

In this issue of Blood, 2 manuscripts, 1 and ,2 report on the development of T-cell lymphomas derived from CD19-specific chimeric antigen receptor (CD19-CAR) T cells that were generated with the piggyBac transposon system, and perform detailed mechanistic studies revealing no apparent evidence of insertional mutagenesis, but widespread copy number variations (CNVs), including gains and losses (see figure).
Adoptive immunotherapy with CAR T cells had remarkable success for B-cell lineage hematological malignancies highlighted by the US Food and Drug Administration approval of 4 CD19-CARs and 1 BCMA-CAR T-cell product since 2017. These commercial T-cell products are generated with retroviral or lentiviral vectors similar to the majority of cell products for ongoing early-phase clinical studies conducted by academic institutions or biotech. Although retroviral and lentiviral vector-modified T cells have an outstanding safety record,3 nonviral gene transfer technologies are being developed to replace viral vectors because of costs, logistics, and risk of insertional mutagenesis. Transposon-based gene-delivery systems were developed and refined for more than 20 years. They have been used to genetically modify T cells to express CARs in preclinical models as well as early-phase clinical studies. Results of early-phase clinical studies using the Sleeping Beauty transposon system have demonstrated efficacy and safety with no evidence of malignant transformation.4,5 The clinical trial reported by Bishop et al and Micklethwaite et al are the first peer-reviewed clinical reports of the piggyBac transposon system. They describe 10 patients who received donor-derived CD19-CAR T cells with relapsed/refractory B-cell malignancies after matched sibling donor hematopoietic stem cell transplant. Two patients developed T-cell lymphoma derived from CD19-CAR T cells, which was unexpected because piggyBac transposons integration sites overlap to a great extend with integration sites of retroviral vectors,6 which have a proven safety record in the context of CAR T-cell therapies.
In detailed mechanistic studies of the index case, Micklethwaite et al found a high number of integration sites (24), but no transposon insertion into typical oncogenes. Lymphoma cells harbored structural variants (SVs) and numerous CNVs, including gains and losses, with the latter having the dominant influence on the observed global changes in gene expression. Gene ontology analysis revealed upregulation of genes associated with primitive embryonic development and cell–cell adhesion and downregulation of genes associated with regulation of cell adhesion, T-cell activation, and activation of phospholipase C. Gene expression analysis revealed transcriptional readthrough from the 3′ end of the transgene with 4 genes (FAM11D, COL8A1, HIVEP1, and FYN) being in frame and overexpressed. Of these, FYN modulates TCR signaling and T-cell differentiation.7 However, the authors found no evidence of downstream effects of FYN overexpression in their detailed analysis. The second lymphoma had fewer integration sites (4), but also displayed numerous SVs with dysregulated gene expression, which correlated again with CNVs. However, gene expression by gene ontology analysis differed from the gene expression found in the index lymphoma. Likewise, although transcriptional readthrough was observed, the authors identified increased expression of a noncoding RNA (LOC107985043) instead of the aforementioned genes. Of interest, both lymphomas shared integration sites in the BACH2 locus (introns 3 and 4), resulting in decreased gene expression. BACH2 is a transcription factor that plays a prominent role in T-cell biology by regulating T-cell plasticity.8 Thus, additional studies seem warranted to exclude BACH2 integration of piggyBac transposons as a contributing factor in lymphomagenesis. However, BACH2 haploinsufficiency in humans is associated with immune dysfunction and not T-cell lymphoma.9
What induced the marked SVs and CNVs in both lymphomas? Because CAR T-cell products in this clinical study were generated from healthy donors, previous exposure of genotoxic chemotherapy can be excluded as a contributing factor. It is unlikely that these changes were caused by the transposon because it encoded a standard expression cassette with a second-generation CD19.41BBζ CAR driven by the hEF1α promoter flanked by chicken b-globin hypersensitivity site 4 insulators.
Micklethwaite et al discuss in detail that their CAR T-cell production protocol might have contributed to the overserved SNs and CNVs. Their protocol used standard techniques including electroporation of peripheral blood mononuclear cells with transposase messenger RNA and CAR transposon plasmid followed by expansion on irradiated peripheral blood mononuclear cells in the presence of interleukin-15 in G-Rex10 tissue culture devices. Could the observed SVs and CNVs be a result of “transposon hopping” during transposase expression, which can last for up to 4 days after messenger RNA transfection?10 The piggyBac transposase mediates seamless and precise excision with TTAA site repair, so this should be less of an issue compared with other transposon systems such as Sleeping Beauty, which leave behind a footprint after excision. Nonetheless, this question has so far not been experimentally addressed and based on the authors findings should be examined in the future. The piggyBac transposase is known to interact with bromodomain-containing protein 4,6 a transcriptional regulator and keeper of genome stability. Thus, additional studies are warranted because transposase interaction with bromodomain-containing protein 4 could possibly have contributed to the observed high incidence of SVs and CNVs. In human cells, piggyBac transposon plasmid backbone integration can occur at a high rate.11 Although the study shows that the CAR transposons did not integrate into known oncogenes, the possibility of plasmid backbone altering the genome also needs to be evaluated.
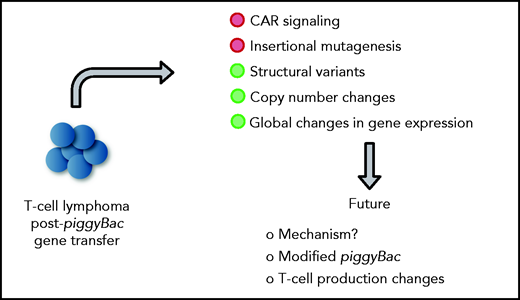
Mechanistic insight into lymphomagenesis post-piggyBac gene transfer and proposed future studies. Green circles highlight contributing factors.
Mechanistic insight into lymphomagenesis post-piggyBac gene transfer and proposed future studies. Green circles highlight contributing factors.
Although the authors’ findings are of great concern, the findings should not preclude the future clinical development of piggyBac-based transposon-based gene delivery systems. Instead, these findings should fuel discovery research leading to safer transposon systems, similar to the development of safer viral vectors more than a decade ago after reports of retroviral-induced leukemias on clinical gene therapy studies for immunodeficiencies.12 Fewer integrations per cell and targeted transposon insertion should improve safety. The recent resolution of the piggyBac transpososome structure by cryo-electron microscopy should help facilitate such studies.13
Conflict-of-interest disclosure: S.G. has patent applications in the fields of T-cell and/or gene therapy for cancer; consults for Catamaran Bio, Nektar Therapeutics, TESSA Therapeutics; is on the Scientific Advisory Board of Tidal; and is a data safety and monitoring board member of Immatics. M.H.W. has patent applications involving T-cell modification and is on the Scientific Advisory Board of Saliogen.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal