

Key Points
Patients with ICUS and isolated neutropenia display low frequency of mutations.
Clonal neutropenic patients have a significantly higher risk of developing a myeloid neoplasm than those with no evidence of clonality.

Abstract
The incidence and prognosis of clonal hematopoiesis in patients with isolated neutropenia among patients with idiopathic cytopenia of undetermined significance (ICUS), known as ICUS-N or chronic idiopathic neutropenia (CIN) patients, is poorly defined. The current study sought to investigate the frequency and clinical significance of mutations of genes implicated in myeloid malignancies using next-generation sequencing in patients with CIN (n = 185) with a long follow-up. We found that 21 (11.35%) of 185 patients carried a total of 25 somatic mutations in 6 genes with a median variant allele frequency of 12.75%. The most frequently mutated genes were DNMT3A and TET2 involving >80% of patients, followed by IDH1/2, SRSF2, and ZRSR2. The frequency of transformation to a myeloid malignancy was low in the total group of patients (5 of 185 patients [2.70%]). However, from the transformed patients, 4 belonged to the clonal group (4 of 21 [19.05%]) and 1 to the nonclonal group (1 of 164 [0.61%]), indicating that the presence of mutation(s) confers a relative risk for transformation of 31.24 (P = .0017). The variant allele frequency of the mutant clones in the transformed patients was >10% in all cases, and the genes most frequently associated with malignant transformation were SRSF2 and IDH1. No significant differences were identified between the clonal and nonclonal groups in the severity of neutropenia. Patients with clonal disease were older compared with nonclonal patients. These data contribute to the better understanding of the heterogeneous entities underlying ICUS and highlight the importance of mutation analysis for the diagnosis and prognosis of patients with unexplained neutropenias.
Introduction
The term chronic idiopathic neutropenia (CIN) has long been used to define the persistent reduction of peripheral blood (PB) absolute neutrophil counts that cannot be attributed to any underlying congenital or acquired disease, nutritional deficiency, or exposure to drugs, chemical compounds, or ionizing irradiation.1-3 In adults, CIN affects mainly middle-aged women, usually displays an uncomplicated course, and only rare cases of transformation to a myeloid malignancy have been reported so far.3-9 The pathophysiology of CIN in adults has been associated with increased apoptosis of the granulocytic progenitor cells within an inhibitory bone marrow (BM) microenvironment consisting of myelosuppressive oligoclonal T lymphocytes, activated monocytes, and mesenchymal stem cells with altered properties; all these cells collectively produce proinflammatory mediators that may also induce a degree of dysplasia in the BM.10-16 It has therefore been suggested that CIN shares common pathophysiological features with BM failure syndromes implicating immune mechanisms such as aplastic anemia and myelodysplastic syndromes (MDS).17
In 2005, the MDS study groups introduced the term idiopathic cytopenia of undetermined significance (ICUS), which was then established to describe patients with persistent (>4 months) cytopenia(s) of any degree who are suspected of having MDS but do not fulfill the minimal diagnostic criteria nor have evidence of any underlying disease associated with cytopenia.18,19 Accordingly, there is an overlap between CIN and ICUS terminology. The identification of somatic mutations in MDS-associated genes in the PB or BM of patients with ICUS by using next-generation sequencing (NGS) may change the diagnosis of ICUS to clonal cytopenia of undetermined significance (CCUS) in those patients harboring one or more somatic mutations with a variant allele frequency (VAF) of at least 2%.19-21 The frequency of CCUS among patients with ICUS has been reported between 38% and 64% in different studies.20,22,23 The discrimination between nonclonal ICUS and CCUS is clinically important because patients with CCUS display a markedly increased probability of developing a myeloid neoplasm over time.22 It has been shown that the malignant transformation is largely related to the number and type of mutations, the pattern of comutations, and the size of clone(s) defined by the VAF in NGS analysis.22
The frequency of patients with ICUS and isolated neutropenia, also known as ICUS-N,24 is rare in studies investigating ICUS/CCUS. Isolated neutropenia is also rare among patients with lower risk MDS.25,26 Therefore, the incidence and prognosis of clonal hematopoiesis among ICUS-N patients remain obscure.
In the current study, we performed NGS analysis of genes that are recurrently mutated in myeloid malignancies in a cohort of patients with the diagnosis of CIN according to previously reported criteria that largely overlap with the ICUS-N proposed criteria.19,27 The patients were studied either prospectively or retrospectively and had a long and sequential follow-up. Our aim was to estimate for the first time the frequency of clonal hematopoiesis in patients with CIN/ICUS-N and probe the significance of somatic mutations in disease evolution to myeloid malignancies. We also intended to highlight the heterogeneity that may underlie the ICUS diagnosis in terms of the mutation profile and disease outcome.
Patients and methods
Patients
We studied 185 patients (146 female subjects and 39 male subjects) aged 18 to 89 years (median age, 59 years) with a diagnosis of CIN according to previously established criteria.2,3 The patients had an absolute neutrophil count <1.8 × 109/L (mean, 1.39 ± 0.42 × 109/L; median, 1.6 × 109/L; range, 0.2-1.7 × 109/L) for a period lasting >4 months (median, 168 months; range, 12-408 months), had no clinical/laboratory evidence of any underlying disease associated with neutropenia, absence of a history of exposure to irradiation, use of chemical compounds or intake of drugs that might cause neutropenia, negative antineutrophil antibodies in granulocyte immunofluorescence and agglutination test results, inconclusive results of ΒM biopsy and aspiration for a specific diagnosis with absence/rare dysplastic features, and normal BM karyotype.28,29 Grouped and individual patient data are presented in Table 1 and supplemental Table 1 (available on the Blood Web site), respectively. Congenital neutropenias were excluded by genetic testing (supplemental Table 2) in the Severe Chronic Neutropenia International Registry reference laboratory in Tubingen, Germany.30 Benign ethnic neutropenia, currently characterized as “typical neutrophil count with Fy(a-b-) status,” was excluded by DARC/ACKR1 genotyping for the single-nucleotide polymorphism rs2814778 (-46T > C).31-33
Clinical and hematologic data of the total group of patients with CIN and comparison between the nonclonal and clonal (CCUS) subgroups categorized on the basis of mutation status
| Variable | Total patients with CIN | Nonclonal patients with CIN | Clonal CIN (CCUS) patients | P* |
|---|---|---|---|---|
| No. of patients | 185 | 164 | 21 | |
| Age, y | 57 ± 17 | 56 ± 17 | 67 ± 14 | .0052 |
| Median, range | (59, 18-89) | (58, 18-89) | (68, 44-87) | |
| Sex (female/male) | 146/39 | 129/35 | 17/4 | NS |
| Disease duration, mo† | 171 ± 100 | 170 ± 98 | 176 ± 120 | NS |
| Median, range | (168, 12-408) | (168, 12-408) | (144, 12-396) | |
| WBC count, ×109/L | 3.46 ± 0.60 | 3.48 ± 0.62 | 3.30 ± 0.46 | NS |
| Median, range | (3.50, 1.90-5.70) | (3.50, 1.90-5.70) | (3.30, 2.10-4.00) | |
| ANC, ×109/L | 1.39 ± 0.42 | 1.39 ± 0.43 | 1.36 ± 0.37 | NS |
| Median, range | (1.60, 0.20-1.70) | (1.60, 0.20-1.70) | (1.50, 0.50-1.70) | |
| Monocyte count, ×109/L | 0.33 ± 0.13 | 0.34 ± 0.14 | 0.33 ± 0.12 | NS |
| Median, range | (0.30, 0.10-0.70) | (0.30, 0.10-0.70) | (0.30, 0.20-0.60) | |
| Lymphocyte count, ×109/L | 1.62 ± 0.45 | 1.63 ± 0.45 | 1.54 ± 0.39 | NS |
| Median, range | (1.60, 0.70-3.25) | (1.60, 0.70-3.25) | (1.60, 0.80-2.30) | |
| Hgb, g/dL | 13.31 ± 0.96 | 13.29 ± 0.97 | 13.45 ± 0.89 | NS |
| Median, range | (13.20, 10.60 – 16.20) | (13.20, 10.60 – 16.20) | (13.40, 11.30 – 15.80) | |
| MCV, fL | 88.13 ± 5.56 | 88.31 ± 5.41 | 86.74 ± 6.61 | NS |
| Median, range | (89.0, 65.00-97.70) | (89.00, 65.00-97.70) | (88.90, 66.90-93.30) | |
| Platelet count, ×109/L | 222 ± 47 | 224 ± 47 | 197 ± 42 | .011 |
| Median, range | (221, 150-336) | (226, 150-336) | (185, 150-265) |
| Variable | Total patients with CIN | Nonclonal patients with CIN | Clonal CIN (CCUS) patients | P* |
|---|---|---|---|---|
| No. of patients | 185 | 164 | 21 | |
| Age, y | 57 ± 17 | 56 ± 17 | 67 ± 14 | .0052 |
| Median, range | (59, 18-89) | (58, 18-89) | (68, 44-87) | |
| Sex (female/male) | 146/39 | 129/35 | 17/4 | NS |
| Disease duration, mo† | 171 ± 100 | 170 ± 98 | 176 ± 120 | NS |
| Median, range | (168, 12-408) | (168, 12-408) | (144, 12-396) | |
| WBC count, ×109/L | 3.46 ± 0.60 | 3.48 ± 0.62 | 3.30 ± 0.46 | NS |
| Median, range | (3.50, 1.90-5.70) | (3.50, 1.90-5.70) | (3.30, 2.10-4.00) | |
| ANC, ×109/L | 1.39 ± 0.42 | 1.39 ± 0.43 | 1.36 ± 0.37 | NS |
| Median, range | (1.60, 0.20-1.70) | (1.60, 0.20-1.70) | (1.50, 0.50-1.70) | |
| Monocyte count, ×109/L | 0.33 ± 0.13 | 0.34 ± 0.14 | 0.33 ± 0.12 | NS |
| Median, range | (0.30, 0.10-0.70) | (0.30, 0.10-0.70) | (0.30, 0.20-0.60) | |
| Lymphocyte count, ×109/L | 1.62 ± 0.45 | 1.63 ± 0.45 | 1.54 ± 0.39 | NS |
| Median, range | (1.60, 0.70-3.25) | (1.60, 0.70-3.25) | (1.60, 0.80-2.30) | |
| Hgb, g/dL | 13.31 ± 0.96 | 13.29 ± 0.97 | 13.45 ± 0.89 | NS |
| Median, range | (13.20, 10.60 – 16.20) | (13.20, 10.60 – 16.20) | (13.40, 11.30 – 15.80) | |
| MCV, fL | 88.13 ± 5.56 | 88.31 ± 5.41 | 86.74 ± 6.61 | NS |
| Median, range | (89.0, 65.00-97.70) | (89.00, 65.00-97.70) | (88.90, 66.90-93.30) | |
| Platelet count, ×109/L | 222 ± 47 | 224 ± 47 | 197 ± 42 | .011 |
| Median, range | (221, 150-336) | (226, 150-336) | (185, 150-265) |
Grouped data are expressed as means ± 1 SD.
ANC, absolute neutrophil count; Hgb, hemoglobin; MCV, mean corpuscular volume; NS, nonsignificant; WBC, white blood cell.
P values indicate the statistical difference for the comparison between the nonclonal and clonal CIN using the Student t test.
Time from the identification of neutropenia until the last follow-up or disease transformation.
The study was approved by the ethics committees of the University Hospital of Heraklion and the IRCCS S. Matteo Hospital and University of Pavia. Informed consent was obtained from all subjects.
Genomic DNA isolation
Genomic DNA was isolated from whole PB samples and/or BM aspirates by using the QIAamp Blood DNA Mini Kit (QIAGEN, Hilden, Germany) (supplemental Methods).
Mutation analysis by NGS
A total of 160 patients with CIN were studied for the mutational status of 54 recurrently mutated genes in myeloid malignancies using the Illumina TruSight Myeloid Sequencing panel (Illumina, San Diego, CA) (supplemental Table 3); 25 patients were studied for the mutational status of 40 recurrently mutated genes in myeloid malignancies using the Ion Torrent Oncomine Myeloid Panel (Thermo Fisher Scientific, Waltham, MA) (supplemental Table 4; supplemental Methods).
Statistical analysis
Comparisons of blood cell counts between clonal and nonclonal patients with CIN, based on identification of mutations with a VAF ≥2%,34 were performed by using the Student t test. Comparison of the distribution of qualitative variables between the 2 groups, as well as between clonal groups with different mutations, was performed by using the χ2 test. Generalized linear model for the binomial family and the log link function were used to estimate the age-adjusted relative prevalence of clonal hematopoiesis in patients with CIN vs individuals without hematologic diseases from population-based studies.35-38
Results
Disease characteristics of patients with CIN and comparisons between clonal and nonclonal groups
Mutation analysis for evaluation of the frequency of somatic mutations in myeloid-associated genes in the cohort of patients with CIN was performed at diagnosis in 70 patients and retrospectively in 115 patients. In patients studied retrospectively, the median time from the inclusion in the study (day of confirmation of CIN diagnosis in our department) to DNA sampling was 60 months (range, 12-288 months). The median duration of follow-up of the total group of patients since inclusion in the study was 132 months (range, 8-336 months).
Disease characteristics of patients with CIN in the total group and the subgroups with clonal and nonclonal disease, based on mutation profiling, are presented in Table 1. No statistically significant differences were identified between the 2 groups in disease duration and sex characteristics. A female predominance was observed in both groups. However, the mean age of patients with clonal disease was higher compared with the nonclonal patients (P = .0052). No statistically significant differences were observed between the 2 groups in white blood cell counts and differentials, hemoglobin, and mean corpuscular volume values. Patients with clonal CIN had significantly lower platelet counts compared with patients with nonclonal disease (P = .011) but within normal range.
Mutation analysis
Frequency and spectrum of somatic mutations in CIN
Overall, 21 (11.35%) of 185 patients with CIN carried somatic mutation(s) in ≥1 of the myeloid genes studied. The most frequently mutated genes and their distribution are depicted in Figure 1A. Overall, 25 somatic mutations in 6 genes were identified (supplemental Table 5). The vast majority of patients (18 of 21) carried 1 mutation, 2 of 21 patients harbored 2 mutations (one patient in IDH2 and SRSF2 and another patient a double TET2 mutation), and 1 of 21 patients harbored 3 mutations (one in IDH1 and two in DNMT3A) (Figure 1B). The most frequently mutated genes were DNMT3A (12 of 25 mutations [48%]) and TET2 (7 of 25 mutations [28%]) involving >80% of patients (11 of 21 and 6 of 21 patients [52.4% and 28.6%, respectively]), followed by IDH1/2 (3 of 25 mutations [12%] in 3 of 21 patients [14.3%]), SRSF2 (2 of 25 mutations [8%] involving 2 of 21 patients [9.5%]), and ZRSR2 (1 of 25 mutations [4%] involving 1 of 21 patients [4.8%]). The most common base-pair change in the somatic variants was a cytosine-to-thymine (C > T) transition, followed by cytosine-to-guanine (C > G) and cytosine-to-adenine (C > A) transitions (Figure 1C).
![Somatic mutations identified by NGS profiling in patients with CIN. (A) The graph shows the frequency of somatic mutations identified in the patients. A total of 25 somatic mutations were detected in 21 patients: 12 mutations were detected in DNMT3A and 7 mutations in TET2, followed by 3 mutations in IDH1/IDH2, 2 mutations in SRSF2, and 1 mutation in ZRSR2. The bars show the number of mutations per gene. (B) The bars show the number of patients harboring 1 to 3 mutations in the aforementioned genes. The majority of patients (n = 18) harbored 1 mutation, 2 patients were detected with 2 mutations (in 2 genes [IDH2/SRSF2] and 1 gene [TET2], respectively) and 1 patient harbored 3 mutations (1 in IDH1 and 2 in DNMT3A). (C) The graph shows the base-pair changes in the somatic variants. The bars depict the proportion of variants with a single nucleotide substitution. A proportion of 31.8% of the single-nucleotide sequence corresponded to a C > T substitution.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/14/10.1182_blood.2021010815/4/m_bloodbld2021010815f1.png?Expires=1770130799&Signature=tLnqVTbIA8MzGdtA843gMPiw1G~BVA3SMM~RaKgiU6ycfm7HknguvSLVjwAVh5Yo~y8M0QHkZJY7ym-6xk0fC4ROISRZjyrf~1R7Kf~HqHHM9FHnNP9Ilv~i7KmtIrZOaK5fEkJkJljRMDtb04G0fUAVVh7DoxuaS3W-Y7oMAHaUs~F5gZIguIRFzI8oE4eIbHJLBJE~V~-LtLNDX1n8ChSWE0pL2ejEqzhMRfCGO1-4EZYHYFcNdv6qtGGWjarVZHCXXYvjzPgKk4SXqW7TxW3J0DaBsh6aDoNSvOJhqKWmByB~yiao6QDQkMg6YNR1WbgYIlLDcTJ7EVc5kAYIZA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Somatic mutations identified by NGS profiling in patients with CIN. (A) The graph shows the frequency of somatic mutations identified in the patients. A total of 25 somatic mutations were detected in 21 patients: 12 mutations were detected in DNMT3A and 7 mutations in TET2, followed by 3 mutations in IDH1/IDH2, 2 mutations in SRSF2, and 1 mutation in ZRSR2. The bars show the number of mutations per gene. (B) The bars show the number of patients harboring 1 to 3 mutations in the aforementioned genes. The majority of patients (n = 18) harbored 1 mutation, 2 patients were detected with 2 mutations (in 2 genes [IDH2/SRSF2] and 1 gene [TET2], respectively) and 1 patient harbored 3 mutations (1 in IDH1 and 2 in DNMT3A). (C) The graph shows the base-pair changes in the somatic variants. The bars depict the proportion of variants with a single nucleotide substitution. A proportion of 31.8% of the single-nucleotide sequence corresponded to a C > T substitution.
Somatic mutations identified by NGS profiling in patients with CIN. (A) The graph shows the frequency of somatic mutations identified in the patients. A total of 25 somatic mutations were detected in 21 patients: 12 mutations were detected in DNMT3A and 7 mutations in TET2, followed by 3 mutations in IDH1/IDH2, 2 mutations in SRSF2, and 1 mutation in ZRSR2. The bars show the number of mutations per gene. (B) The bars show the number of patients harboring 1 to 3 mutations in the aforementioned genes. The majority of patients (n = 18) harbored 1 mutation, 2 patients were detected with 2 mutations (in 2 genes [IDH2/SRSF2] and 1 gene [TET2], respectively) and 1 patient harbored 3 mutations (1 in IDH1 and 2 in DNMT3A). (C) The graph shows the base-pair changes in the somatic variants. The bars depict the proportion of variants with a single nucleotide substitution. A proportion of 31.8% of the single-nucleotide sequence corresponded to a C > T substitution.
The identified somatic variants included 17 missense mutations, 4 nonsense mutations, 3 frameshift deletions, and 1 splice site variant (Figure 2A). The most frequently mutated gene group according to the pathway ontology was DNA methylation followed by RNA splicing (Figure 2B). All 17 missense variants are well documented in the literature (supplemental Table 6).
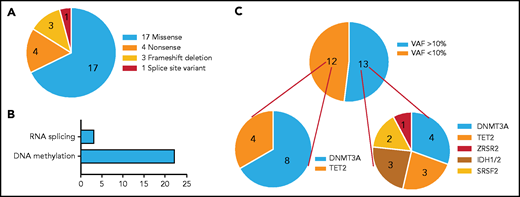
Distribution of mutations identified in patients with CIN according to amino acid changes, reading frameshifts, functional classification, and burden. (A) Distribution of coding somatic non-synonymous variants according to amino acid changes or reading frameshifts. We detected 17 missense mutations, 4 nonsense mutations, 3 frameshift deletions, and 1 splice site mutation in a total of 6 genes. (B) Distribution of coding somatic variants according to functional classification. The majority of the detected mutations affected genes involved in DNA methylation. (C) Distribution of mutations according to mutational burden. Ten patients had 13 mutations with VAF >10% distributed across 6 genes (DNMT3A, n = 4; TET2, n = 3; IDH1/IDH2, n = 3; SRSF2, n = 2; ZRSR2, n = 1). The remaining had 12 mutations with VAF <10% distributed across 2 genes (DNMT3A, n = 8; TET2, n = 4).
Distribution of mutations identified in patients with CIN according to amino acid changes, reading frameshifts, functional classification, and burden. (A) Distribution of coding somatic non-synonymous variants according to amino acid changes or reading frameshifts. We detected 17 missense mutations, 4 nonsense mutations, 3 frameshift deletions, and 1 splice site mutation in a total of 6 genes. (B) Distribution of coding somatic variants according to functional classification. The majority of the detected mutations affected genes involved in DNA methylation. (C) Distribution of mutations according to mutational burden. Ten patients had 13 mutations with VAF >10% distributed across 6 genes (DNMT3A, n = 4; TET2, n = 3; IDH1/IDH2, n = 3; SRSF2, n = 2; ZRSR2, n = 1). The remaining had 12 mutations with VAF <10% distributed across 2 genes (DNMT3A, n = 8; TET2, n = 4).
Variant allele frequencies
The 25 somatic mutations identified in 21 patients displayed a median VAF of 12.75% (range, 2.40%-33.18%). Patients were analyzed on the basis of a cutoff VAF level of 10%, previously reported to be associated with a significantly increased risk of developing a hematologic neoplasm (Figure 2C).36,37 Ten patients had 13 mutations with VAF >10% (median, 22.45%; range, 12.75%-33.18%), distributed across 6 genes (DNMT3A, TET2, SRSF2, IDH1/IDH2, and ZRSR2). The remaining patients had 12 mutations with a median VAF of 3.2% (range, 2.40%-8.67%), distributed across 2 genes (DNMT3A and TET2).
The bioinformatics analysis revealed 13 variants in 13 patients with a VAF frequency within the germline range (median, 48.91%; range, 40.22%-51.73%) (supplemental Table 1). Although patients’ germline DNA was not available for testing, a thorough review of population variant databases (dbSNP, 1000G, ExAc, ESP6500, and gnomAD) (supplemental Methods) indicated the germline nature of these high VAF mutations.
Age-adjusted frequency of clonal hematopoiesis in CIN compared with the general population
It has been shown that clonal hematopoiesis is common in the aging general population; we thus compared the prevalence of clonal hematopoiesis in patients with CIN vs that reported in the general population.36-38 No significant difference was identified in the age-adjusted prevalence of clonal hematopoiesis between patients with CIN and community-dwelling individuals with no known hematologic disease. However, by using the cutoff age of 70 years, we found a significantly higher prevalence of clonal hematopoiesis in patients with CIN aged <70 years compared with age-adjusted individuals from the community (relative prevalence, 2.56; 95% confidence interval [CI], 1.55-4.22; P = .001), whereas no difference was identified between subjects aged ≥70 years (relative prevalence, 1.11; 95% CI, 0.62-1.98; P = .697).
Association of mutation status with CIN transformation to a myeloid malignancy
Five (2.70%) of 185 patients with CIN transformed to a myeloid malignancy, namely chronic myelomonocytic leukemia (CMML) (n = 1), unclassifiable MDS/myeloproliferative neoplasm (MDS/MPN) (n = 1), acute (myelomonocytic) myeloid leukemia (AML) (n = 1), and MDS with multilineage dysplasia (n = 2).28 Four patients belonged to the clonal group (4 of 21 patients [19.05%]), and 1 patient who transformed to MDS belonged to the nonclonal group (1 of 164 patients [0.61%]), indicating a significantly higher proportion of patients who transformed to a myeloid malignancy in the clonal group compared with the nonclonal group (P < .0001). The absence of mutations in the patients had a negative predictive value for the development of a myeloid neoplasm of 0.99 (95% CI, 0.96-0.99). On the contrary, the presence of mutation(s) conferred a relative risk (RR) of 31.24 (95% CI, 3.66-266.50) for transformation to a myeloid malignancy (odds ratio [OR], 38.35; 95% CI, 4.05-363.20) with significantly higher risk in the presence of at least 2 mutations (n = 3) compared with 1 (n = 18) mutation (P = .023). The presence of a mutant clone with a VAF >10% was associated with a higher probability of developing a myeloid malignancy (OR, 28.85; 95% CI, 1.41-588.90; P = .0027); the VAF of individual mutations in the 4 patients who transformed was >10% in all cases (median VAF, 23.19%; range, 12.75%-27.60%).
The patient who transformed to CMML (#11, supplemental Table 1) carried the typical SRSF2 mutation p.P95R commonly associated with this disease and displayed a VAF of 27.60%.28,39SRSF2 mutations have been reported in ∼40% of CMML cases and 5% to 20% of MDS cases.28 CMML in this patient occurred 216 months after the initial CIN diagnosis.
The patient who transformed to unclassifiable MDS/MPN (#16, supplemental Table 1) carried the SRSF2 p.P95R and an IDH2 p.R140Q mutation with a VAF of 25.50% and 23.19%, respectively. Although mainly found in AML (10%-20%), IDH2 p.R140Q mutations have been rarely reported in MDS and MPNs.40,41 MDS/MPN in our patient occurred 96 months after the initial CIN diagnosis.
The patient who transformed to AML (#21, supplemental Table 1) carried the IDH1 p.R132S mutation with a VAF of 12.75%. Recurrent IDH1 p.R132S mutations have been mainly reported in AML (5%-15%).41 AML in this patient occurred 108 months after the initial CIN diagnosis.
The CIN patient who transformed to MDS with multilineage dysplasia (#2, supplemental Table 1) carried 3 mutations: IDH1 p.R132S, DNMT3A p.R736H, and DNMT3A p.Q248fs with VAFs of 20.90%, 24.55%, and 17.3%, respectively. Besides AML, recurrent IDH1 p.R132S mutations have been reported in MDS.42 Heterozygous somatic mutations in DNMT3A have been recurrently reported in ∼5% to 13% of MDS cases.42 MDS in this patient occurred 24 months after the initial CIN diagnosis. The only CIN patient without mutation(s) who developed MDS with multilineage dysplasia (#80, supplemental Table 1) transformed 96 months after the initial CIN diagnosis.
Among the variants detected in the cohort of patients with CIN, DNMT3A p.R882H had the highest prevalence, with 3 occurrences (VAFs of 22.45%, 18%, and 2.5%), followed by 2 occurrences of SRSF2 p.P95R (VAFs of 27.6% and 25.5%) and IDH1 p.R132S (VAFs of 12.5% and 20.9%). Both patients with mutations in the splicing factor SRSF2, either as isolated or as comutation with IDH2 p.R140Q, progressed to a myeloid neoplasm (CMML and MDS/MPN, respectively), validating its high predictive value.22 The presence of both IDH2 p.R140Q and SRSF2 p.P95R mutations conferred an RR of 6.67 (95% CI, 2.35-18.93) for the development of a myeloid malignancy (OR, 15.00; 95% CI, 0.50-449.90). Also, both patients detected with IDH1 p.R132S, either as isolated or as comutated with DNMT3A, developed a myeloid neoplasm (AML and MDS). The presence of SRSF2 p.P95R or IDH1 p.R132S mutations (as isolated or comutations) conferred an RR of 9.50 (95% CI, 2.56-35.25) for the development of a myeloid malignancy in patients with CIN (OR, 35.00; 95% CI, 1.27-962.10). Conversely, subjects with the DNMT3A p.R882H mutations displayed a stable course, thus confirming the low predictive value of isolated DNMT3A mutations.22,43 The presence of any DNMT3A mutation did not increase the risk for the development of a myeloid malignancy in patients with CIN (RR, 0.30 [95% CI, 0.04-2.46]; OR, 0.23 [95% CI, 0.02-2.73]).
Follow-up analyses by NGS
Follow-up analysis by NGS was performed in 34 patients with CIN (9 clonal, 25 nonclonal) (supplemental Table 1) (Figure 3). In patients with clonal disease, the median time between the first and subsequent analysis was 23 months (range, 6-111 months). Six of these patients carried the initial somatic mutations with no substantial change in VAF and displayed an absence of additional mutations and stable disease course (Figure 3A-E, 3I). Two patients who progressed to MDS/MPN and AML (patients #16 and #21, respectively; supplemental Table 1) displayed a notable clonal expansion with additional mutations at the time of progression. Specifically, the patient who progressed to MDS/MPN acquired a mutation in JAK2 and ASXL1, whereas the patient who progressed to AML acquired the typical NPM1 p.L287fs mutation (Figure 3F-G). The patient who developed MDS with multilineage dysplasia (patient #2, supplemental Table 1), carrying 3 mutations in DNMT3A and IDH1, exhibited a moderate increase in the VAF of these mutations at follow-up (Figure 3H). No additional mutations were detected at the time of transformation, further confirming the high predictive value of the presence of 3 mutations for evolution to a myeloid malignancy without acquisition of additional mutations.22 The median time between the first and sequential analysis in the 23 patients with nonclonal disease was 30 months (range, 9-112 months). No mutations were identified, and none of these patients showed signs of disease progression.
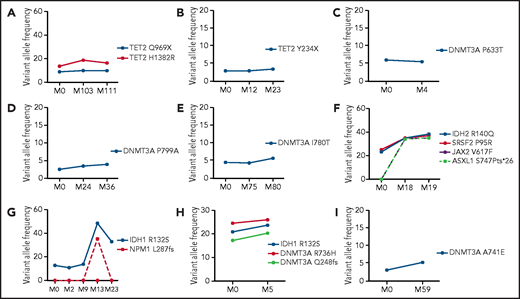
Follow-up NGS analysis in patients with CIN with clonal disease. The graphs show the mutational status and size of clones in 9 patients with CIN with clonal disease in whom follow-up NGS studies were available (shown in supplemental Table 1). The detected mutations and the respective VAFs (%) at the first evaluation (M0) and subsequent analysis in different time points shown in months (M) are depicted. In 6 patients, the clonal size (VAF) of the acquired mutations remained relatively stable between the first and subsequent NGS analyses (A-E, I). Of the 3 patients with available follow-up NGS who progressed to myeloid neoplasm, the clonal size was increased and additional mutations were detected at the time of progression in 2 of them (panels F and G); in the third patient, only a modest increase was detected in the VAF of mutations, with no additional mutations acquired at the time of progression (H). Panels A-I correspond to patients #7, #15, #1, #20, #8, #16, #21, #2, and #12, respectively, as presented in supplemental Table 1.
Follow-up NGS analysis in patients with CIN with clonal disease. The graphs show the mutational status and size of clones in 9 patients with CIN with clonal disease in whom follow-up NGS studies were available (shown in supplemental Table 1). The detected mutations and the respective VAFs (%) at the first evaluation (M0) and subsequent analysis in different time points shown in months (M) are depicted. In 6 patients, the clonal size (VAF) of the acquired mutations remained relatively stable between the first and subsequent NGS analyses (A-E, I). Of the 3 patients with available follow-up NGS who progressed to myeloid neoplasm, the clonal size was increased and additional mutations were detected at the time of progression in 2 of them (panels F and G); in the third patient, only a modest increase was detected in the VAF of mutations, with no additional mutations acquired at the time of progression (H). Panels A-I correspond to patients #7, #15, #1, #20, #8, #16, #21, #2, and #12, respectively, as presented in supplemental Table 1.
Discussion
ICUS patients with clonal hematopoiesis, also characterized as CCUS, display an increased risk of developing a myeloid malignancy compared with nonclonal ICUS patients.22,44,45 The group of ICUS patients with isolated neutropenia, also characterized as ICUS-N or CIN, is underrepresented in studies investigating ICUS, and therefore limited data are available for the mutation status and outcome of these patients.26 In the current study, we performed for the first time NGS mutation analysis of genes associated with myeloid malignancies in a cohort of well-defined CIN/ICUS-N patients with a long follow-up. We also assessed longitudinally the mutational pattern and role of mutations in disease evolution to a myeloid malignancy.
We found that 21 of 185 patients with CIN (11.35%) carried somatic mutations in at least 1 of the myeloid genes studied. This proportion is significantly lower than the mutation frequency of 38% to 64% previously reported in ICUS patients, suggesting that CIN probably represents a distinct ICUS group with a more favorable mutation profile.20,22,23 No significant differences were identified between patients with CIN with clonal and nonclonal disease in the severity of neutropenia, white blood cell counts, hemoglobin levels, and MCV. A lower platelet count was observed in patients with clonal disease, albeit within normal range, that might reflect subtle inflammatory processes and/or an age-related effect given the older age of the clonal CIN group.46-48 Furthermore, a female predominance was observed in both the clonal and nonclonal CIN groups, which is in contrast to the male predominance previously reported in ICUS, corroborating further the distinct characteristics of CIN.22,23
The 21 clonal patients with CIN carried 25 somatic mutations in 6 myeloid genes with a median VAF of 12.75%; the most frequently mutated genes were DNMT3A and TET2, followed by IDH1/2, SRSF2, and ZRSR2. However, mutations in genes implicated in hematologic malignancies with a VAF of at least 2% have also been reported in hematologically normal individuals with increasing frequency by age, a condition known as clonal hematopoiesis of indeterminate potential (CHIP).21DNMT3A and TET2 have been reported among the most common mutated myeloid genes in CHIP.36,37,49 We may thus hypothesize that mutations in DNMT3A and TET2 in our study population is an age-related, incidental finding rather than an abnormality with major pathophysiological significance, especially when VAF is <10%. A similar conclusion has also been reached in individuals with anemia of the elderly.43 In favor of the assumption of an age-related phenomenon is the fact that clonal patients with CIN were significantly older compared with nonclonal patients and that the age-adjusted prevalence of clonal hematopoiesis in patients with CIN was similar to that of the general population in older ages. Furthermore, one patient aged 70 years among 7 patients with a typical neutrophil count with Fy(a-b-) status presented also a DNMT3A mutation with 4% VAF (these 7 patients have been excluded from the study). None of the patients carrying an isolated mutation in the aforementioned genes transformed to a myeloid malignancy, whereas all the mutations with a VAF <10% were detected in DNMT3A and TET2. Overall, our data on the frequency and minimal effect of isolated mutations of DNMT3A and TET2 in MDS/AML evolution in patients with CIN are in accordance with studies on age-related clonal hematopoiesis.21,36,37,50
In the current study, 5 of 185 patients with CIN (2.70%) transformed to a myeloid malignancy, with a median time to progression from the initial diagnosis of 96 months. This proportion is significantly lower than the 25% transformation proportion previously reported in ICUS and indicates further that patients with CIN represent a distinct ICUS group with more benign clinical features.22 Among the 5 patients who transformed to a myeloid malignancy, 4 patients carried 1 or more mutations in SRSF2, IDH2, IDH1, and DNMT3A, and the detection of mutations was associated with a RR of 31.24 for transformation. These data are in accordance with a previous study in ICUS showing that patients with clonal disease (CCUS) display a 14-fold higher probability of developing a myeloid neoplasm compared with nonclonal patients; the transformation was higher in the presence of spliceosome gene mutations, including SRSF2 or in comutations of DNMT3A with additional genes, similar to our patients.22 They are also in agreement with a recent study of patients with clonal hematopoiesis who progressed to AML, in which it was shown that in addition to spliceosome genes (eg, SRSF2), IDH1/IDH2 mutations are high-risk mutations.51 In our study, the presence of SRSF2 or IDH1 mutations (isolated or comutations) conferred a RR of 9.50 for the development of a myeloid malignancy. Furthermore, in accordance with studies showing that a VAF >10% is associated with increased risk for the development of a myeloid malignancy in CHIP,36,37 the patients with CIN who transformed in our study had clonal disease with individual VAFs >10%, and overall a VAF >10% was associated with higher probability for transformation. Although the VAF size is not the only parameter associated with disease evolution, these patients need to be carefully followed up for the early identification of a myeloid malignancy. In our practice, we monitor clonal patients with CIN using serial PB evaluation (every 3-6 months depending on the type of mutation and size of clone) combined with NGS analysis, and we perform BM examination upon indication (changes in PB counts and/or morphology).45 However, longitudinal and sequential NGS data in patients with CIN with clonal disease, particularly with VAF >10%, in combination with PB and BM morphology studies, will allow the introduction of specific recommendations for the monitoring of these patients.
The absence of mutations in our patients had a negative predictive value for malignant transformation of 0.99. Longitudinal studies are important not only for patients with clonal disease but also for those with nonclonal disease at baseline, for the evaluation of differences in outcome, for the early detection of clonal aberrations during the course of the disease, and for investigation of their clinical significance. In the majority of patients tested for clonal evolution over time, most single mutant clones seemed to be remarkably stable, with minimal VAF change and stable disease status. The same observation has been reported in individuals with anemia of the elderly.43 Two of three patients with CIN who had available follow-up studies and transformed to a myeloid malignancy displayed a clonal expansion, as was reflected by the increase in VAF and the development of additional mutations, whereas in the third patient only a modest VAF increase was identified.
Thirteen patients in our study population were identified with mutations within germline range. Although germline NGS testing was not performed, the germline, nonpathogenic nature of the mutations was inferred by the thorough review of population-specific databases, our functional annotation pipeline, and the stable neutropenia and disease state time course.
The majority of patients included in the study had mild neutropenia. However, of the MDS/AML transformed patients with CIN, only 1 had severe neutropenia (#80, supplemental Table 1), whereas 2 patients had moderate neutropenia (#2, #21, supplemental Table 1) and 2 patients had mild neutropenia (#11, #16, supplemental Table 1), suggesting that all patients with CIN need a thorough investigation, irrespective of the severity of neutropenia.
In summary, we studied the clinical characteristics and mutation profile of myeloid genes in a cohort of well-characterized ICUS patients with isolated neutropenia displaying the longest follow-up reported thus far. We have shown that this group of patients, appearing in the literature as CIN or ICUS-N, display a female predominance and a more benign clinical course compared with patients with ICUS and other type of cytopenia(s), according to available data published so far. The CIN/ICUS-N patients display a 11.35% frequency of mutations in myeloid genes, with the most commonly mutated genes being DNMT3A and TET2, representing also the frequently mutated genes in age-related CHIP; a low proportion of patients (2.70%) progressed to an overt myeloid malignancy and the risk is associated with the type of mutations and clone(s) size, with SRSF2 and IDH1 being highly implicated in this process. This study contributes to a better understanding of the heterogeneous entities underlying ICUS and also highlights the importance of mutation analysis for the diagnosis and follow-up of patients with unexplained neutropenias.
Acknowledgments
This study was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC), Milan, Italy (Investigator Grant #20125, AIRC 5x1000 Project #21267, International Accelerator project #22796). The part of work performed in the Hellenic Network of Precision Medicine on Cancer was supported by the initiative of the Research and Innovation Department of the Ministry of Education, Research and Religion, funded by the Framework of the Hellenic Republic-Siemens Settlement Agreement. The study is based on work from COST (European Cooperation in Science and Technology) Action CA18233 “European Network for Innovative Diagnosis and treatment of Chronic Neutropenias, EuNet-INNOCHRON” supported by COST.
Authorship
Contribution: H.A.P. and L.M. designed the research, analyzed/interpreted data, and wrote the manuscript; G. Tsaknakis designed the research, performed experiments, analyzed/interpreted data, prepared figures and tables, and contributed to the writing of the manuscript; A.G. performed experiments and analyzed/interpreted data; S.P., P.K., and C.P. contributed to patient material and provided clinical data; C.E., G. Todisco, and E.B. collected clinical data; E.R. performed bioinformatics analysis; E.M. analyzed sequencing data; I.M., I.F., E.L., A.B., and S.M. performed experiments; and N.T. supervised part of the NGS analysis and critically reviewed the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Helen A. Papadaki, University Hospital of Heraklion, P.O. Box 1352, Heraklion, Crete, Greece; e-mail: e.papadaki@uoc.gr.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

Comments
Questionable recommendation for mutational analysis in CIN patients..