

Key Points
CMV exposure results in a persistently expanded CD57+/CD27− CD4+ Th1 subset expressing granzyme B, interferon-γ, and tumor necrosis factor.
CD57+/CD27− CD4+ T cells are associated with impaired T-cell receptor diversity and a reduction in MHC class II+ antigen-presenting cells.

Abstract
Donor and recipient cytomegalovirus (CMV) serostatus correlate with transplant-related mortality that is associated with reduced survival following allogeneic stem cell transplant (SCT). Prior epidemiologic studies have suggested that CMV seronegative recipients (R–) receiving a CMV-seropositive graft (D+) experience inferior outcomes compared with other serostatus combinations, an observation that appears independent of viral reactivation. We therefore investigated the hypothesis that prior donor CMV exposure irreversibly modifies immunologic function after SCT. We identified a CD4+/CD57+/CD27– T-cell subset that was differentially expressed between D+ and D– transplants and validated results with 120 patient samples. This T-cell subset represents an average of 2.9% (D–/R–), 18% (D–/R+), 12% (D+/R–), and 19.6% (D+/R+) (P < .0001) of the total CD4+ T-cell compartment and stably persists for at least several years post-SCT. Even in the absence of CMV reactivation post-SCT, D+/R– transplants displayed a significant enrichment of these cells compared with D–/R– transplants (P = .0078). These are effector memory cells (CCR7–/CD45RA+/−) that express T-bet, Eomesodermin, granzyme B, secrete Th1 cytokines, and are enriched in CMV-specific T cells. These cells are associated with decreased T-cell receptor diversity (P < .0001) and reduced proportions of major histocompatibility class (MHC) II expressing classical monocytes (P < .0001), myeloid (P = .024), and plasmacytoid dendritic cells (P = .0014). These data describe a highly expanded CD4+ T-cell population and putative mechanisms by which prior donor or recipient CMV exposure may create a lasting immunologic imprint following SCT, providing a rationale for using D– grafts for R– transplant recipients.
Introduction
Cytomegalovirus (CMV ) remains an important cause of morbidity and mortality after allogeneic hematopoietic stem cell transplantation (SCT).1,2 Despite advances in antiviral prophylaxis and treatment, recipient and donor CMV serological status remain important risk factors that are correlated with transplant-related complications and long-term survival, and the observed adverse impact of CMV on transplant-related mortality is well documented.3,4 For example, in a study of >49 000 allogeneic SCT patients, seronegative patients receiving seropositive unrelated donor grafts showed decreased overall survival compared with those receiving grafts from seronegative donors.5 In another study of 1750 non–T-cell-depleted transplants, CMV seropositive donors with CMV seronegative recipients had a significantly higher risk for mortality, including a higher incidence of bacterial and fungal infections, compared with other donor/recipient CMV serostatus combinations, even after controlling for CMV disease.6 Thus, CMV seropositivity may generate broad defects in pathogen-specific immunity that persists even after clinically significant CMV infection has resolved.
CMV reactivation after SCT is known to skew the immune repertoire. A study of 17 allogeneic SCT patients demonstrated that CMV reactivation affects CD8+ T-cell reconstitution and is associated with a large expansion of CD8+ effector memory cells and subsequent reduction in overall T-cell receptor β (TCR-β) diversity.7 Another study demonstrated that in 20 allogeneic SCT patients following CMV reactivation after transplant, CMV-specific CD4+ T cells also reconstituted in parallel with CMV-specific CD8+ T, but at a notably lower magnitude, and included an expanded effector memory subset.8 Thus, CMV reactivation appears to significantly alter both CD4+ and CD8+ T-cell immune reconstitution after SCT.
Here, we explore the immunologic phenomenon that may explain prior clinical observations demonstrating a negative association between donor CMV seropositivity and long-term clinical outcomes independent of CMV disease and reactivation. In this context, we demonstrate that seronegative recipients receiving grafts from seropositive donors displayed significant enrichment of a highly differentiated effector memory CD4+ T-cell subset characterized by the expression of CD57+ that persists over many years after SCT. We functionally characterize this CD4+ T-cell subset, demonstrate its enrichment in CMV-specific T cells, and show that it is associated with a reduction in TCR-β diversity and the frequency of major histocompatibility complex (MHC) class II expressing antigen-presenting cells.
Materials and methods
Human samples
Human peripheral blood mononuclear cell (PBMC) and plasma samples were obtained from the Chronic Graft-vs-Host Disease Consortium, which includes patients with and without chronic graft-versus-host disease (cGVHD), and QIMR Berghofer and the Royal Brisbane and Women’s Hospital. Ethics approval was obtained from the Human Research Ethics Committees at respective institutions, and the study was conducted in accordance with the Declaration of Helsinki.
Multiparameter flow cytometry
A comprehensive list of antibody reagents used is shown in supplemental Table 1, available on the Blood Web site. Intracellular staining was performed using the Foxp3/Transcription Factor Staining Buffer Set (eBiosciences). Cytokine staining was performed following 4-hour stimulation with phorbol myristate acetate (Sigma-Aldrich) 50 ng/mL, ionomycin (Sigma-Aldrich) 1 μg/mL, and Brefeldin A (BioLegend). Flow cytometry was performed on a BD FACSymphony A3 (BD Biosciences) or a BD LSR Fortessa (BD Biosciences) using BD FACSDiva (8.0.1) software and analyzed using FlowJo.
CMV serology and PCR
Plasma samples from donors and recipients were tested for CMV-specific immunoglobulin G (IgG) by automated CMV enzyme-linked immunosorbent assay before transplant, and CMV polymerase chain reaction (PCR) assays following transplant were performed by respective institutions. CMV screening was conducted weekly until day 100 posttransplant, with follow-up screening depending on presence or absence of GVHD, use of steroids, and prior need for CMV-directed therapy. CMV PCR threshold for detection ranged from 50 to 183 IU/mL across all institutions, below which CMV may be detected, but not quantified. CMV reactivation was defined as any detectable result in the PCR assay. The threshold for treatment varied across institutional practices but ranged from 150 to 1000 IU/mL.
Time-of-flight mass cytometry
Time-of-flight mass cytometry was performed by the Vanderbilt University Cancer and Immunology Core (CIC) using the CIC-002 Human T-cell and CIC-004 Human Myeloid Panels (for antibody list, see supplemental Figure 1A-B). Visualization of single-cell data was performed using uniform manifold approximation and projection (UMAP).9 Cluster analysis was performed using Phenograph,10 and cell subset characterization was performed using Marker Enrichment Modeling.11 Implementation of these methods were executed on R Statistical Software (version 3.5.3; Foundation for Statistical Computing, Vienna, Austria).
Plasma cytokine analysis
All human plasma samples were obtained from the Chronic GVHD Consortium. Plasma cytokine analysis was performed using BD Biosciences Cytometric Bead Array kits and included granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-1β (IL-1β), IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12p40, IL-13, IL-17A, interferon-γ (IFN-γ), monocyte chemoattractant protein-1, and tumor necrosis factor (TNF).
Peptide pool stimulation and degranulation analysis
pp65, EBNA1, and EBNA2 peptide pools were purchased from JPT. All peptides were dissolved in dimethyl sulfoxide and stored at −20°C. Degranulation assays were performed by treating PBMC samples with brefeldin A, monensin, and CD107a antibody before stimulation with 1 μg/mL of relevant peptide. Multiparameter flow cytometry and intracellular cytokine staining was performed following a 4-hour incubation at 37°C as described previously.
MHC class II tetramer analysis
HLA-DRB1 tetramers were custom made by the National Institutes of Health tetramer core facility and assembled with CMV- and Epstein-Barr virus (EBV)-derived peptides (Genscript). These include HLA-DRB1*0701 bound to CMV-glycoprotein B-derived peptide DYSNTHSTRYV (HCMV gB 217-227), which is known to induce a strong CMV-CD4 response,12 HLA-DRB1*0701 bound to EBNA1-drived peptide VYGGSKTSLYNLRRGTALAI (EBNA1 509-528),13 HLA-DRB1*0301 bound to EBV EBNA2-drived peptide PAQPPPGVINDQQLHHLPSG (EBNA2 301-320),13 and HLA-DRB1*0101 bound to EBV EBNA1-drived peptide TSLYNLRRGTALA (EBNA1 515-527).14,15 Control tetramers with peptide PVSKMRMATPLLMQA (human class II-associated invariant chain peptide [CLIP] 87-101) was also provided. Tetramer staining was performed using a 1:750 dilution.
TCR/BCR sequencing and analysis
Total RNA was extracted and quantified (Qiagen) from unsorted peripheral blood mononuclear cells. TCR/B-cell receptor (BCR) sequencing was performed using the Archer Immunoverse TCR-β/γ and BCR IgH kits. Analysis was performed using molecular identifier groups-based error correction16 pipeline, VDJTools,17 and the Archer Analysis platform. Diversity scores were calculated using the inverse Simpson Index,18 which was implemented in R Statistical Software via the Immunarch package.
Statistical analysis
Analysis of clinical characteristics and flow cytometry was performed using GraphPad Prism v8.0.1. Baseline transplant characteristics were compared across CMV serostatus groups using 1-way analysis of variance (age) and χ2 tests (sex, disease, donor type, graft source, conditioning regimen, severity of acute, and cGVHD). The fraction of CD57+/CD27− T cells across CMV serostatus and with respect to CMV reactivation or treatment was analyzed using Kruskal-Wallis and Mann-Whitney tests. Temporal and repeated sample correlation of T cells was calculated using Spearman’s ρ (rs). Comparison of antibody staining intensity between groups was performed using Mann-Whitney tests. Correlation of T cells with other PBMC subsets and with TCR/BCR diversity was also calculated using Spearman’s ρ. Plasma cytokine profiling across CMV serostatus and immune cell reconstitution were analyzed using Kruskal-Wallis tests, and correlation of cytokine levels with fraction of T cells was calculated using Spearman’s ρ.
Results
Identification of CD57+/CD27– CD4+ T cells after transplantation
We screened peripheral blood samples from 12 patients from the Chronic GVHD Consortium at a median time point of 358 days after SCT (7 with CMV seropositive donors, 5 with CMV seronegative donors; supplemental Table 2) using time-of-flight mass cytometry panels CIC-002 (human T-cell emphasis; supplemental Figure 1A) and CIC-004 (human myeloid cell emphasis; supplemental Figure 1B). Only 1 cluster in the T-cell panel and none in the myeloid panel met a false discovery rate cutoff <0.2 (human T-cell panel cluster 15, P = .00051, q = 0.15, Figure 1A; myeloid panel, supplemental Figure 1C-D). This cluster was characterized by high expression of CD4 and CD57 and low expression of CD27 (Figure 1B), which was found in larger numbers in CMV-seropositive donors compared with CMV-negative donors (Figure 1C). CMV seropositivity in the non-SCT setting has been associated with the expansion of a CD4+/CD57+ T-cell subset,19 and thus, in the context of our screening results, we hypothesized that this cellular subset may play an important role in transplants with prior donor CMV exposure.
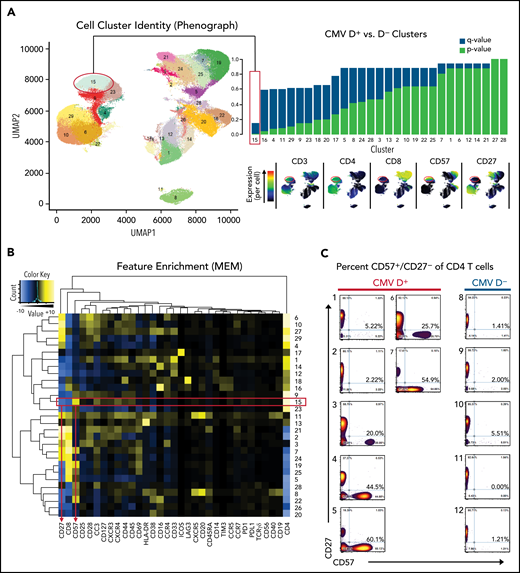
Identification of a CD57+/CD27− CD4+ T-cell population. (A) Clustered UMAP plot generated using 33 antibodies from the human T-cell panel (CIC-002) characterizing PBSC samples from 7 CMV donor-positive (D+) and 5 CMV donor-negative (D–) transplants show 29 distinct cell clusters across all samples. Differential frequency for each cluster between D+ and D– samples are shown with corresponding P (blue) and q values (red). Only 1 cluster (15) met an false discovery rate threshold of 0.2. Selected antibody results are shown to the right of the UMAP plot. (B) For each cluster, normalized antibody intensity is shown using Marker Enrichment Modeling (MEM). (C) 2 × 2 gating was performed for CD4+ T cells to emphasize the CD27/CD57 profile for D+ and D– groups.
Identification of a CD57+/CD27− CD4+ T-cell population. (A) Clustered UMAP plot generated using 33 antibodies from the human T-cell panel (CIC-002) characterizing PBSC samples from 7 CMV donor-positive (D+) and 5 CMV donor-negative (D–) transplants show 29 distinct cell clusters across all samples. Differential frequency for each cluster between D+ and D– samples are shown with corresponding P (blue) and q values (red). Only 1 cluster (15) met an false discovery rate threshold of 0.2. Selected antibody results are shown to the right of the UMAP plot. (B) For each cluster, normalized antibody intensity is shown using Marker Enrichment Modeling (MEM). (C) 2 × 2 gating was performed for CD4+ T cells to emphasize the CD27/CD57 profile for D+ and D– groups.
CD57+/CD27– donor T cells are associated with CMV serostatus after transplantation
We subsequently analyzed 120 patient samples, including 35 paired samples taken at follow-up, collected at a median time of 318 days post-SCT (25th-75th quartile, 155-585 days), with repeat samples taken at an average of 94 days after the initial sample (range, 28-218 days). Of the 85 unique patients evaluated, 23 were donor and recipient CMV-serostatus negative (D–/R–), 20 were donor-negative recipient-positive (D–/R+), 18 were donor-positive recipient-negative (D+/R–), and 24 were donor- and recipient-positive (D+/R+). Baseline transplant characteristics show that across the 4 donor/recipient CMV serostatus combinations, there were no significant differences with respect to recipient age, sex, disease, donor type, graft source, conditioning regimen, and severity of acute or cGVHD (supplemental Table 3).
A comparison of all donor and recipient CMV serostatus combinations show that the donor/recipient seronegative group had a significantly lower fraction of CD57+/CD27– cells (Figure 2A) in the CD4+ T-cell compartment compared with all other serostatus combinations with mean fractions of 2.9 ± 1.1% (D–/R–), 18 ± 8.2% (D–/R+), 12 ± 8.2% (D+/R–), and 19.6 ± 5.5% (D+/R+) (mean ± 95% confidence interval, P < .0001) (Figure 2B). A similar but less penetrant pattern was seen in the CD8+ T-cell compartment, and CD57+/CD27– T cells comprised a significantly larger overall fraction of the CD8+ population with mean fractions of 39 ± 10.2% (D–/R–), 57 ± 11.0% (D–/R+), 45 ± 11.6% (D+/R–), and 66 ± 6.0% (D+/R+) (P = .0036) (Figure 2B). Notably, the fraction of CD57+/CD27– T cells in any given patient comprised up to 71% and 86% in the CD4+ and CD8+ compartments, respectively, suggesting that this cellular subset can be present in a substantial proportion of T cells larger than that observed in healthy seropositive individuals in the non-SCT setting, where only up to 15% of T cells have been shown to harbor this phenotype.19 In addition, the presence or absence of cGVHD did not influence the proportion of CD57+/CD27– CD4+ T cells seen in peripheral blood (supplemental Figure 2A-B).
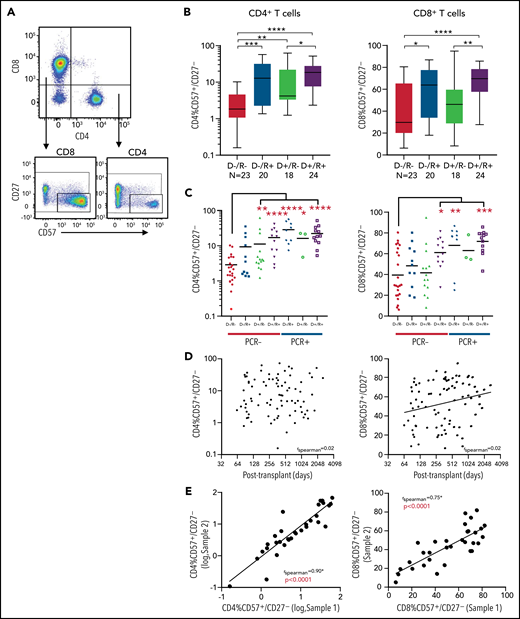
CD57+/CD27– T cells with respect to donor and recipient CMV serostatus. (A) Flow cytometry gating strategy use to identify CD57+/CD27– T cells. (B) Fraction of CD57+/CD27– population of CD4+ and CD8+ T cells with respect to CMV serostatus (D–/R–, D–/R+, D–/R–, D+/R+). (C) Fraction of CD57+/CD27– population of CD4+ and CD8+ T cells broken down by both CMV serostatus and posttransplant CMV reactivation status (PCR− or PCR+). (D) Fraction of CD57+/CD27– T-cell populations in relation to time of sample drawn from day of transplant. No significant temporal correlation was seen for the CD4 subset (rs = 0.024, P > .05). There was a marginal increase in expression over time for the CD8 subset (rs = 0.23, P = .035). (E) The fraction of CD57+/CD27– CD4+ and CD8+ T cells from baseline (sample 1, x-axis) to repeat sample (sample 2, y-axis) taken at a mean of 93 days (28-218) later were compared for 35 patients with repeat samples. There was a significant correlation between the 2 populations over time for both the CD4+ (log-log axis, rs = 0.90, P < .0001) and CD8+ T-cell subsets (rs = 0.75, P < .0001). Statistical significance is denoted by *P < .05, **P < .01, ***P < .001, ****P < .0001.
CD57+/CD27– T cells with respect to donor and recipient CMV serostatus. (A) Flow cytometry gating strategy use to identify CD57+/CD27– T cells. (B) Fraction of CD57+/CD27– population of CD4+ and CD8+ T cells with respect to CMV serostatus (D–/R–, D–/R+, D–/R–, D+/R+). (C) Fraction of CD57+/CD27– population of CD4+ and CD8+ T cells broken down by both CMV serostatus and posttransplant CMV reactivation status (PCR− or PCR+). (D) Fraction of CD57+/CD27– T-cell populations in relation to time of sample drawn from day of transplant. No significant temporal correlation was seen for the CD4 subset (rs = 0.024, P > .05). There was a marginal increase in expression over time for the CD8 subset (rs = 0.23, P = .035). (E) The fraction of CD57+/CD27– CD4+ and CD8+ T cells from baseline (sample 1, x-axis) to repeat sample (sample 2, y-axis) taken at a mean of 93 days (28-218) later were compared for 35 patients with repeat samples. There was a significant correlation between the 2 populations over time for both the CD4+ (log-log axis, rs = 0.90, P < .0001) and CD8+ T-cell subsets (rs = 0.75, P < .0001). Statistical significance is denoted by *P < .05, **P < .01, ***P < .001, ****P < .0001.
We next asked if the observed expansion of CD57+/CD27– T cells seen in D+ and/or R+ transplants was dependent on CMV reactivation posttransplant. Even in the absence of CMV reactivation (as defined by PCR positivity at any time point posttransplant), recipients with a CMV-seropositive donor had a higher fraction of CD57+/CD27– CD4+ T cells (P < .01, Figure 2C). CMV reactivation (PCR+) appeared to further increase the fraction of these cells. A similar, albeit weaker relationship was seen in the CD8+ population, though unlike in the CD4+ population, this relationship was absent in donor-seropositive to recipient-seronegative transplants. Of those who had CMV detectable by PCR, administration of antiviral treatment (ganciclovir or foscarnet) did not modify the fraction of CD57+/CD27– T cells in either the CD4+ or CD8+ compartments (supplemental Figure 3A-B).
We then evaluated if the fraction of CD57+/CD27– T cells changes over time following transplant. There was no significant correlation between the fraction of these cells in the CD4+ T-cell compartment and time of sample taken after transplant (Figure 2D; rs = 0.02, P = .83). A weakly positive correlation was seen over time in the CD8+ T-cell compartment (rs = 0.23, P = .035). Within any given patient, this population appeared temporally stable because there was a strong correlation between the initial and follow-up samples taken from the same patient with an average interval of 94 days between samples (CD4+: rs = 0.90, P < .0001; CD8+: rs = 0.75, P < .0001; Figure 2E). We also compared the fraction of CD4+/CD57+/CD27− T cells in PBMC samples present before and after receiving therapy for cGVHD and found that the composition of CD4+/CD57+/CD27− T cells between the 2 timepoints within each individual were highly correlated (r = 0.78, P < .0043), suggesting that treatment, including prednisone and calcineurin inhibitors, did not significantly affect the fraction of these cells (supplemental Figure 4A-B).
Multivariate analysis including donor and recipient CMV serostatus, prior CMV reactivation, time of sample acquisition, presence or absence of cGVHD, and presence or absence of relapse disease also confirmed that positive donor or recipient CMV serostatus and presence of prior CMV reactivation were significantly associated with a higher proportion of CD57+/CD27– T cells in the CD4+ compartment (P < .05; supplemental Figure 5A-B).
Together, these data demonstrate that the CD57+/CD27– CD4+ T-cell compartment is stably expanded in transplant patients with prior CMV exposure, including exposures limited to CMV seropositive donors (D+/R–), even in the absence of clinically detectable viral reactivation posttransplant.
CD57+/CD27– CD4+ T cells are a functional effector memory population
We next sought to further characterize the CD57+/CD27– CD4+ T-cell subset. We show that these T cells exhibit a predominantly CCR7–/CD45RAlow-int/CD45ROint-high/CD28– effector memory phenotype with low expression of exhaustion/inhibitory markers (TIGIT–/TIM3–/KLRG1–/CTLA4–) and upregulation of activation markers (PD-1+/DNAM-1+) (Figure 3A). There was also a striking increase in the expression of granzyme B and no change in FasL compared with the remaining CD4+ T-cell population (Figure 3A). Transcription factor profiling demonstrated the CD57+/CD27– CD4+ T-cell subset to be FOXP3–, RORγT–, and suggested a Th1 polarized population with higher T-bet and Eomesodermin (EOMES) and lower expression of GATA3 and BCL6 compared with the remaining CD4+ T cells (Figure 3B). Although EOMES has traditionally been associated with CD8+ T-effector function,20 it is also capable of upregulating the granzyme-perforin axis in CD4 populations,21 which is consistent with the phenotype seen here. We also show that this CD4+ T-cell subset is functional and secretes IFN-γ and TNF in response to phorbol myristate acetate/ionomycin stimulation (Figure 3C). Approximately 80% of CD57+/CD27– CD4+ T cells expressed IFN-γ or TNF following stimulation, significantly higher than seen in the remaining CD4+ T-cell population (P < .0001; Figure 3D). There was also less IL-10 secretion in the CD57+/CD27– CD4+ T-cell subset, consistent with the lack of FOXP3-expressing cells.
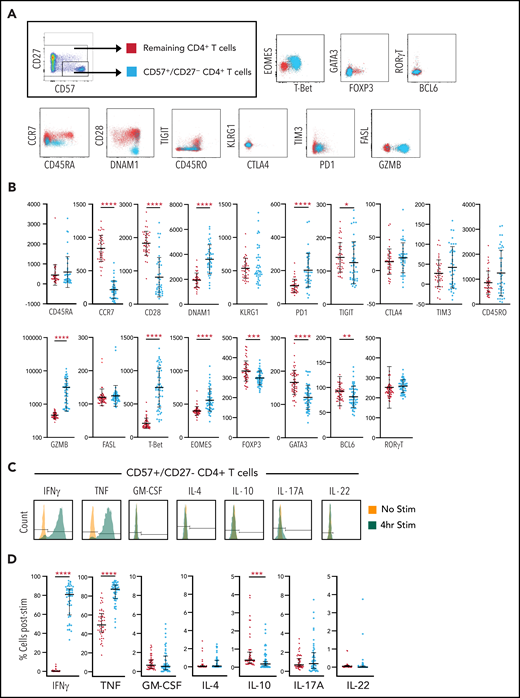
Characterization of CD4+/CD57+/CD27– T cells. (A) CD4+ lymphocytes were gated on the CD57+/CD27– profile for each patient sample and compared with the remaining bulk CD4+ population for that patient with respect to expression of various markers, including CCR7, CD45RA, CD28, DNAM1, TIGIT, CD45RO, KLRG1, CTLA4, TIM3, PD1, FASL, Granzyme B, and transcription factors EOMES, T-bet, GATA3, FOXP3, RORγT, and BCL6. Representative plots for markers are shown. (B) Respective geometric mean of fluorescent intensity (MFI) comparing CD57+/CD27– CD4+ T cells to remaining bulk CD4+ T cells. (C) Production of IFN-γ, TNF, GM-CSF, IL-4, IL-10, IL-17A, and IL-22 are shown for a representative sample of the CD57+/CD27– CD4+ T-cell population. The gating strategy used to identify positive or negative expression is illustrated. (D) Representative profile for each cytokine is shown for both CD57+/CD27– CD4+ T cells and remaining CD4+ T cells with respect to percent of positive cells identified by the gating strategy shown in panel C. Median with interquartile range are illustrated. Statistical significance is denoted by *P < .05, **P < .01, ***P < .001, ****P < .0001.
Characterization of CD4+/CD57+/CD27– T cells. (A) CD4+ lymphocytes were gated on the CD57+/CD27– profile for each patient sample and compared with the remaining bulk CD4+ population for that patient with respect to expression of various markers, including CCR7, CD45RA, CD28, DNAM1, TIGIT, CD45RO, KLRG1, CTLA4, TIM3, PD1, FASL, Granzyme B, and transcription factors EOMES, T-bet, GATA3, FOXP3, RORγT, and BCL6. Representative plots for markers are shown. (B) Respective geometric mean of fluorescent intensity (MFI) comparing CD57+/CD27– CD4+ T cells to remaining bulk CD4+ T cells. (C) Production of IFN-γ, TNF, GM-CSF, IL-4, IL-10, IL-17A, and IL-22 are shown for a representative sample of the CD57+/CD27– CD4+ T-cell population. The gating strategy used to identify positive or negative expression is illustrated. (D) Representative profile for each cytokine is shown for both CD57+/CD27– CD4+ T cells and remaining CD4+ T cells with respect to percent of positive cells identified by the gating strategy shown in panel C. Median with interquartile range are illustrated. Statistical significance is denoted by *P < .05, **P < .01, ***P < .001, ****P < .0001.
Overall cytokine and transcription factor profiles of CD57+/CD27– CD4+ T cells in our transplant population suggests that these cells are a highly differentiated effector memory subset that express increased levels of Th1 cytokines TNF and IFN-γ in conjunction with the cytolytic molecule granzyme B. This raises the possibility that these cells may play a functional role in response to CMV.
CD57+/CD27– CD4+ T cells are enriched for CMV specificity
pp65 is one of several CMV proteins that comprise a major target of the cytotoxic T-cell response against CMV.22 To specifically assess whether CMV-reactive CD4+ T cells express the CD57+/CD27– profile, we performed degranulation analysis and cytokine profiling on 12 patient PBMC samples following exposure to pp65 peptide pools. Compared with nonpeptide controls, pp65 induced TNF/IFN-γ and granzyme B/CD107a coexpression in a subset of CD4+ T cells (Figure 4A). Gating on these activated CD4+ T cells, as defined by either TNF+/IFN-γ+ or granzyme B+/CD107a+ coexpression, we evaluated the fraction of CD57+/CD27– T cells (Figure 4B). Compared with the overall CD4 population, TNF+/IFN-γ+ and granzyme B+/CD107a+ CD4+ T cells were significantly enriched for the CD57+/CD27– fraction (Figure 4C; bulk CD4+: μ = 22.4%; TNF+/IFN-γ+: μ = 79.1%, P < .0001; granzyme B+/CD107a+: μ = 68.0%, P < .0001). Of note, up to 15% of CD57+/CD27– CD4+ T cells were reactive to pp65, as defined by either TNF+/IFN-γ+ or granzyme B+/CD107a+ responses, though there were some samples with <1% pp65 reactivity (supplemental Figure 6A-B).
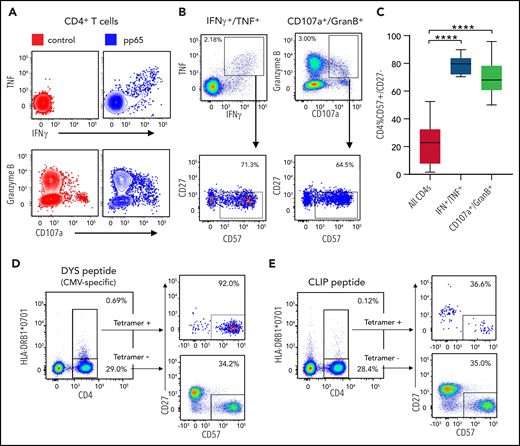
Profiling of pp65 peptide-stimulated CD4 T cells. (A) Twelve patient PBMC samples were treated with pp65 peptide or control (no peptide). Representative sample shows results of pp65-stimulated samples compared with control with respect to TNF and IFN-γ staining as well as Granzyme B and CD107a staining. (B) The fraction of CD57+/CD27– cells out of the TNF+/IFN-γ+ CD4+ subset as well as out of the GranB+/CD107a+ CD4+ subset is shown in a representative sample treated with pp65. (C) The fraction of CD57+/CD27– cells out of bulk CD4+ subsets, TNF+/IFNγ+ CD4+ subsets, and GranB+/CD107a+ CD4+ subsets show a higher percentage of CD57+/CD27– cells in the latter groups compared with the former (P < .0001, both groups). (D) Profiling of CD4+ T cells in a patient carrying class II allele HLA-DRB1*0701 with respect to CMV-specific peptide. CD4+ T cells in a patient carrying the class II HLA-DRB1*0701 allele profiled with tetramer HLA-DRB1*0701 loaded with the DYS peptide. (E) The same patient in panel D profiled with tetramer HLA-DRB1*0701 loaded with the irrelevant CLIP peptide. Statistical significance is denoted by ****P < .0001.
Profiling of pp65 peptide-stimulated CD4 T cells. (A) Twelve patient PBMC samples were treated with pp65 peptide or control (no peptide). Representative sample shows results of pp65-stimulated samples compared with control with respect to TNF and IFN-γ staining as well as Granzyme B and CD107a staining. (B) The fraction of CD57+/CD27– cells out of the TNF+/IFN-γ+ CD4+ subset as well as out of the GranB+/CD107a+ CD4+ subset is shown in a representative sample treated with pp65. (C) The fraction of CD57+/CD27– cells out of bulk CD4+ subsets, TNF+/IFNγ+ CD4+ subsets, and GranB+/CD107a+ CD4+ subsets show a higher percentage of CD57+/CD27– cells in the latter groups compared with the former (P < .0001, both groups). (D) Profiling of CD4+ T cells in a patient carrying class II allele HLA-DRB1*0701 with respect to CMV-specific peptide. CD4+ T cells in a patient carrying the class II HLA-DRB1*0701 allele profiled with tetramer HLA-DRB1*0701 loaded with the DYS peptide. (E) The same patient in panel D profiled with tetramer HLA-DRB1*0701 loaded with the irrelevant CLIP peptide. Statistical significance is denoted by ****P < .0001.
We also evaluated the reactivity of CD4+ T cells in a patient carrying the class II HLA-DRB1*0701 allele by using HLA-DRB1*0701 tetramers loaded with the DYS peptide (part of CMV glycoprotein B), which is known to induce a strong MHC class II–dependent response12 (Figure 4D). CLIP-loaded tetramer, which provides a stable MHC II–peptide complex, was used as a control. In this patient sample, 35% of CD4+ T cells expressed the CD57+/CD27– phenotype at baseline. Of the tetramer-positive CD4+ T cells (DYS+: 2.32% of CD4s, CLIP+: 0.42% of CD4s), 92% of the CD4+ T cells in the DYS peptide group were CD57+/CD27– compared with 37% in the CLIP peptide group (Figure 4D-E). These results suggest that the majority of CMV-reactive CD4+ T cells express the CD57+/CD27– phenotype.
Because other viruses including lymphocytic choriomeningitis virus, EBV, hepatitis C, and HIV-1 have been associated with “cytolytic” CD4+ T cells,23-27 we also examined if CD57+/CD27– CD4+ T cells may be EBV-specific, particularly given the ubiquitous nature of this virus in our transplant population. Results of peptide pool stimulation with pp65 (CMV), EBNA1 (EBV), and EBNA2 (EBV) showed that CD57+/CD27– CD4+ T cells have a notably stronger response to CMV compared with EBV peptide pools, as denoted by IFN-γ+/TNF+ (supplemental Figure 7A) (P < .0001 pp65 vs EBNA1; P < .0001 pp65 vs EBNA2; supplemental Figure 7B). In an additional 2 patients carrying DRB1 alleles (DRB1*0701/DRB1*0101 and DRB1*0701/DRB1*0301) that have known CMV- and EBV-specific class II MHC tetramer-peptide complexes, 1.3% and 17.1% of CD57+/CD27– CD4+ T cells were positive for CMV-DYS tetramer, whereas <0.1% were positive for EBV-VYG,13 EBV-TSL,14,15 or EBV-PAQ13 tetramers (supplemental Figure 7C). Furthermore, in 36 patients with donor EBV serostatus available, there was no difference in the frequency of CD57+/CD27– CD4+ T cells between EBV donor-seropositive and EBV donor-seronegative subsets (supplemental Figure 7D). Together, these data provide further support for the notion that the CD57+/CD27– CD4+ T-cell fraction is enriched in T cells reactive to CMV.
CD57+/CD27− CD4+ T cells are associated with reduced TCR diversity and decreased MHC class II expressing APCs
We next asked if the presence of CD57+/CD27– CD4+ T cells was associated with systemic immune modulation that might in turn inform putative mechanisms underlying the association between the use of CMV-seropositive donor grafts and inferior transplant outcomes.
Given the significant proportion of CD57+/CD27– cells that can comprise the CD4+ T-cell subset, we asked if there were any notable differences in systemic cytokines with respect to CMV donor/recipient serostatus and across patients with different fractions of CD57+/CD27– CD4+ T cells. Of 65 unique patient samples with serum cytokines measured, there were no differences in 15 cytokine levels, including granulocyte colony-stimulating factor, GM-CSF, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-12p40, IL-13, IL-17A, IFN-γ, monocyte chemoattractant protein-1, and TNF with respect to recipient/donor CMV serostatus (supplemental Figure 8A). There was also no significant correlation between fraction of CD57+/CD27− T cells in the CD4+ subpopulation for each cytokine expressed (supplemental Figure 8B-P).
We also asked if there was an association between the fraction of CD57+/CD27– CD4+ T cells with overall TCR and BCR diversity, with the hypothesis that an expansion of these cells is presumably generated as a result of prior or current CMV exposure that may constrict the immune repertoire. Twenty-one patient samples with known fractions of CD57+/CD27– T cells had TCR-β and BCR IgH sequencing performed on bulk PBMC samples. There appeared to be a strong negative correlation between the fraction of CD57+/CD27– CD4+ T cells with respect to TCR diversity (rs = −0.88, P < .0001; Figure 5A). A similar, but slightly weaker correlation was also seen for the CD8+ T-cell subset (rs = −0.65, P = .0014; Figure 5B), an observation that is consistent with recent reports of immune repertoire skewing in the context of CMV reactivation post-SCT.7 There was no significant association between CD57+/CD27– CD4+ or CD8+ T cells with respect to BCR diversity (Figure 5C-D).
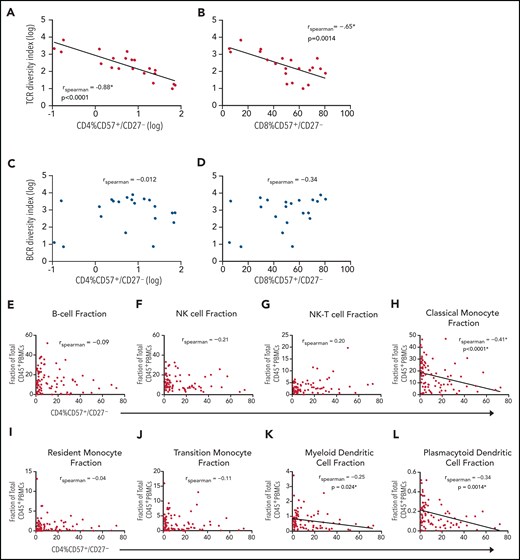
Association of CD57+/CD27– CD4+ T cells with lower TCR diversity and decreased MHC class II-expressing APCs. (A-D) Correlation between fraction of CD57+/CD27– T cells and TCR and BCR diversity. Twenty-one patient samples with known fraction of CD57+/CD27– T cells had TCR and BCR sequencing performed on bulk T and B cells. Diversity (inverse Simpson) indices expressed on a log axis for T and B cells are plotted with respect to the fraction of CD57+/CD27– CD4+ T cells as well as CD57+/CD27– CD8+ T cells. There is a negative association between the fraction of CD57+/CD27– CD4+ (rs = −0.88, P < .00001, expressed on a log axis) and CD8+ T cells (rs = −0.65, P = .0014) with (A-B) TCR diversity but not with (C-D) BCR diversity. (E-L) Composition of PBMCs. For each cell type shown, the proportion of the cell type of total CD45+ PBMCs is plotted against the fraction CD4+ T cells that exhibit the CD57+/CD27– phenotype. Cell populations that had a significant correlation to the phenotype included (H) classical monocytes (rs = −0.41, P < .0001), (K) myeloid dendritic cells (rs = −0.25, P = .024), and (L) plasmacytoid dendritic cells (rs = −0.34, P = .0014).
Association of CD57+/CD27– CD4+ T cells with lower TCR diversity and decreased MHC class II-expressing APCs. (A-D) Correlation between fraction of CD57+/CD27– T cells and TCR and BCR diversity. Twenty-one patient samples with known fraction of CD57+/CD27– T cells had TCR and BCR sequencing performed on bulk T and B cells. Diversity (inverse Simpson) indices expressed on a log axis for T and B cells are plotted with respect to the fraction of CD57+/CD27– CD4+ T cells as well as CD57+/CD27– CD8+ T cells. There is a negative association between the fraction of CD57+/CD27– CD4+ (rs = −0.88, P < .00001, expressed on a log axis) and CD8+ T cells (rs = −0.65, P = .0014) with (A-B) TCR diversity but not with (C-D) BCR diversity. (E-L) Composition of PBMCs. For each cell type shown, the proportion of the cell type of total CD45+ PBMCs is plotted against the fraction CD4+ T cells that exhibit the CD57+/CD27– phenotype. Cell populations that had a significant correlation to the phenotype included (H) classical monocytes (rs = −0.41, P < .0001), (K) myeloid dendritic cells (rs = −0.25, P = .024), and (L) plasmacytoid dendritic cells (rs = −0.34, P = .0014).
Of note, samples with a relatively high fraction of CD57+/CD27– CD4+ T cells also had a higher fraction of CD57+/CD27– CD8+ T cells, though the inverse was not true (supplemental Figure 9). TCR diversity of CD57+/CD27– T cells vs the remaining T-cell subset suggest that the relative reduction in diversity seen in the CD57+/CD27– subcompartment appears to be greater for CD4+ T cells compared with CD8+ T cells (supplemental Figure 10A-B). This may explain the stronger negative correlation with overall TCR diversity seen in the CD4 vs CD8 compartment.
Because CD4+/CD57+/CD27– T cells have been hypothesized to assume a cytolytic capability and target MHC class II expressing antigen-presenting cells (APCs),23 we next examined the association between these T cells and other subsets of PBMCs that express MHC class II, including B cells; classical, transitional, and resident monocytes; myeloid dendritic cells (DCs); and plasmacytoid DCs (Figure 5E-L). Cell populations that had a statistically significant correlation with the CD4+/CD57+/CD27– T-cell phenotype included classical monocytes (CD45+/CD3−/CD19−/CD56−/HLA-DR+/CD11c+/CD123−/CD14+/CD16−), myeloid DCs (CD45+/CD3−/CD19−/CD56−/HLA-DR+/CD11c+/CD123−/CD14−/CD16−), and plasmacytoid DCs (CD45+/CD3−/CD19−/CD56−/HLA-DR+/CD11c−/CD123+), with classical monocytes showing the strongest negative association (rs = −0.41, P < .0001; Figure 5H). The negative association observed in the classical monocyte fraction was also seen at earlier timepoints (posttransplant day +60, +90, and +180) upon subsequent analysis of 31 additional patients (supplemental Figure 11).
These data confirm an inverse association between the frequency of CD57+/CD27− CD4+ T cells and class II-expressing APCs, raising the possibility that this CD4+ T-cell subset may affect immunity after bone marrow transplantation by modulating antigen presentation.
Discussion
Prior studies of small patient numbers have demonstrated skewing of the CD4+ and CD8+ T-cell repertoire in response to CMV reactivation after SCT.7,8 Here, in a large patient series of 120 samples, we demonstrate that even in the absence of detectable CMV reactivation, the peripheral blood of CMV– recipients (R–) transplanted with CMV+ donor (D+) grafts is highly enriched in a CD57+/CD27– subset of CD4+ effector memory T cells that are associated with other immunomodulatory effects. These include a reduction in T-cell diversity and a decrease in specific MHC class II-expressing APC subpopulations.
Allogeneic SCT is associated with significant morbidity and mortality, principally GVHD, opportunistic infection, and disease relapse. These negative outcomes reflect aberrant immunity (to alloantigens) or a failure of pathogen and leukemia-specific immunity that are potentially modified by CMV.28 Long-term immune imprinting by CMV represents one of the most dominant effects described to date,29 and the precise effect and mechanisms by which the CD57+/CD27– CD4+ T-cell subset described here may generate downstream defects in immunity after transplantation deserves further study. CD57 is a surface marker described on “cytolytic” CD4+ T cells, a less-well studied subset of CD4+ T cells that has been shown ex vivo to kill in a granzyme- or FasL ligand-dependent manner and is associated with certain viral infections.23-27 As seen in our study, CD57+/CD27– CD4+ T cells represent a late effector memory subset that express high levels of granzyme B and CD107a, consistent with high cytolytic capacity. In concordance with this, there was a strong negative correlation between this fraction of CD4+ T cells and MHC class II-expressing APCs including classical monocytes and myeloid/plasmacytoid DCs. It is well established that MHC class II-dependent CMV-specific T cell immunity requires presentation of viral antigens by donor DCs and that this pathway is grossly perturbed during GVHD and CMV infection.30 Furthermore, prior reports have suggested in lymphocytic choriomeningitis virus models that “cytolytic” CD4+ T cells can kill in an MHC class II-dependent manner.31 The data here provide correlative evidence between the expansion of donor CD4+ cytotoxic T lymphocytes after bone marrow transplantation and the loss of donor APC capable of presenting cognate antigen, which may lead to a broader defect in pathogen-specific immunity. This paradigm will require interrogation in preclinical systems to ascertain true cause and effect relationships.
Because of the magnitude of expansion in this T-cell subset (in some cases comprising up to 70% of the total CD4+ population), there may also be an additional immunologic “cost” in the generation and/or maintenance of these CD57+ memory CD4+ T cells. Consistent with this hypothesis, we demonstrate an inverse relationship between the magnitude of this T-cell fraction and overall TCR diversity. CD57+ T cells may accumulate over time with latent CMV infection, a process that has been described as memory inflation.19,32 Prior reports have suggested that this CD57+ memory T-cell differentiation is associated with inferior immune reconstitution and protective immunity in part because of weaker T-cell signaling, increased senescence, poor proliferative capacity, and lower IL-2 production.33-36 It is thus unclear whether the expanded fraction of CD4+ T cells in our context provides a direct and necessary immune surveillance role against CMV, or whether it accumulates over time as a result of low-level persistent or periodic CMV escape from latency.
There are some limitations to this study. The absence of detectable CMV reactivation does not exclude subclinical viral reactivation that may be present below the assay detection threshold. Thus, it is possible that CD57+ memory CD4+ T-cell differentiation occurs as a result of chronic low-level, clinically undetectable, or transient CMV reactivation. This is supported by our findings that, for D–/R+ transplants without detectable CMV reactivation, there was a trend for increased proportions of CD57+/CD27– CD4+ T cells compared with D–/R– transplant pairs. We cannot distinguish whether the expanded CD57+ memory CD4+ T cells seen are predominantly donor–graft derived or are generated de novo after transplantation and are currently phenotyping donors and transplant recipients after SCT in an attempt to define this.
Our study is also not powered to detect differences in clinical outcomes between D+/R– and D–/R– transplants, which have been studied in epidemiologic reports including several thousand patients.5,6 Although these studies show a negative association between donor CMV seropositivity and survival in CMV recipient-seronegative transplants, other studies have suggested that donor CMV seropositivity may have a protective effect on relapsed disease particularly in the acute myeloid leukemia population.37,38 We demonstrate that CD57+/CD27– CD4+ T cells are associated with reduced numbers on MHC class II-expressing monocytes, a cellular subset known to disseminate CMV in vivo.39 We thus postulate that CD57+/CD27– CD4+ T cells may function to kill CMV-infected APCs. Because CMV also has the ability to latently infect leukemic myeloid precursors,40 our findings raise the possibility that these expanded memory CD4+ T cells have the potential to regulate both CMV-infected leukemic and nonleukemic myeloid cells that may explain an apparent “protective effect” of CMV on acute myeloid leukemia relapse, offset by an increased overall risk for infection from a profound impairment in immune repertoire diversity. This hypothesis also requires testing in preclinical systems.
Of note, our long-term posttransplant patient cohort was derived from the Chronic GVHD Consortium, which enabled us to obtain results up to nearly 10 years post-SCT. However, about three-quarters of these patients had at least mild cGVHD, with the remaining having no cGVHD, which is an overrepresentation of samples with cGVHD relative to that seen in typical clinical transplant cohorts. Nevertheless, our data in regard to the CD57+ CD4+ T-cell fraction was not influenced by cGVHD per se.
Donor/recipient CMV serostatus remains a powerful (negative) predictive marker of transplant outcome. Although our study is not powered to determine if CD57+ memory CD4+ T-cell differentiation is associated with relapse or nonrelapse mortality, it nevertheless demonstrates a potentially important lasting change in the CD4+ T-cell repertoire that may result in the compromise of transplant recipients’ immune fitness. Notably, in the absence of CMV reactivation after SCT, this CD57+ memory CD4+ T-cell differentiation is most penetrant in transplant recipients of CMV-seropositive donors (D+/R–, D+/R+), although a trend is also observed in recipients transplanted with seronegative donors (D–/R+). The observation in D+/R– transplants is the most clinically relevant because of the potential to prioritize a seronegative donor. Given the highly immunocompromised state of transplant recipients by virtue of endogenous and pharmacological T-cell defects, the repercussions from additional potential long-lasting effects of CMV imprinting via seropositive grafts should be considered.
Acknowledgments
The authors thank Michael Boeckh for providing insight in data interpretation.
This work was supported by National Institutes of Health, National Cancer Institute grants R01 CA118953 (S.J.L.), T32 CA009515 (A.C.Y.), and CA226833 (J.M.I.; S.M.B.).
Authorship
Contribution: A.C.Y., A.V., K.B.D., M.H.J., S.J.L., and G.R.H., designed research studies; A.C.Y., A.V., A.R., K.S.E., L.S., and S.D.O. performed experimental work; K.B.D., M.H.J., S.J.L., and J.M.I. planned the mass cytometry study and antibody panels; A.C.Y., S.M.B., and J.M.I. performed mass cytometry data analysis; A.R. analyzed TCR/BCR sequencing data; A.C.Y., S.M.B., T.B., J.M.I., and E.W.N. developed R scripts for data analysis and visualization; K.C., L.E.O., and A.S.H. compiled patient data; A.C.Y., A.V., A.R., C.A.J., K.B.D., P.Z., M.A.D.-E., T.K.K., E.W.N., J.M.I., S.J.L., and G.R.H. provided intellectual input and helped write the manuscript; and all authors contributed to reviewing the manuscript.
Conflict-of-interest disclosure: K.B.D. received unrelated research support from Kadmon. M.H.J. received unrelated research support from Mallinckrodt and Janssen. J.M.I. was a cofounder and a board member of Cytobank Inc. and received unrelated research support from Incyte Corp, Janssen, and Pharmacyclics. S.J.L. received unrelated research support from AstraZeneca, Incyte, Amgen, Kadmon, Novartis, Pfizer, Syndax, and Takeda. G.R.H. has consulted for Generon and NapaJen and received unrelated research funding from Roche Pharmaceuticals, Compass Pharmaceuticals, iTeos Pharmaceuticals, Applied Molecular Transport Pharmaceuticals, and Syndax Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Albert C. Yeh, Clinical Research Division, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N S2-204, Seattle, WA 98109; e-mail: ayeh@fredhutch.org.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal