

Key Points
In aging, B-cell regeneration is regulated by cross-talk between peripheral B cells and progenitors in the BM.
This repression is mediated by a TNF-α/IGFBP-1/IGF-1 immune-endocrine axis; perturbation of this axis restores B-cell lymphopoiesis in aging.

Abstract
Loss of B lymphocyte regeneration in the bone marrow (BM) is an immunologic hallmark of advanced age, which impairs the replenishment of peripheral B-cell subsets and results in impaired humoral responses, thereby contributing to immune system dysfunction associated with aging. A better understanding of the mechanism behind this loss may suggest ways to restore immune competence and promote healthy aging. In this study, we uncover an immune-endocrine regulatory circuit that mediates cross-talk between peripheral B cells and progenitors in the BM, to balance B-cell lymphopoiesis in both human and mouse aging. We found that tumor necrosis factor α (TNF-α), which is increasingly produced by peripheral B cells during aging, stimulates the production of insulin-like growth factor-binding protein 1 (IGFBP-1), which binds and sequesters insulin-like growth factor 1 (IGF-1) in the circulation, thereby restraining its activity in promoting B-cell lymphopoiesis in the BM. Upon B-cell depletion in aging humans and mice, circulatory TNF-α decreases, resulting in increased IGF-1 and reactivation of B-cell lymphopoiesis. Perturbation of this circuit by administration of IGF-1 to old mice or anti–TNF-α antibodies to human patients restored B-cell lymphopoiesis in the BM. Thus, we suggest that in both human and mouse aging, peripheral B cells use the TNF-α/IGFBP-1/IGF-1 axis to repress B-cell lymphopoiesis. This trial was registered at www.clinicaltrials.govas#NCT00863187.
Introduction
With advancing age, changes in the cellular composition and competence of the immune system to respond to immunogenic insult result in increased susceptibility to infectious diseases, poor responsiveness to new or evolving pathogens, and reduced efficacy of vaccinations.1-3 A major cause of reduced immunity with aging is reduced replenishment of the peripheral compartment with new and diverse naïve lymphocytes, which are generated in primary lymphoid organs.4,5 Within the bone marrow (BM), this process is characterized by altered hematopoiesis, including altered function of hematopoietic stem cells, which shift from a balanced-differentiated state into a myeloid-biased state, thereby reducing the generation of new B cells.6,7 The altered function of hematopoietic stem cells with aging is still poorly understood and is thought to involve both intrinsic and microenvironmental effects.8
Despite the age-associated loss of B-cell lymphopoiesis in the BM, the actual number of B cells in the periphery remains largely unchanged,3,9 suggesting the existence of cellular homeostasis. This process, which balances between cell generation and cell death, changes with aging to adapt to physiological changes. Indeed, aging is associated with a decline in the frequency of naïve B cells, along with the accumulation of atypical antigen-experienced oligoclonal B cells in the periphery, which are thought to extend survival.9-12 The mechanism for this alteration in the cellular composition of B lineage cells remains poorly understood and is an important challenge in attempts to prolong healthy life expectancy. Although rejuvenation of the peripheral compartment with new naïve B cells is desirable for improved immunity in the older population, this process is limited by the markedly reduced B-cell lymphopoiesis in the older BM.
In earlier studies, we found that the age-related changes in B-cell homeostasis can be prevented or even reversed by inducing a change in cellular homeostasis. We have shown this in mouse models with chronic B-cell deficiency from birth,13 and in older mice and humans that have undergone peripheral B-cell depletion.14,15 In particular, we found that removal of old B cells alters cellular homeostasis to reactivate B-cell lymphopoiesis in the BM, a paradigm that was later confirmed in other lineages and tissues,16,17 and this is in agreement with studies showing that removal of senescent cells improves tissue function and increases lifespan.18 Moreover, our studies provided evidence that B-cell homeostasis in aging is in fact regulated by cross-talk between peripheral B cells and BM progenitors, but the nature of this cross-talk has remained elusive. In this study, we uncover a novel immune-endocrine regulatory circuit that mediates this cross-talk to balance B-cell lymphopoiesis in aging. We found that tumor necrosis factor α (TNF-α), which is increasingly produced by peripheral B cells in aging, stimulates the production of insulin-like growth factor-binding protein 1 (IGFBP-1), which binds insulin-like growth factor 1 (IGF-1) in the circulation and sequesters its activity in promoting B-cell lymphopoiesis in the BM. Upon B-cell depletion, TNF-α in the circulation decreases, resulting in increased IGF-1 and reactivation of B-cell lymphopoiesis. Thus, we suggest that in aging, peripheral B cells use the TNF-α/IGFBP-1/IGF-1 axis to suppress B-cell lymphopoiesis.
Methods
Mice
Male and female mice used in these experiments were either young (age 2-4 months) or old (age 19-24 months). Mice were C57BL/6 (The Jackson Laboratory, Bar Harbor, ME), C57BL/6 human CD20 transgenic (hCD20Tg), or Rag-2fl/fl mice,19 crossed with Mx-cre transgenic mice.20 Details regarding the in vivo experimental procedures are provided in the supplemental Data, available on the Blood Web site.
B-cell purification and culture conditions
BM cultures for interleukin-7 (IL-7)-driven differentiation of B-cell precursors were prepared as previously described.21 Cells were grown for 5 to 7 days, harvested, and used as described. In some experiments, fetal calf serum was replaced with 1% fresh mouse serum with or without IGF-1 (20 ng/mL), growth hormone (GH; 40 ng/mL), or added goat anti–IGF-1 (50 ng/mL) (all from Peprotech). Splenic B cells were purified by magnetic depletion of CD43+ cells using the MACS MicroBeads and MACS columns (Miltenyi Biotec) (≥90% purity). For cell death analysis, 2 × 105 to 4 × 105 purified splenic B cells were cultured in 24-well plates for 24 to 48 hours. At the indicated time points (as shown in each figure legend), cells were collected and apoptotic cells were quantified by propidium iodide staining.
Human blood collection
Plasma samples collected from healthy young volunteers (age 18-35 years), healthy older volunteers (age 55 years or older), and older patients (age 55 years or older) with B-cell non-Hodgkin lymphoma who were previously treated with rituximab B-cell depletion therapy were used to monitor B-cell–mediated age-related alterations in soluble peripheral blood molecules. The study was approved by the Institutional Review Board (IRB 3097). Inclusion and exclusion criteria were previously reported.14
To study the role of TNF-α and anti–TNF-α treatments in regulating B-cell lymphopoiesis in humans, clinical data collected from 32 patients at the Rheumatology Institute at Rambam Health Care Campus (Haifa, Israel) were analyzed using cross-sectional and longitudinal approaches. The study was observational, with no clinical decision making, and all patients provided informed consent to participate in the study (IRB 0643-15 RMB) (details are provided in the supplemental Data). Statistical significance of the difference between experimental groups was determined by using an unpaired, 2-tailed Student t test with differences considered significant at P < .05.
Results
B cells from old mice are resistant to apoptosis and have extended survival
We previously showed that generation of B cells in the BM is regulated by cellular homeostasis in the peripheral compartment and that this homeostasis changes with aging.14,22 A possible explanation for this change is the extended survival of atypical B cells in the peripheral compartment. Consistent with this notion, splenic B cells from young mice died at a rate that was twofold to threefold higher than that for splenic B cells from old mice in an in vitro test of spontaneous death over 24 to 48 hours (Figure 1A-B). AnnexinV staining confirmed that most of the dead cells were apoptotic (supplemental Figure 1). To test whether these differences reflect an intrinsic property of old B cells or are a property of old mice, we examined the characteristics of newly generated B cells in old mice. After B cells were depleted in old hCD20Tg mice and the B cell compartment was reconstituted by de novo B-cell lymphopoiesis in the BM (old-depleted),14,15 the newly generated splenic B cells underwent spontaneous apoptosis at a rate that was not different from that of splenic B cells from young mice (Figure 1A-B). To test this concept in vivo, we used mice homozygous for an RAG2-floxed allele, and bearing an Mx-cre transgene (Mx-cre/RAG2fl/fl). Administration of polyinosinic:polycytidylic acid [poly(I:C)] resulted in ablation of both RAG2 alleles, thereby aborting B-cell lymphopoiesis in the BM at the pro-B stage and enabling us to monitor the survival rate of peripheral B cells over time19 (supplemental Figure 1). Within 17 weeks, splenic B-cell numbers in young Mx-cre/RAG2fl/fl mice dropped by 50% relative to a reduction of only 20% to 30% in old Mx-cre/RAG2fl/fl mice (Figure 1C-D). Together with our previous findings,14 these results suggest that peripheral B cells in old mice have extended survival and are able to modify cellular homeostasis in the B-cell compartment.
![Peripheral B cells in old mice have extended survival. (A-B) Splenic B cells from young, old, or old hCD20Tg mice treated for B-cell depletion that have reconstituted their peripheral B-cell compartment from de novo B-cell lymphopoiesis in the BM (old depleted) were cultured for the indicated time intervals. Cells were collected and fixed, and spontaneous death was determined by propidium iodide (PI) staining. Shown are representative results after 24 hours (A) and kinetic measurements of accumulated spontaneous apoptosis (B). Significance of differences between groups of young and old mice or between old-depleted mice and old mice are marked with stars (n = 6 mice from each group in a total of 4 independent experiments). (C-D) Young and old Mx-cre/RAG-2fl/fl or control mice were injected intraperitoneally with polyinosinic:polycytidylic acid [poly(I:C)] to ablate floxed alleles, and 17 weeks later, spleens were analyzed to quantify B cells. Shown are representative plots for individual mice with the indicated genotypes (C) and absolute cell numbers (D). Graph depicts means from 5 mice in each group ± standard error (SE). **P < .01; ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/19/10.1182_blood.2021012428/4/m_bloodbld2021012428f1.png?Expires=1769092441&Signature=y0kIXVLYoGEwqYfo2tnp60yTHgTBnv-r2OdLWhr-VV3BmpbPAy4UHPU4tvm6A4DWWpMojpXTJK2qWDLH2J-4v8xD7dBsA0dJJF2ymu-eQFgP1IyAd3N5~2fpwiwOl2COce~dKUc5wB1lIW3eHlbOqJXh~HwiBclQ-VAiMs9bjD-gYbzlOZThAAOAQ1dFu5soSCbpvMwr-rdcvBammdCj~T4UDXf49a1zlLA0B1sLhxYwAQRXD0TR-qvm~DeNMGXBj9fYlzE82iMcO9S8EQle3Ey8pXV5DdUGKVxSx~XVG6unTH28lND~CHo1vkdBQ0cJezhCIFJvqwxR9ZxuQq5Q3g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Peripheral B cells in old mice have extended survival. (A-B) Splenic B cells from young, old, or old hCD20Tg mice treated for B-cell depletion that have reconstituted their peripheral B-cell compartment from de novo B-cell lymphopoiesis in the BM (old depleted) were cultured for the indicated time intervals. Cells were collected and fixed, and spontaneous death was determined by propidium iodide (PI) staining. Shown are representative results after 24 hours (A) and kinetic measurements of accumulated spontaneous apoptosis (B). Significance of differences between groups of young and old mice or between old-depleted mice and old mice are marked with stars (n = 6 mice from each group in a total of 4 independent experiments). (C-D) Young and old Mx-cre/RAG-2fl/fl or control mice were injected intraperitoneally with polyinosinic:polycytidylic acid [poly(I:C)] to ablate floxed alleles, and 17 weeks later, spleens were analyzed to quantify B cells. Shown are representative plots for individual mice with the indicated genotypes (C) and absolute cell numbers (D). Graph depicts means from 5 mice in each group ± standard error (SE). **P < .01; ***P < .001.
Peripheral B cells in old mice have extended survival. (A-B) Splenic B cells from young, old, or old hCD20Tg mice treated for B-cell depletion that have reconstituted their peripheral B-cell compartment from de novo B-cell lymphopoiesis in the BM (old depleted) were cultured for the indicated time intervals. Cells were collected and fixed, and spontaneous death was determined by propidium iodide (PI) staining. Shown are representative results after 24 hours (A) and kinetic measurements of accumulated spontaneous apoptosis (B). Significance of differences between groups of young and old mice or between old-depleted mice and old mice are marked with stars (n = 6 mice from each group in a total of 4 independent experiments). (C-D) Young and old Mx-cre/RAG-2fl/fl or control mice were injected intraperitoneally with polyinosinic:polycytidylic acid [poly(I:C)] to ablate floxed alleles, and 17 weeks later, spleens were analyzed to quantify B cells. Shown are representative plots for individual mice with the indicated genotypes (C) and absolute cell numbers (D). Graph depicts means from 5 mice in each group ± standard error (SE). **P < .01; ***P < .001.
B cells from old mice actively suppress B-cell lymphopoiesis in the BM
Because B-cell depletion reactivates B-cell lymphopoiesis in the BM of old mice,15 we hypothesized that age-related alterations in B-cell homeostasis are mediated by cross-talk mechanisms between peripheral B cells and progenitors in the BM. To test this notion, we adoptively transferred splenic B cells from old or young wild-type mice into young hCD20Tg recipient mice, which were pretreated to deplete the peripheral B-cell compartment15,23 (supplemental Figure 2). Recipient mice were then analyzed for B-cell lymphopoiesis in the BM 28 days after depletion (scheme for the experiment shown in Figure 2A). In mice reconstituted with splenic B cells from old mice, we observed significant suppression of B-cell lymphopoiesis. Numbers of pro-B, pre-B, and immature B cells were reduced by fourfold to fivefold, relative to levels in mice reconstituted with splenic B cells from young mice (Figure 2B-C; populations are indicated by arrows). Consistent with this finding, we observed a threefold reduction in the accumulation of newly generated B cells in the spleens in mice that received B cells from old mice (Figure 2D-E). Similarly, in a different experimental approach, we transferred splenic B cells from young or old mice into green fluorescent protein (GFP)-Tg BM chimera mice and found that both B-cell lymphopoiesis and accumulation in the spleen were suppressed in chimeric mice receiving B cells from old mice (supplemental Figure 3).
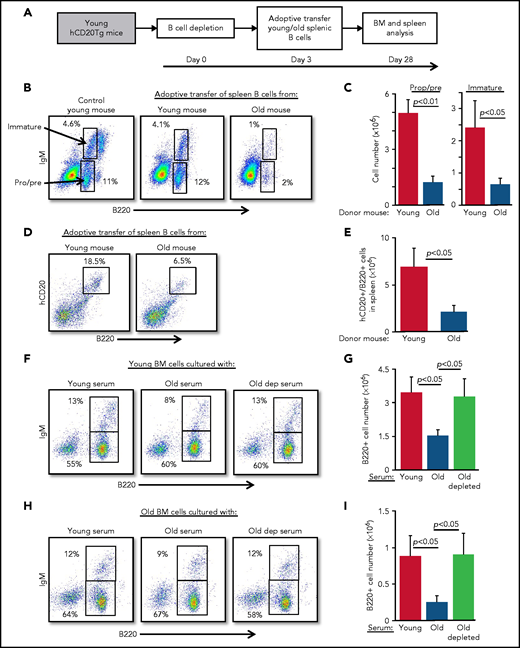
Peripheral B cells from old mice suppress B-cell lymphopoiesis. Splenic B cells from young and old mice were adoptively transferred to young hCD20Tg mice that were treated (and confirmed by blood stain) for B-cell depletion. Bone marrow and spleen from recipient mice were quantified for B-cell lymphopoiesis 28 days after depletion. (A) Schematic diagram showing details of the kinetics of the experiment. (B-C) Analysis of BM cells for the indicated mice. For analysis, viable lymphocytes were defined by forward scatter and light side scatter, and gates were set to analyze pro-B (pro) and pre-B (pre) cells (B220+CD93+IgM–) and immature B cells (B220+CD93+IgM+). Shown are representative plots for a single mouse from each group; arrows indicate populations (B). Absolute cell numbers (C). Graph depicts mean from 4 mice in each group ± SE. (D-E) Analysis of spleen cells for the indicated mice. Gates were set to quantify newly generated host B cells as B220+hCD20+. Shown are representative plots for individual mice from each group (D) and absolute cell numbers (E). Graph depicts means from 4 mice in each group ± SE. (F-I) IL-7–driven BM cultures to grow B cells in vitro were prepared from young (F-G) or old (H-I) mice. In these experiments, the fetal calf serum (FCS) in culture media was replaced with 1% fresh mouse serum from the indicated mice. After 5 days, cells were harvested, counted, and stained for surface markers and analyzed for pro-B and pre-B (B220+/IgM–) and immature (B220+/IgM+) B cells. Shown are representative results from a single experiment (F,H) and absolute B-cell counts (G,I) in the cultures. Graphs depict means from 5 experiments ± SE. dep, depleted.
Peripheral B cells from old mice suppress B-cell lymphopoiesis. Splenic B cells from young and old mice were adoptively transferred to young hCD20Tg mice that were treated (and confirmed by blood stain) for B-cell depletion. Bone marrow and spleen from recipient mice were quantified for B-cell lymphopoiesis 28 days after depletion. (A) Schematic diagram showing details of the kinetics of the experiment. (B-C) Analysis of BM cells for the indicated mice. For analysis, viable lymphocytes were defined by forward scatter and light side scatter, and gates were set to analyze pro-B (pro) and pre-B (pre) cells (B220+CD93+IgM–) and immature B cells (B220+CD93+IgM+). Shown are representative plots for a single mouse from each group; arrows indicate populations (B). Absolute cell numbers (C). Graph depicts mean from 4 mice in each group ± SE. (D-E) Analysis of spleen cells for the indicated mice. Gates were set to quantify newly generated host B cells as B220+hCD20+. Shown are representative plots for individual mice from each group (D) and absolute cell numbers (E). Graph depicts means from 4 mice in each group ± SE. (F-I) IL-7–driven BM cultures to grow B cells in vitro were prepared from young (F-G) or old (H-I) mice. In these experiments, the fetal calf serum (FCS) in culture media was replaced with 1% fresh mouse serum from the indicated mice. After 5 days, cells were harvested, counted, and stained for surface markers and analyzed for pro-B and pre-B (B220+/IgM–) and immature (B220+/IgM+) B cells. Shown are representative results from a single experiment (F,H) and absolute B-cell counts (G,I) in the cultures. Graphs depict means from 5 experiments ± SE. dep, depleted.
To test whether this cross-talk is mediated by soluble factors in the blood, we modified an IL-7–driven BM culture system for B-cell lymphopoiesis21 by replacing the 10% fetal calf serum that is usually used in culture media with 1% mouse serum collected from young, old, or old B cell-depleted mice. In this system, serum from old mice suppressed B-cell lymphopoiesis compared with cultures containing serum from young mice. Suppression was reflected by a twofold to 2.5-fold reduction in total B-cell numbers and delayed differentiation to the immunoglobulin M (IgM)-expressing immature stage, because the frequency of immature B cells was reduced by about 40% (from 13% to 8%). In contrast, no suppression was observed in BM cultures containing sera from B-cell–depleted old mice (Figure 2F-G). To exclude the possibility that these inhibitory effects reflect unique properties of young BM cells, we repeated the experiment using BM cells from old mice and found similar results (Figure 2H-I). Collectively, these findings suggest that peripheral B cells in old mice suppress B-cell lymphopoiesis in the BM and that this suppression is mediated by soluble factors in the blood.
B-cell lymphopoiesis in aging is regulated by the GH/IGF-1 endocrine axis
To identify soluble factors that may mediate the suppression of B-cell lymphopoiesis in aging, we searched the published literature and identified IGF-1, an endocrine anabolic hormone that is abundant in the plasma at young ages but decreases dramatically with advancing age.24-28 IGF-1 was previously shown to support the growth and differentiation of progenitor B cells in vitro and in vivo.29-31 In agreement with the published literature, plasma IGF-1 was threefold lower in old mice compared with that in young mice; however, after B-cell depletion in old mice, IGF-1 increased and was comparable to that in young mice (Figure 3A). Moreover, addition of anti–IGF-1 antibody to BM cultures grown in the presence of serum from young mice abolished the growth of B lymphoid progenitors (Figure 3B), suggesting that the IGF-1 found in the serum of young mice is important for B-cell lymphopoiesis.
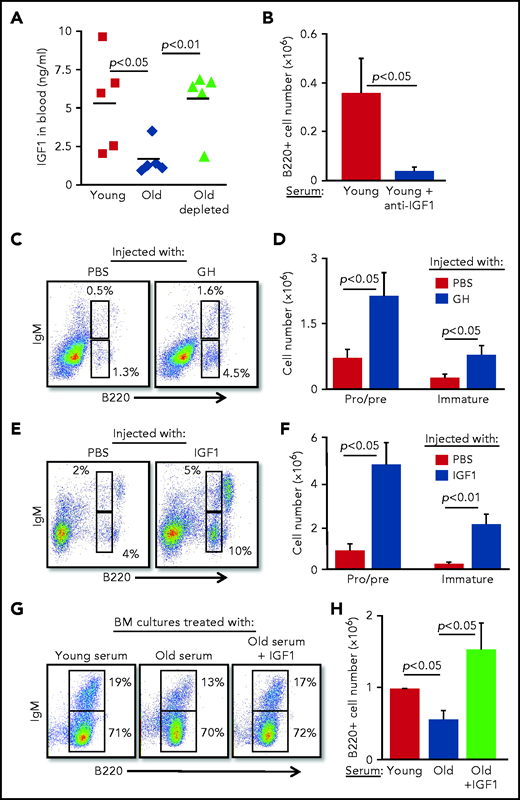
B-cell lymphopoiesis in aging is regulated by IGF-1. (A) Sera collected from the indicated mice were analyzed for IGF-1 by enzyme-linked immunosorbent assay (ELISA). Shown are results for individual mice and group means (n = 5 for each group). (B) IL-7–driven BM cultures were prepared, replacing the FCS in media with 1% serum from young mice in the presence or absence of goat–anti-mouse IGF-1 (50 ng/mL). After 5 days, cells were harvested, counted, and stained to quantify B-cell numbers. Graph depicts mean from 4 experiments ± SE. (C-F) Old mice were subcutaneously injected with human GH (hGH) (C-D) or with human IGF-1 (hIGF-1) (E-F) for 10 days. Control mice were injected with phosphate-buffered saline (PBS). One day after the last injection, we analyzed the BM of the mice for B-cell lymphopoiesis (described in the Figure 2 legend) with gates marked for pro-B , pre-B, and immature B cells. Shown are representative plots for a single mouse from each group (C,E) and absolute cell numbers (D,F) for pro-B , pre-B, and immature B-cell populations. Graphs depict means from 5 mice in each group ± SE (in 2 different experiments). Reference values for pro-B , pre-B, and immature B cell numbers in young mice are shown in Figure 2. (G-H) IL-7–driven BM cultures containing 1% fresh serum from young or old mice in the absence or presence of hIGF-1 were prepared. After 5 days, cells were harvested, counted, and stained to quantify B-cell numbers. Shown are representative results from a single experiment (G) and absolute B-cell counts (H) in the cultures. Graph depicts mean from 4 experiments ± SE.
B-cell lymphopoiesis in aging is regulated by IGF-1. (A) Sera collected from the indicated mice were analyzed for IGF-1 by enzyme-linked immunosorbent assay (ELISA). Shown are results for individual mice and group means (n = 5 for each group). (B) IL-7–driven BM cultures were prepared, replacing the FCS in media with 1% serum from young mice in the presence or absence of goat–anti-mouse IGF-1 (50 ng/mL). After 5 days, cells were harvested, counted, and stained to quantify B-cell numbers. Graph depicts mean from 4 experiments ± SE. (C-F) Old mice were subcutaneously injected with human GH (hGH) (C-D) or with human IGF-1 (hIGF-1) (E-F) for 10 days. Control mice were injected with phosphate-buffered saline (PBS). One day after the last injection, we analyzed the BM of the mice for B-cell lymphopoiesis (described in the Figure 2 legend) with gates marked for pro-B , pre-B, and immature B cells. Shown are representative plots for a single mouse from each group (C,E) and absolute cell numbers (D,F) for pro-B , pre-B, and immature B-cell populations. Graphs depict means from 5 mice in each group ± SE (in 2 different experiments). Reference values for pro-B , pre-B, and immature B cell numbers in young mice are shown in Figure 2. (G-H) IL-7–driven BM cultures containing 1% fresh serum from young or old mice in the absence or presence of hIGF-1 were prepared. After 5 days, cells were harvested, counted, and stained to quantify B-cell numbers. Shown are representative results from a single experiment (G) and absolute B-cell counts (H) in the cultures. Graph depicts mean from 4 experiments ± SE.
Because IGF-1 is secreted in response to GH, we next tested whether intervening in the GH/IGF-1 axis enhances B-cell lymphopoiesis in old mice. B-cell lymphopoiesis was significantly enhanced in old mice upon administration of GH (Figure 3C-D) and upon administration of IGF-1 (Figure 3E-F), as reflected by the increased frequency and absolute cell number of pro-B, pre-B, and immature B cells. Further analysis revealed increased numbers of both pro-B and pre-B cells in old mice injected with IGF-1 (supplemental Figure 4). We show that supplementing serum from old mice with IGF-1 circumvents its inhibitory effects in BM cultures (Figure 2F-I) and significantly enhances B-cell lymphopoiesis, as revealed by both cell number and percentage of IgM-expressing cells (Figure 3G-H). These findings suggest that suppression of B-cell lymphopoiesis in aging results from a reduced level of IGF-1, whose level in the circulation is regulated by peripheral B cells.
Regulation of IGF-1 by peripheral B cells in aging is mediated by TNF-α through IGFBP-1
We next sought a mechanism by which peripheral B cells might downregulate the abundance of IGF-1 in old mice. One possible mechanism is through secretion of the pro-inflammatory cytokine TNF-α, because production of this cytokine by unstimulated B cells is markedly increased in old mice32-34 (Figure 4). Consistent with this notion, administration of TNF-α in rodents suppresses circulatory IGF-1,35 whereas treating patients with anti–TNF-α enhances IGF-1.36,37 In agreement with previous studies,38,39 we found that TNF-α was significantly increased in plasma of old mice relative to that of young mice. However, in old mice that were treated for B-cell depletion, levels of TNF-α in plasma were significantly reduced and were not different from those found in young mice (Figure 4A). A longitudinal study of individual old mice revealed a dramatic decrease in plasma TNF-α within 14 days of B-cell depletion (Figure 4B). To further confirm this, we quantified production of TNF-α by peripheral B cells. In agreement with earlier studies,32,34 we found that B cells from old mice express larger amounts of TNF-α messenger RNA and secrete more TNF-α when cultured in vitro compared with B cells from young mice. However, the abundance of TNF-α messenger RNA and TNF-α secretion by B cells from old B-cell–depleted mice were significantly reduced and were not different from those of B cells from young mice (Figure 4C-D). We concluded that TNF-α produced by peripheral B cells controls IGF-1 production and consequentially tunes B-cell lymphopoiesis in the BM of old mice. Depletion of old B cells is followed by repopulation of the peripheral B-cell compartment with newly generated B cells that produce smaller amounts of TNF-α, which may account for the increase in IGF-1 and reactivation of B-cell lymphopoiesis.
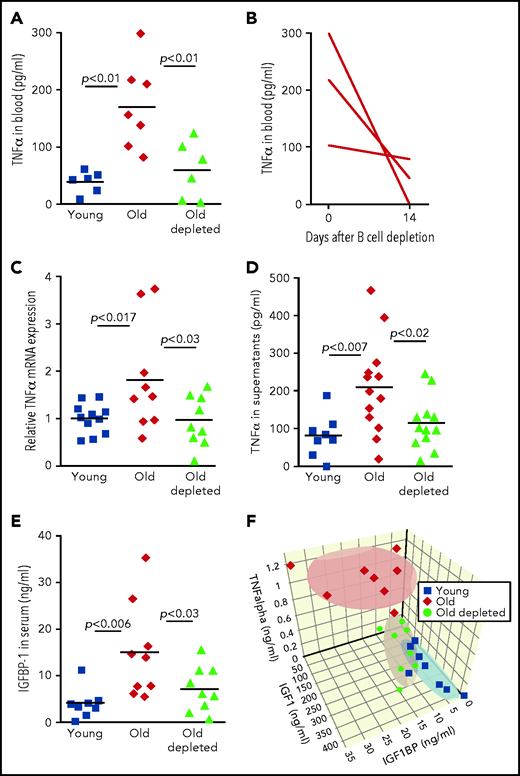
Regulation of IGF-1 by peripheral B cells in aging is mediated by TNF-α through IGFBP-1. (A) Sera collected from the indicated mice were analyzed for TNF-α by ELISA. Shown are results for individual mice and group means (n = 6-7 for each group). (B) Sera were collected from old hCD20Tg mice before and 14 days after B-cell depletion and analyzed for levels of TNF-α by ELISA. Shown are longitudinal results for individual mice (n = 3). (C) Purified splenic B cells from the indicated mice were analyzed for relative expression of TNF-α messenger RNA (mRNA) by quantitative polymerase chain reaction (qPCR) normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Graph depicts results for individual mice (n = 9-11) and group mean ± SE. (D) Purified splenic B cells from the indicated mice were cultured in vitro for 24 hours. Supernatants were collected and analyzed for TNF-a by ELISA. Shown are results for individual mice and group means (n = 8-13 for each group). (E) Sera collected from the indicated mice were analyzed for IGFBP-1 by ELISA. Shown are results for individual mice and group means (n = 8-9 for each group). (F) Measurements of IGF-1, TNF-α, and IGFBP-1 for individual mice from the indicated groups were plotted in a 3-dimensional chart. Each group is clustered with a color-matched covariance ellipsoid centered around the mean of each group.
Regulation of IGF-1 by peripheral B cells in aging is mediated by TNF-α through IGFBP-1. (A) Sera collected from the indicated mice were analyzed for TNF-α by ELISA. Shown are results for individual mice and group means (n = 6-7 for each group). (B) Sera were collected from old hCD20Tg mice before and 14 days after B-cell depletion and analyzed for levels of TNF-α by ELISA. Shown are longitudinal results for individual mice (n = 3). (C) Purified splenic B cells from the indicated mice were analyzed for relative expression of TNF-α messenger RNA (mRNA) by quantitative polymerase chain reaction (qPCR) normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Graph depicts results for individual mice (n = 9-11) and group mean ± SE. (D) Purified splenic B cells from the indicated mice were cultured in vitro for 24 hours. Supernatants were collected and analyzed for TNF-a by ELISA. Shown are results for individual mice and group means (n = 8-13 for each group). (E) Sera collected from the indicated mice were analyzed for IGFBP-1 by ELISA. Shown are results for individual mice and group means (n = 8-9 for each group). (F) Measurements of IGF-1, TNF-α, and IGFBP-1 for individual mice from the indicated groups were plotted in a 3-dimensional chart. Each group is clustered with a color-matched covariance ellipsoid centered around the mean of each group.
Yet abundance of bioactive IGF-1 in the circulation is primarily regulated by IGF-1–binding proteins (IGFBPs).40 Of the 6 IGFBPs, expression of only IGFBP-1 is increased upon infusion of TNF-α.41 IGFBP-1 is produced primarily in the liver,42 and in several developmental models, it was found to bind IGF-1 and to inhibit its activity.43,44 To test for a role of IGFBP-1 in mediating the TNF-α/IGF-1 crosstalk, we quantified IGFBP-1 levels in the circulation of young, old, and old B-cell–depleted mice. We found that circulatory IGFBP-1 significantly increases by about threefold with aging. After B-cell depletion, IGFBP-1 decreased and was not significantly different from that in young mice (Figure 4E). Collectively, these findings suggest that in old mice, B-cell lymphopoiesis in the BM is regulated by peripheral B cells through a TNF-α/IGFBP-1/IGF-1 axis. To further support this notion, we created a 3-dimensional chart (Figure 4F), in which individual mice were plotted on the basis of their abundance of IGF-1, TNF-α, and IGFBP-1; we note that mice clustered in ellipsoids for each group (young, old, or old B-cell–depleted). Figure 4F reveals that high levels of TNF-α correlate with high levels of IGFBP-1 and low levels of IGF-1 and that although clusters of young and old B-cell–depleted groups overlapped, the old cluster was distinct and stands alone.
A role for the TNF-α/IGFBP-1/IGF-1 axis in regulating B-cell lymphopoiesis in older humans
To explore the role of the TNF-α/IGFBP-1/IGF-1 axis in regulating B-cell lymphopoiesis in older humans, we examined human plasma samples collected from 3 cohorts: healthy young volunteers (age 18-35 years), healthy older volunteers (age 55 years or older), and older patients with B-cell non-Hodgkin lymphoma (age 55 years or older) who were previously treated with rituximab B-cell depletion therapy without subsequent disease progression (old-depletion). Plasma from these individuals was collected in the course of a clinical trial (IRB 3097), and details of patients, as well as the inclusion and exclusion criteria have been described.14 In agreement with previous publications,24-28 plasma IGF-1 decreased with aging. However, as we observed for old mice (Figure 3), plasma IGF-1 increased in older patients after B-cell depletion to become almost the same as that in the young cohort (Figure 5A). Furthermore, peripheral B cells from older humans produced larger amounts of TNF-α compared with B cells from young humans, whereas peripheral B cells from older humans with B-cell depletion produced TNF-α in an amount that was almost the same as that in the young group (Figure 5C). In accordance with these observations, we found that IGFBP-1 increased with aging in human plasma but decreased after B-cell depletion and became similar to that detected in plasma from the young humans (Figure 5B). On the basis of these results, we suggest that in both mouse and human aging, peripheral B cells regulate B-cell lymphopoiesis in the BM through cross-talk in which the TNF-α/IGFBP-1/IGF-1 axis has a major role.
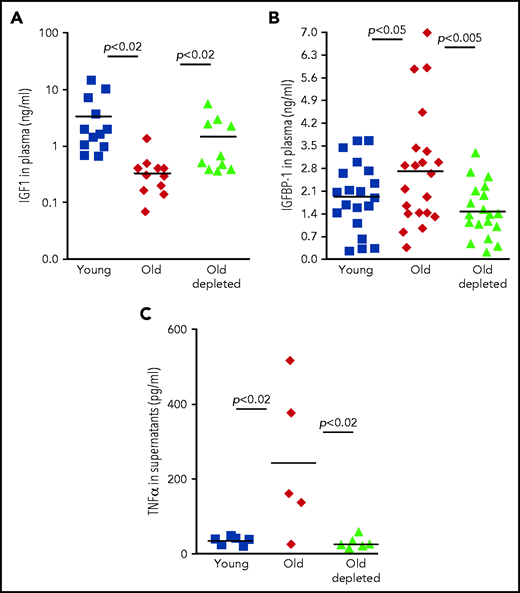
The TNF-α/IGFBP-1/IGF-1 axis in regulating B-cell lymphopoiesis in older humans. (A-B) Plasma samples were collected from healthy young or older humans or older patients with lymphoma treated for B-cell depletion (old depleted) (see the Methodology section of NCT00863187). Plasma samples were analyzed for IGF-1 (A) and IGFBP-1 (B) by ELISA. Shown are results for individual patients and group means (indicated by thick solid horizontal line) (n = 9-18 for each group). (C) Peripheral blood B cells were collected from patients from the indicated human cohorts and cultured in vitro for 24 hours. Supernatants were collected and analyzed for TNF-a by ELISA. Shown are results for individual patients and group means (n = 5-6 for each group).
The TNF-α/IGFBP-1/IGF-1 axis in regulating B-cell lymphopoiesis in older humans. (A-B) Plasma samples were collected from healthy young or older humans or older patients with lymphoma treated for B-cell depletion (old depleted) (see the Methodology section of NCT00863187). Plasma samples were analyzed for IGF-1 (A) and IGFBP-1 (B) by ELISA. Shown are results for individual patients and group means (indicated by thick solid horizontal line) (n = 9-18 for each group). (C) Peripheral blood B cells were collected from patients from the indicated human cohorts and cultured in vitro for 24 hours. Supernatants were collected and analyzed for TNF-a by ELISA. Shown are results for individual patients and group means (n = 5-6 for each group).
B-cell lymphopoiesis and IGF-1 are suppressed in patients with chronic inflammation, whereas treatment with anti–TNF-α is associated with increased IGF-1 and restored B-cell lymphopoiesis
To confirm the role of the TNF-α/IGFBP-1/IGF-1 axis in regulating B-cell lymphopoiesis in humans, we performed a cross-sectional study to quantify the fraction of transitional B cells in blood samples from patients with inflammatory joint disease (IJD). We chose to focus on this group of patients, because chronic inflammation is known to promote premature aging of tissues and age-related diseases.45-47 Moreover, transitional B cells may be used to quantify lymphopoiesis, because they represent newly generated B cells in transition from the BM to the spleen to complete their maturation.48,49 Patients with chronic IJD such as rheumatoid arthritis, psoriatic arthritis, or spondyloarthropathy are characterized by an abundance of TNF-α in the blood, especially in the active stage of IJD.50 Patients with IJD, either naïve to anti–TNF-α (IJD-NB [nonbiologic agents]) or treated with anti–TNF-α (IJD–TNF-α), were compared with controls who did not have inflammatory disease, including healthy volunteers and patients with degenerative osteoarthritis whose disease is not associated with a systemic inflammatory response. Data in Table 1 include clinical details, number of patients, age, and sex. There were no significant differences in age and sex between groups. Blood cells were analyzed by flow cytometry, and the frequency of transitional and mature cells was determined as shown in Figure 6A. In a comparison of patients with IJD-NB and controls, the IJD was associated with a 2.9-fold reduction in the transitional B-cell compartment (P = .001). In this comparison, we observed that treatment of patients who had IJD with anti-TNF-α therapy was associated with a 2.3-fold increase in the transitional B-cell compartment (P < .005) relative to those treated with non-biologic agents alone (IJD-NB) (Figure 6B).
Baseline characteristics of controls and patients with IJD with or without anti-TNF-α therapy
| Characteristic | Healthy (n = 4) | IJD-NB (n = 14) | IJD-TNF-α (n = 19) | |||
|---|---|---|---|---|---|---|
| Parameter | No. | Parameter | No. | Parameter | No. | |
| Median age, y (range) | 58 (40-76) | 52 (44-60) | 51 (43-59) | |||
| Sex (F:M) | 3:1 | 4:1 | 2:1 | |||
| Basic condition | Osteoarthritis | 2 | RA | 9 | RA | 15 |
| PsA | 3 | PsA | 3 | |||
| No joint disease | 2 | SPA | 2 | SPA | 1 | |
| Characteristic | Healthy (n = 4) | IJD-NB (n = 14) | IJD-TNF-α (n = 19) | |||
|---|---|---|---|---|---|---|
| Parameter | No. | Parameter | No. | Parameter | No. | |
| Median age, y (range) | 58 (40-76) | 52 (44-60) | 51 (43-59) | |||
| Sex (F:M) | 3:1 | 4:1 | 2:1 | |||
| Basic condition | Osteoarthritis | 2 | RA | 9 | RA | 15 |
| PsA | 3 | PsA | 3 | |||
| No joint disease | 2 | SPA | 2 | SPA | 1 | |
Baseline characteristic of all patients included in cross-sectional comparison of percent of transitional B cells. The IJD patients had rheumatoid arthritis (RA) or psoriatic arthritis (PsA), or spondyloarthropathy (SPA). Differences in patient age were not found to be a statistically significant confounder.
F, female; M, male.
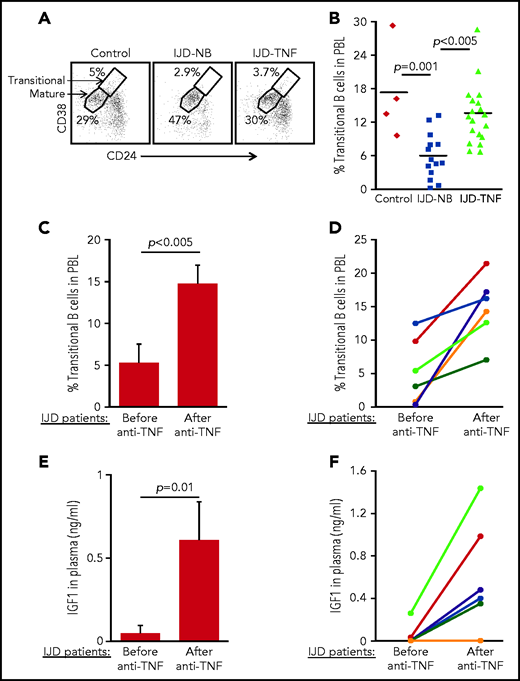
Transitional B cells in patients with IJD treated or not treated with anti–TNF-α. (A-B) Cross-sectional analysis of transitional B cells in healthy controls (n = 4) and patients with IJD treated with TNF-α (IJD-TNF-α, n = 19) or naïve to anti-TNF-α (IJD-NB, n = 14). Blood cells were analyzed by flow cytometry and mature (CD19+/CD24lo/med/CD38lo/med) and transitional (CD19+/CD24hi/CD38hi) B-cell frequency was determined. (A) Representative analysis for a single patient from each group gated on CD19+ cells. Shown are gates for mature and transitional B cells indicated by arrows. (B) Frequency of transitional B cells. Quantity of transitional B cells was calculated as the fraction of transitional B cells divided by the sum of transitional and mature B cells. Shown are results for individual patients and group means. (C-D) Longitudinal analysis of transitional B-cell ratio of patients with IJD undergoing treatment with anti–TNF-α. (C) Transitional B-cell ratio averages in patient subset (n = 6) analyzed upon initiation of anti–TNF-α therapy and again after 3 months of anti–TNF-α therapy. Results are expressed as mean ± SE. (D) Longitudinal analysis for individual patients with IJD transitional B-cell ratios before and after anti–TNF-α therapy. (E-F) Longitudinal analysis of plasma IGF-1 in patients with IJD undergoing anti–TNF-α therapy. (E) Plasma IGF-1 averages in the patient subset (n = 6) analyzed upon initiation of anti–TNF-α therapy and again after 3 months of anti–TNF-α therapy. Results are expressed as mean ± SE. (F) Longitudinal analysis for individual patients with IJD transitional B-cell ratios before and after anti–TNF-α therapy.
Transitional B cells in patients with IJD treated or not treated with anti–TNF-α. (A-B) Cross-sectional analysis of transitional B cells in healthy controls (n = 4) and patients with IJD treated with TNF-α (IJD-TNF-α, n = 19) or naïve to anti-TNF-α (IJD-NB, n = 14). Blood cells were analyzed by flow cytometry and mature (CD19+/CD24lo/med/CD38lo/med) and transitional (CD19+/CD24hi/CD38hi) B-cell frequency was determined. (A) Representative analysis for a single patient from each group gated on CD19+ cells. Shown are gates for mature and transitional B cells indicated by arrows. (B) Frequency of transitional B cells. Quantity of transitional B cells was calculated as the fraction of transitional B cells divided by the sum of transitional and mature B cells. Shown are results for individual patients and group means. (C-D) Longitudinal analysis of transitional B-cell ratio of patients with IJD undergoing treatment with anti–TNF-α. (C) Transitional B-cell ratio averages in patient subset (n = 6) analyzed upon initiation of anti–TNF-α therapy and again after 3 months of anti–TNF-α therapy. Results are expressed as mean ± SE. (D) Longitudinal analysis for individual patients with IJD transitional B-cell ratios before and after anti–TNF-α therapy. (E-F) Longitudinal analysis of plasma IGF-1 in patients with IJD undergoing anti–TNF-α therapy. (E) Plasma IGF-1 averages in the patient subset (n = 6) analyzed upon initiation of anti–TNF-α therapy and again after 3 months of anti–TNF-α therapy. Results are expressed as mean ± SE. (F) Longitudinal analysis for individual patients with IJD transitional B-cell ratios before and after anti–TNF-α therapy.
To show the direct effect of anti–TNF-α treatment in restoring B-cell lymphopoiesis in patients with IJD, we performed a longitudinal study wherein we analyzed transitional B cells in a subset of patients with IJD who had not been exposed to anti-TNF-α (Table 2) before initiation of anti–TNF-α therapy and 3 months after initiation of anti–TNF-α therapy. In this study, a 3-month course of anti–TNF-α was associated with a 2.8-fold increase in the transitional B-cell compartment (Figure 6C). Moreover, an increase in the transitional B-cell compartment was observed in all patients (100%) and ranged from 1.3- to 52-fold (Figure 6D). Consistent with these results, anti–TNF-α treatment in patients with IJD was associated with a 12.6-fold increase in plasma IGF-1 (Figure 6E), which was observed in 5 (83%) of 6 patients, whereas 1 patient showed no change (Figure 6F). These results suggest that B-cell lymphopoiesis is suppressed in patients with chronic inflammation and that treatment with anti–TNF-α enhances plasma IGF-1 to restore B-cell lymphopoiesis in patients with IJD.
Baseline characteristics of patients with IJD analyzed before and after treatment with anti–TNF-α
| Characteristic | IJD-NB/IJD-TNF-α (n = 6) |
|---|---|
| Age, y | 45 |
| Sex (F:M) | 3:1 |
| RA | 4 |
| PsA | 1 |
| SPA | 1 |
| Characteristic | IJD-NB/IJD-TNF-α (n = 6) |
|---|---|
| Age, y | 45 |
| Sex (F:M) | 3:1 |
| RA | 4 |
| PsA | 1 |
| SPA | 1 |
Baseline characteristic of all patients included in longitudinal comparison of percent of transitional B cells before and 3 months after initiation of anti–TNF-α therapy.
Discussion
Regulation of B-cell hematopoiesis is not yet well understood. This question is of particular importance in aging, because declining B-cell hematopoiesis in the BM and dramatic alterations in peripheral B-cell subsets and the B-cell repertoire in older individuals result in increased morbidity and mortality among people in this age group. In this study, we used older mice and a cohort of older humans to show that in aging, peripheral B cells suppress B-cell lymphopoiesis in the BM by reducing the amount of IGF-1 in the circulation, and that this alteration is mediated by an axis composed of a pro-inflammatory cytokine (TNF-α) and an important endocrine system (IGFBP-1/IGF-1). We suggest that B-cell homeostasis in both human and mouse aging is regulated by cross-talk between peripheral B cells and progenitors in the BM, and that this cross-talk is mediated by a TNF-α/IGFBP-1/IGF-1 immune-endocrine axis.
We show here that in old mice, peripheral B cells have extended survival and are able to suppress B-cell lymphopoiesis. By using a B-cell depletion mouse model23 and our cohort of older human patients with lymphoma who were treated with B-cell depletion,14 we identified IGF-1 as a mediator of the cross-talk between peripheral B cells and progenitors in aging. We found that IGF-1, whose level in plasma of both human and rodents decreases with aging,26-28 is significantly increased after B-cell depletion, suggesting that the decrease of IGF-1 in aging is mediated by peripheral B cells. IGF-1 signaling is known to promote hematopoiesis in general51,52 and to potentiate IL-7–mediated growth and differentiation of B lineage cells.29-31 However, because the competence of BM progenitors to respond to IGF-1 does not change with aging,53 it seems that reduction of IGF-1, but not responsiveness to IGF-1, is a major cause for suppressing B-cell lymphopoiesis in aging. This argument is supported by our results showing that administration of GH or IGF-1 to old mice reactivated B-cell lymphopoiesis and that supplementing BM cultures containing serum from old mice with IGF-1 restored growth and expansion of B-cell lineage progenitors (Figure 4).
Ageing is associated with increased inflammatory activity in the blood, and this study proposes that this inflammatory activity is a major cause for the reduced IGF-1. TNF-α is a pro-inflammatory cytokine that increases in the circulation with aging.54 On the basis of our results, we suggest that, in older humans and mice, peripheral B cells are a major source of the age-related increase of TNF-α in the circulation. Supporting this claim are earlier studies showing that age-associated B cells (ABCs) that accumulate with aging11 produce TNF-α,32-34 that about 50% of TNF-α–producing lymphocytes in the spleen are B cells, and that B-cell–specific ablation of TNF-α ameliorates TNF-α levels in an inflammatory mouse model.55 TNF-α produced locally in the BM by ABCs was previously shown to suppress B-cell lymphopoiesis through direct induction of apoptosis in pro-B cells.34 Our study suggests that in addition to this local effect, the increased production of TNF-α by peripheral B cells in aging has a major systemic effect in regulating B-cell lymphopoiesis by modifying levels of IGF-1. Several studies show that administration of TNF-α in rodents suppresses IGF-1 in circulation35,56,57 and that treatment with anti–TNF-α in patients enhances IGF-1,36,37 thereby supporting the existence of a regulatory systemic feedback between TNF-α and IGF-1 in aging. In accordance with this notion, we found that B-cell depletion significantly reduces TNF-α in the circulation in both mice and humans (Figures 5 and 7). This finding is consistent with our previous study showing that B-cell depletion eliminates ABCs and replaces the peripheral compartment with newly generated B cells.14 Moreover, we show that increased TNF-α is associated with suppression of IGF-1 and B-cell lymphopoiesis in a clinical study performed in patients with IJD, and both are restored upon treatment with anti-TNF-α (Figure 6). These findings further support the existence of major systemic effects for TNF-α in regulating B-cell lymphopoiesis through altering levels of IGF-1.
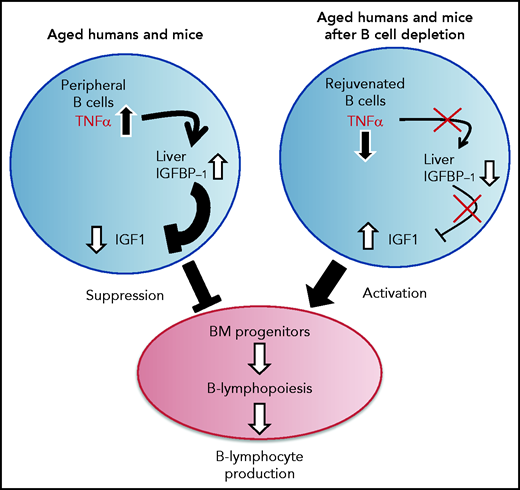
A proposed model for the function of the TNF-α/IGFBP-1/IGF-1 axis in regulating B-cell lymphopoiesis in aging. B-cell lymphocyte production in the BM is regulated in aging by the level of IGF-1 in the circulation. The long-lived B cells, which accumulate in the periphery with aging, produce large amounts of the pro-inflammatory cytokine TNF-α, which stimulates the liver to increase production of IGFBP-1. The increased IGFBP-1 binds IGF-1 and sequesters its activity in the BM, resulting in a suppressed B-cell lymphopoiesis. Upon B-cell depletion, the peripheral compartment is replenished with newly generated naïve B cells that secrete small amounts of TNF-α, resulting in a decline in the plasma level of TNF-α. The reduced TNF-α is followed by a decrease in IGFBP-1 and a consequential increase in IGF-1 and reactivation of B-cell lymphopoiesis in the BM.
A proposed model for the function of the TNF-α/IGFBP-1/IGF-1 axis in regulating B-cell lymphopoiesis in aging. B-cell lymphocyte production in the BM is regulated in aging by the level of IGF-1 in the circulation. The long-lived B cells, which accumulate in the periphery with aging, produce large amounts of the pro-inflammatory cytokine TNF-α, which stimulates the liver to increase production of IGFBP-1. The increased IGFBP-1 binds IGF-1 and sequesters its activity in the BM, resulting in a suppressed B-cell lymphopoiesis. Upon B-cell depletion, the peripheral compartment is replenished with newly generated naïve B cells that secrete small amounts of TNF-α, resulting in a decline in the plasma level of TNF-α. The reduced TNF-α is followed by a decrease in IGFBP-1 and a consequential increase in IGF-1 and reactivation of B-cell lymphopoiesis in the BM.
The regulation of circulatory IGF-1 is primarily mediated by IGFBPs, of which IGFBP-1 displays rapid, dynamic regulation in vivo and functions as a primary determinant of free IGF-1 levels in serum.58 IGFBP-1 is primarily produced in the liver,42 and its expression increases in vivo upon administration of TNF-α.41,57,59 Our study proposes that TNF-α, through induction of IGFBP-1 synthesis, suppresses circulatory IGF-1 and the consequent B-cell lymphopoiesis. This is supported by earlier studies showing that IGFBP-1 increases with aging60 and that hepatocytes from old rats produce more IGFBP-1 in response to inflammatory signals.61 In agreement with this, we found that IGFBP-1 is increased in older humans and mice but is significantly reduced after B-cell depletion (Figures 4 and 5).
Our study proposes that an immune-endocrine regulatory circuit is activated in aging to mediate the cross-talk between peripheral B cells and progenitors in the BM. A schematic diagram suggesting how this TNF-α/IGFBP-1/IGF-1 axis controls B-cell hematopoiesis in aging and upon B-cell depletion is shown in Figure 7. Accordingly, peripheral B cells in older humans and mice secrete increased amounts of TNF-α, which acts to increase production of IGFBP-1. The increased IGFBP-1 binds IGF-1 and sequesters its activity in promoting B-cell lymphopoiesis in the BM. Upon B-cell depletion and rejuvenation of the peripheral compartment with newly generated naïve B cells that secrete small amounts of TNF-α, plasma TNF-α decreases, followed by a decrease in IGFBP-1 and a consequential increase in IGF-1 and reactivation of B-cell lymphopoiesis. Importantly, identifying the TNF-α/IGFBP-1/IGF-1 axis and its function in both humans and mice signifies this finding and suggests that an external intervention to rejuvenate the peripheral B-cell compartment in aging can be pursued by targeting components of this axis rather than depleting the entire peripheral B-cell compartment. Indeed, as we show here, administration of IGF-1 to old mice stimulated a significant increase in B-cell hematopoiesis, and administration of anti–TNF-α therapy to patients with IJD restored B-cell lymphopoiesis and relieved the TNF-α–mediated suppression of B-cell regeneration.
Paradoxically, in contrast to its positive effect on B-cell regeneration, as shown here, the blood concentration of IGF-1 negatively correlates with lifespan.62 It is now thought that reduced activity of the GH/IGF-1 axis protects against cancer and diabetes,63 both major age-related diseases. Reducing the GH/IGF-1 signaling in an attempt to delay aging is achieved by restricting calories64 as well as through pharmaceutical inhibition,26 although the effects are controversial.65 However, increasing lifespan by reducing IGF-1 may be costly. One major cost that we show here is the suppression of B-cell lymphopoiesis. This is in agreement with an earlier study showing that dietary restriction suppresses lymphoid differentiation in the BM in aging.66 Thus, the benefits of reducing IGF-1 may turn into an increased risk of morbidity and mortality because of infectious diseases or other evolving or new pathogens. Aging is also associated with increased production of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1 in the circulation, a process referred to as “inflammaging,” which is thought to accelerate biological aging and worsen age-related diseases.67 Because we have showed that peripheral B cells are major producers of TNF-α in aging, it would be interesting to determine whether B-cell depletion ameliorates inflammaging and prevents or delays the development of age-related diseases.
Acknowledgments
The authors thank Angel Porgador, and Roi Gazit, from Ben-Gurion University for thoughtful discussions and assistance, Rita Erlich and Tsofnat Margi for coordinating the clinical study 0643-15 RMB, Eleyah Melamed for graphical assistance, and Garrett Friedman, from Technion for technical assistance and human data collection in clinical study IRB 0643-15 RMB.
This work was supported by grants from the Israel Ministry of Science, Technology and Space (MOST), the Wolens Gerontology Research Fund, the Colleck Research Fund, and the D. Dan & Betty Kahn Foundation’s gift to the University of Michigan–Weizmann Institute–Technion–Israel Institute of Technology Collaboration for Research.
Authorship
Contribution: R.D. and D.M. performed and designed experiments, analyzed the data, and wrote the manuscript; D.B., E.B., S.Z.-R., A.N., D.B., and T.W.-C. performed experiments and/or contributed to data analysis; and Y.B.-M., A.B.-G., I.A., and A.S. contributed to acquisition and analysis of clinical data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Doron Melamed, Rappaport Faculty of Medicine, Department of Immunology, Technion-Israel Institute of Technology, Bat Galim, Haifa 31096, Israel; e-mail: melamedd@technion.ac.il.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal