cascade is known to be deregulated in various cancers and represents a major regulator of cell survival, cell proliferation, and angiogenesis (Figure 2).58
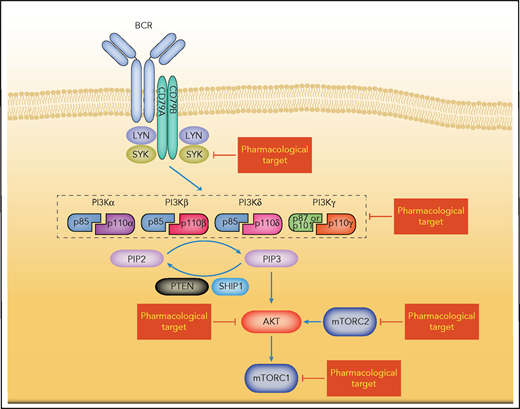
Simplified depiction of BCR-induced PI3K pathway activation. BCR signaling activates the PI3K/AKT-mTOR pathways. The composition of class I isoforms is shown. Potentially targetable vulnerabilities are highlighted.
Simplified depiction of BCR-induced PI3K pathway activation. BCR signaling activates the PI3K/AKT-mTOR pathways. The composition of class I isoforms is shown. Potentially targetable vulnerabilities are highlighted.
Based on their structure and specific substrates, PI3Ks can be divided into 3 classes (I-III).60 The class I isoforms are of therapeutic interest in many cancers and can be further divided into class IA and IB PI3Ks.60 The class IA PI3Ks form a heterodimer comprising a p85 regulatory subunit and a catalytic subunit p110α, p110β, or p110δ (encoded by PIK3CA, PIK3CB, and PIK3CD, respectively), and also referred to as PI3Kα, PI3Kβ, and PI3Kδ. The class IB PI3K contains the catalytic subunit p110γ (encoded by PIK3CG) and a regulatory subunit p101 or p87 (Figure 2).60 PI3Kα and PI3Kβ are ubiquitously expressed, whereas the expression of PI3Kδ and PI3Kγ is mainly restricted to leukocytes.39 Both p110α and p110β deficiencies have been shown to be lethal to mouse embryos,61,62 whereas a p110δ deficiency results in a reduced number of B cells, but is not lethal.63,64 In addition, p110δ has a nonredundant role in BCR signaling: p110δ has been shown to be essential for the activation of BTK and PLCγ2, although the deficiency of p110δ does not alter the expression of p110α and p110β in splenic lymphocytes.63,64
Activated PI3K isoforms phosphorylate PIP3, which in turn activates AKT.59 AKT activity promotes cell survival and cell proliferation, as well as the dysregulation of key effectors in control of cell metabolism.59 Previous studies investigating primary DLBCL samples by immunohistochemical staining of AKT phosphorylation have suggested the activation of the PI3K/AKT pathway in these tumors.65,66 However, the molecular mechanisms of PI3K/AKT activation differ between the molecular DLBCL subtypes. As described in the “B-cell receptor signaling in DLBCL” section, PI3K/AKT activation in ABC DLBCL is associated with constitutive BCR signaling.15,67 Conversely, in GCB DLBCLs loss of the major negative regulator of PI3K/AKT signaling PTEN seems to be the predominant mechanism of PI3K/AKT activation.68 PTEN functions as a lipid phosphatase that dephosphorylates the 3' position of PIP3 and thus mitigates AKT activation.69 Loss of PTEN is a frequent event in GCB DLBCL, which was detectable in more than 50% of primary samples.68 PTEN expression correlates inversely with PI3K/AKT activation in GCB DLBCL models and primary samples as determined by measuring phosphorylated AKT levels.68 Even though the exact molecular mechanisms responsible for PTEN deficiency are not fully understood, both predominantly heterozygous deletions, and less frequently, somatic mutations have been identified.68,70 In addition, recurrent amplifications of the miR-17-92 microRNA locus in GCB DLBCL represent another mechanism for downregulation of PTEN.70,71 Recurrent mutations affecting other essential PI3K pathway components, such as PIK3CA or PIK3CD, have also been described in recent sequencing studies.5,72 Interestingly, in the molecular classification as defined by Chapuy et al, in cluster 3 DLBCLs, both focal deletions and truncating mutations of PTEN were detectable.4 In contrast, cluster 4 DLBCLs were characterized by mutations in BCR/PI3K pathway members (RHOA, GNA13, and SGK1).4
Loss of PTEN seems to predict the response to AKT inhibition, as PTEN-deficient GCB DLBCL models specifically responded to the AKT inhibitor AZD5363.73 In contrast, ABC DLBCLs are largely resistant to AKT inhibition, but are sensitive to PI3Kα/PI3Kδ inhibition in both in vitro and in vivo DLBCL models.73 Intriguingly, inhibition of PI3Kδ is largely ineffective, suggesting that both PI3K isoforms have to be blocked in ABC DLBCL.73,74 Notably, the antilymphoma effect of dual inhibition of PI3Kα and PI3Kδ is mainly a result of decreased AKT and NF-κB signaling.73,74
Clinical implications
BTK inhibitors
Constitutive BCR signaling represents an attractive molecular target. Ibrutinib is an orally available, selective and irreversible BTK inhibitor that disrupts signaling from upstream BCR to NF-κB (Figure 1).75 Preclinical data suggest that especially ABC DLBCLs without a CARD11 mutation respond to BTK inhibition.15 In an early phase 1/2 clinical trial of 80 patients with r/r DLBCL, single-agent ibrutinib showed selective activity in patients with ABC DLBCL. An overall response rate (ORR) of 37% was reported in patients with ABC DLBCL, whereas only 5% of patients with GCB DLBCL responded, confirming the preclinical data.56 Of note, patients with lymphomas with concurrent CD79B and MYD88 mutations showed a higher response rate with an ORR of 80%.56 After a successful phase 1b trial in which ibrutinib in combination with R-CHOP as first-line treatment of patients with DLBCL was evaluated, the randomized phase 3 PHOENIX trial compared ibrutinib plus R-CHOP vs R-CHOP alone in patients with non-GCB DLBCL to improve first-line treatment.76,77 Although, in the overall cohort, the addition of ibrutinib did not improve the primary end point event-free survival or the secondary end point overall survival, a survival benefit was detectable in patients <60 years of age, suggesting a potential role for an intensified R-CHOP regimen that includes a BTK inhibitor in these patients.77 Obviously, these results must be confirmed in additional studies. Such trials are currently being conducted; for example, the second-generation BTK inhibitor acalabrutinib is combined with R-CHOP and compared with R-CHOP alone in patients with untreated DLBCL (registered on www.clinicaltrials.gov as NCT04529772).
PI3K inhibitors
Various PI3K inhibitors have been evaluated for the treatment of patients with DLBCL (Table 1). Idelalisib is a first-in-class PI3Kδ-specific inhibitor that was approved for the treatment of patients with r/r follicular lymphoma, small lymphocytic lymphoma, and chronic lymphocytic leukemia (CLL).78,79 However, it showed only modest activity in preclinical DLBCL models, and correspondingly, no responses were detectable in a small trial conducted in patients with r/r DLBCL.73,80
Efficacy of PI3K inhibitors in patients with r/r DLBCL
| Drug | Targets | n | Treatment | ORR, % | Reference |
|---|---|---|---|---|---|
| Buparlisib | Pan-class I PI3K | 26 | Monotherapy | 11.5 | 93 |
| Copanlisib | Pan-PI3K, with preferential inhibition of PI3Kα and PI3Kδ | 15 | Monotherapy | 6.7 | 110 |
| Copanlisib | Pan-PI3K, with preferential inhibition of PI3Kα and PI3Kδ | 67 | Monotherapy | 19.4 (31.6 in ABC, 13.3 in GCB) | 90 |
| CUDC-907 | PI3Kα, β, and δ, histone deacetylase | 37 | Monotherapy or combined with rituximab | 37 | 97 |
| Umbralisib | PI3Kδ | 26 | Combined with ublituximab | 23 | 102 |
| Parsaclisib | PI3Kδ | 60 | Monotherapy | 25.5 | 94 |
| Drug | Targets | n | Treatment | ORR, % | Reference |
|---|---|---|---|---|---|
| Buparlisib | Pan-class I PI3K | 26 | Monotherapy | 11.5 | 93 |
| Copanlisib | Pan-PI3K, with preferential inhibition of PI3Kα and PI3Kδ | 15 | Monotherapy | 6.7 | 110 |
| Copanlisib | Pan-PI3K, with preferential inhibition of PI3Kα and PI3Kδ | 67 | Monotherapy | 19.4 (31.6 in ABC, 13.3 in GCB) | 90 |
| CUDC-907 | PI3Kα, β, and δ, histone deacetylase | 37 | Monotherapy or combined with rituximab | 37 | 97 |
| Umbralisib | PI3Kδ | 26 | Combined with ublituximab | 23 | 102 |
| Parsaclisib | PI3Kδ | 60 | Monotherapy | 25.5 | 94 |
Despite the encouraging efficacy data for idelalisib in patients with r/r CLL and indolent lymphoma, severe toxic side effects and treatment-related deaths occurred in several clinical trials that tested idelalisib in combination with antibodies alone or with antibodies and chemotherapy, leading to the premature discontinuation of some of these studies.81-83 These adverse events comprised hematologic toxicities such as neutropenia, as well as nonhematologic side effects, including transaminitis, colitis/diarrhea, and infections including sepsis and pneumonia, some of which were caused by a Pneumocystis jirovecii.81-84 Additional studies investigating idelalisib in combination with lenalidomide and rituximab or the SYK inhibitor entospletinib in patients with r/r CLL or lymphoma were halted because of overwhelming, immune-mediated pulmonary and/or hepatic toxicities.85-87
Copanlisib is an intravenous pan-class I PI3K inhibitor with preferential inhibition of PI3Kα and PI3Kδ that has been approved for the treatment of patients with r/r follicular lymphoma by the US Food and Drug Administration.88,89 Preclinical data on the use of copanlisib or other PI3Kα/δ inhibitors suggest activity predominantly in ABC DLBCL.73,74 To test this hypothesis, a phase 2 trial using copanlisib as monotherapy was performed in patients with r/r DLBCL. In this trial, copanlisib achieved an overall response of roughly 20% in patients with r/r DLBCL.90 The response rates were numerically higher in ABC DLBCL (32%) compared with those in patients with GCB DLBCL (13%), confirming preclinical data that suggest that PI3Kα/δ inhibition is predominantly effective in ABC DLBCL (Table 1).73,74 Compared with idelalisib, copanlisib appears to have a more favorable toxicity profile, with a lower incidence of severe complications. Frequently observed adverse events include hyperglycemia, diarrhea, hypertension, and neutropenia.90-92 Based on the promising efficacy of copanlisib in DLBCL, a phase 2 trial evaluating copanlisib plus R-CHOP as first-line therapy for patients with DLBCL is currently underway (NCT04263584).94
Buparlisib is a pan-class I PI3K inhibitor that has been evaluated in a phase 2 study in patients with relapsed or refractory B-cell lymphoma. In the DLBCL subcohort (n = 26), buparlisib monotherapy showed low response rates of only 11.5% (Table 1).93 Parsaclisib is a next-generation inhibitor with specificity to the PI3Kδ isoform. It showed efficacy as a monotherapy in patients with r/r DLBCL, with an ORR of 25.5% in a phase 2 trial (Table 1).94
Further PI3K inhibitors with additional inhibitory effects are currently under clinical development (Table 1). For instance, CUDC-907 (fimepinostat) is a small-molecule inhibitor targeting both HDAC (class I and II) and PI3Ks (class Iα, Iβ, and Iδ). Preclinical results have shown that CUDC-907 decreases MYC expression and induces apoptosis in double-hit DLBCL cells.95 As MYC signaling plays an important role in the pathogenesis of different lymphoma subtypes, a phase 1 trial was initiated.96,97 In this trial of 37 patients with r/r DLBCL, 25 patients received CUDC-907 monotherapy, and 12 patients were treated with CUDC-907 plus rituximab. An ORR of 37% was achieved with a median duration of response of 11.2 months.97 Specifically in those patients with an MYC alteration (translocations or amplifications affecting the MYC locus), the ORR was 64% with a median duration of response of 13.6 months.97 A phase 2 trial in patients with r/r DLBCLs with MYC alterations is currently ongoing (NCT02674750).
Duvelisib represents an orally available PI3Kγ and PI3Kδ inhibitor that was approved by the US Food and Drug Administration in 2018 for the treatment of patients with r/r CLL, small lymphocytic lymphoma and follicular lymphoma after at least 2 prior lines of therapy.98,99 In the initial phase 1 study that also included patients with aggressive r/r lymphoma (n = 26), duvelisib achieved an ORR of 19%.100
Umbralisib (TGR-1202) is a PI3Kδ and casein kinase-1ε inhibitor with improved selectivity for the PI3Kδ isoform when compared with idelalisib or duvelisib.101 A phase 1/1b trial of umbralisib in combination with the novel anti-CD20 antibody ublituximab in r/r B-cell non-Hodgkin lymphoma (B-NHL) reported an ORR of 23% for the DLBCL subcohort.102 A phase 2b randomized study (UNITY-NHL) to evaluate the efficacy of umbralisib alone or in combination with ublituximab in previously treated patients with NHL including DLBCL is currently ongoing (NCT02793583).
Mechanisms of resistance to PI3K inhibitors
Despite the preclinical activity of PI3K inhibitors in DLBCL in vitro and in vivo models, the clinical success of PI3K inhibitors seems to be largely dependent on the oncogenic addictions of the different molecular DLBCL subtypes. Various molecular mechanisms of resistance to PI3K inhibitors have been described preclinically and clinically.
To predict whether patients with DLBCL would respond to PI3K inhibition, an unbiased exploratory analysis was performed in samples from patients treated with copanlisib to correlate clinical response to the occurrence or absence of specific mutations.90 These analyses identified a 16-gene mutation signature that separated responders from nonresponders, further suggesting that genetic aberrations dictate response to PI3K inhibitors.90 This 16-gene signature included TNFAIP3, CREBBP, and PRDM1, which are known to be important in the molecular pathogenesis of DLBCL.90,103 A composite score reflecting the numerical presence or absence of mutations in this gene set was calculated. Patients with a high composite score had a significantly higher ORR and longer progression-free survival compared with patients with a lower score.90
Idelalisib treatment resulted in a feedback activation of PI3Kα in ABC DLBCL cells.104 This rebound of PI3K activity was overcome by subsequent PI3Kα inhibition in preclinical DLBCL models, further underscoring the necessity of inhibiting both PI3Kα and PI3Kδ to achieve responses in ABC DLBCLs.73,74,104 In ABC DLBCL models treated with the PI3Kα/PI3Kδ inhibitor AZD8835, activated CARD11 mutations were identified as a mechanism of resistance.73
A recent preclinical study indicated that levels of C-X-C chemokine receptor type 4 (CXCR4) expression correlate with primary resistance to idelalisib in ABC DLBCL cells, whereas an additional study showed that upregulation of CXCR4 in BCR-dependent DLBCL cell lines and primary tumors was detectable after chemical PI3K blockade.105,106
To overcome resistance to PI3K inhibition and to further improve the efficacy of targeted approaches, especially in patients who have r/r DLBCL, various drug combinations have been investigated in preclinical models. A high-throughput combinatorial screening showed, among other findings, that PI3K-AKT-mTOR inhibitors act synergistically with ibrutinib in ABC DLBCL cells.107 These results were confirmed in independent analyses that showed that a combination of the PI3Kα/δ inhibitor AZD8835 and ibrutinib was highly synergistic and effective in both in vitro and in vivo ABC DLBCL models.73 Using functional BH3 profiling, Bojarczuk and colleagues found that the cytotoxic activity of copanlisib was primarily mediated through BCL-xL– and MCL-1–dependent mechanisms.108 A combination of copanlisib and the BCL-2 inhibitor venetoclax showed synergistic activity in BCR-dependent DLBCLs, with genetic bases for BCL-2 dysregulation in vitro and in vivo.108 Combination treatment of umbralisib and the proteasome inhibitor carfilzomib showed synergistic cytotoxicity in B-cell lymphoma models.109 This combination is currently being evaluated in patients with r/r NHLs, including DLBCL (NCT02867618).
Conclusions and future directions
A substantial fraction of DLBCLs are addicted to oncogenic BCR and PI3K signaling caused by different stimuli and various genetic aberrations. Despite their promising preclinical activity, single-agent PI3K and BCR inhibitors have shown only limited clinical efficacy in patients with DLBCL when nonselected patients were treated. These results underscore the necessity to use targeted agents in the context of oncogenic addictions within the lymphoma cells, and a thorough molecular analysis is necessary before the use of specific agents. In addition, these results illustrate that single-agent regimens are most likely not efficient enough to substantially improve the outcome of patients with DLBCL. To this end, novel combinations with BCR and PI3K inhibitors are needed. These combinations should include conventional chemoimmunotherapeutic regimens, but also other targeted agents and novel immunologic approaches. These novel combinations most likely will overcome mechanisms of resistance and increase cure rates of affected patients.
Acknowledgment
This work was supported by a research grant from the Deutsche Krebshilfe (grant 70113427) (G.L.).
Authorship
Contribution: W.X., P.B., and G.L. wrote the manuscript.
Conflict-of-interest disclosure: G.L. has received research grants from AQUINOX, Acerta, AGIOS, AstraZeneca, Bayer, Celgene, Gilead, Janssen, Morphosys, Novartis, Roche, and Verastem and honoraria from Abbvie, AstraZeneca, Bayer, BMS, Celgene, Gilead, Incyte, Janssen, Karyopharm, Morphosys, Novartis, Nanostring, Roche and Takeda. W.X. and P.B. declare no competing financial interests.
Correspondence: Georg Lenz, Department of Medicine A for Hematology, Oncology, and Pneumology, University Hospital Münster, Albert-Schweitzer-Campus 1, 48149 Münster, Germany; e-mail: georg.lenz@ukmuenster.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal