

Key Points
Platelets internalize and present exogenous antigens through MHC-I during sepsis.
Increased MHC-I on platelets during sepsis reduces antigen-specific CD8+ T-cell numbers and functions.

Abstract
Circulating platelets interact with leukocytes to modulate host immune and thrombotic responses. In sepsis, platelet-leukocyte interactions are increased and have been associated with adverse clinical events, including increased platelet–T-cell interactions. Sepsis is associated with reduced CD8+ T-cell numbers and functional responses, but whether platelets regulate CD8+ T-cell responses during sepsis remains unknown. In our current study, we systemically evaluated platelet antigen internalization and presentation through major histocompatibility complex class I (MHC-I) and their effects on antigen-specific CD8+ T cells in sepsis in vivo and ex vivo. We discovered that both human and murine platelets internalize and proteolyze exogenous antigens, generating peptides that are loaded onto MHC-I. The expression of platelet MHC-I, but not platelet MHC-II, is significantly increased in human and murine platelets during sepsis and in human megakaryocytes stimulated with agonists generated systemically during sepsis (eg, interferon-γ and lipopolysaccharide). Upregulation of platelet MHC-I during sepsis increases antigen cross-presentation and interactions with CD8+ T cells in an antigen-specific manner. Using a platelet lineage–specific MHC-I–deficient mouse strain (B2Mf/f-Pf4Cre), we demonstrate that platelet MHC-I regulates antigen-specific CD8+ T-cell proliferation in vitro, as well as the number and functional responses of CD8+ T cells in vivo, during sepsis. Loss of platelet MHC-I reduces sepsis-associated mortality in mice in an antigen-specific setting. These data identify a new mechanism by which platelets, through MHC-I, process and cross-present antigens, engage antigen-specific CD8+ T cells, and regulate CD8+ T-cell numbers, functional responses, and outcomes during sepsis.
Introduction
T cells are lymphocytes that enable a host to combat pathogens. In sepsis, a systemic inflammatory condition with organ dysfunction in response to invading infectious pathogens, CD8+ T-cell numbers decrease, and their functional responses are impaired, leading to an increased risk of secondary infections and adverse outcomes.1-6 Many promising preclinical therapeutics for sepsis have failed.7 Therefore, a deeper understanding of the pathophysiological cellular responses in sepsis is needed to develop better strategies to improve the care of patients with sepsis.
Platelets are canonically known for their roles in mediating hemostasis and thrombosis. In addition to these well-established roles, platelets are dynamic effector cells across immune and inflammatory continuums. Platelets are capable of sensing exogenous pathogens and contain Toll-like receptors and other pattern recognition receptors that directly bind exogenous pathogens, including viruses, bacteria, and parasites.8-12 In sepsis, our group and others have found platelets play a number of important roles in sepsis.8,13-15 For example, platelets directly interact with other innate and adaptive immune cells during sepsis, such as monocytes and neutrophils.14,16 Platelets also possess the capacity to engage T cells. During ischemia reperfusion, platelets enhance CD4+ T-cell recruitment to liver sinusoids through CD62P (P-selectin).17 In viral hepatitis, activated platelets promote intrahepatic accumulation of CD8+ T cells.18 In HIV and SARS-CoV-2 infections, platelets conjugate with CD8+ T cells.19,20 In malaria, in vitro activated platelets stimulate antigen-specific CD8+ T cells.21 In addition, megakaryocytes, platelet progenitor cells, elicit antigen-specific CD8+ T-cell responses in immune thrombocytopenia.22 However, antigen-specific platelet–CD8+ T-cell interactions are almost completely unexplored in bacterial sepsis.
Antigen-specific CD8+ T-cell responses depend on major histocompatibility complex class I (MHC-I). MHC-I complexes are composed of an α chain, a β2-microglobulin (B2M) chain, and an antigen peptide loaded in the antigen-binding groove of the α chain in both humans and mice, although the nomenclature differs slightly.23 Classic MHC-I alleles in humans include HLA-A, -B, and –C, whereas MHC-I alleles in mice are termed H-2D, -K, and -L. MHC-I genes are highly polymorphic in humans and can generate millions of different MHC-I genotypes.23 The antigen peptides presented on MHC-I are typically 8 to 10 amino acids long and are extremely heterogeneous.24 Once the peptide–MHC-I complexes are shuttled to the cell surface, they engage antigen-specific CD8+ T-cell receptors (TCRs) on CD8+ T cells. Under physiological situations, cells generally present endogenous intracellular-derived peptides (ie, self-antigens) through MHC-I. However, during infectious settings, antigen-presenting cells (APCs), such as dendritic cells, internalize, process, and present antigens of pathogen origin via MHC-I (a process called cross-presentation) that can either elicit or suppress antigen-specific CD8+ T-cell responses, depending on other factors, including costimulatory and coinhibitory molecules.25
MHC-I molecules are found on the surface of all nucleated cells as well as on platelets.26 Platelet MHC-I molecules have some unique features compared with MHC-I on nucleated cells. For example, platelet transfusions are generally well tolerated clinically, whereas organ transplantations cause rejection, mostly because allogeneic MHC-I on platelets induces CD8+ T-cell tolerance and suppression rather than activation.27,28 Nevertheless, whether platelets internalize and process exogenous antigens and whether the antigen presentation and cross-presentation through MHC-I shape CD8+ T-cell responses during sepsis remain unclear.
We identified that both human and mouse platelets actively internalize and process exogenous antigen. Using a murine cecal ligation and puncture (CLP)–induced sepsis model and complementary clinical studies in patients with sepsis, we found that platelet MHC-I expression and antigen cross-presentation by MHC-I are significantly upregulated in sepsis. This resulted in upregulated antigen-specific platelet–CD8+ T-cell interactions, reduced antigen-specific CD8+ T-cell proliferation in vitro, decreased CD8+ T-cell numbers and interferon-γ (IFN-γ) production in vivo, and impaired antigen-specific CD8+ T-cell responses in vivo. In platelet MHC-I–deficient CD8+ transgenic mice, we identified that loss of MHC-I may improve survival in sepsis. These data identify a previously unknown mechanism by which upregulation of platelet MHC-I during sepsis negatively regulates CD8+ T-cell responses and outcomes.
Materials and methods
Human participants
Patients admitted to the intensive care unit with a primary diagnosis of sepsis were prospectively enrolled within 48 (±24) hours of intensive care unit admission. Sepsis was defined using SEPSIS-3 consensus criteria.3 Patients with systemic infection and life-threatening organ dysfunction were diagnosed with sepsis when total Sequential Organ Failure Assessment score was ≥2. Those with septic shock were a subset of patients with sepsis who could be identified as having clinical sepsis with persisting hypotension requiring vasopressors to maintain mean arterial pressure ≥65 mm Hg and a serum lactate level >2 mmol/L (18 mg/dL). For comparison, healthy donors were also recruited. Healthy donors were defined as medication-free men and women age >21 years without any acute or chronic medical conditions, illness, or hospitalization within 30 days before enrollment. All healthy donors and patients provided informed consent before sample collection (Institutional Review Board #51506 and #102638). Supplemental Table 1 (available on the Blood Web site) shows demographic information for healthy donors and patients with sepsis.
Mice
Wild-type (WT) C57BL/6J male and female mice were obtained from The Jackson Laboratory. B6.SJL-Ptprc (B6 Cd45.1) mice were obtained either from The Jackson Laboratory or from the laboratory of Peter Jensen (Department of Pathology, University of Utah, Salt Lake City, UT). B2mfl/fl-Pf4Cre mice were obtained from Craig N. Morrell (University of Rochester School of Medicine, Rochester, NY).29 OT-I mice were obtained from The Jackson Laboratory and crossed with B2mfl/fl-Pf4Cre and littermate control B2mfl/fl mice to generate the B2mfl/fl–Pf4Cre–OT-I (knockout [KO]–OT-I and WT-OT-I) mice. Rag2−/− OT-I mice were obtained from Taconic Biosciences. All mice were bred and maintained in the University of Utah animal facility under pathogen-free conditions according to National Institutes of Health guidelines. All animal experiments were performed according to protocols approved by the institutional Animal Care and Use Committee (protocol #18-10012). Male and female mice between ages 9 and 16 weeks were used in all experiments.
Platelet isolation
Platelets were isolated as previously described.30 Briefly, blood was collected into a one‐sixth volume of acid/citrate/dextrose buffer. Platelet‐rich plasma was obtained by centrifugation at 100g for 20 minutes or for 10 minutes using minicentrifuge at room temperature. Platelets were isolated from plasma by centrifugation at 500g for 10 minutes and resuspended in PIPES/saline/glucose buffer containing 20 nmol/L of prostaglandin E1, CD45+ depleted following the Miltenyi Biotec manufacturer protocol, centrifuged at 500g for 10 minutes again in PIPES/saline/glucose buffer containing 20 nmol/L of prostaglandin E1, and resuspended in Medium 199.
Megakaryocyte culture and stimulation
CD34+-derived human megakaryocytes were cultured as previously described.31 CD34+ hematopoietic stem and progenitor cells were isolated from human cord blood and cultured in serum-free expansion media supplemented with 25 ng/mL of stem cell factor and 20 ng/mL of thrombopoietin (TPO) for 6 days. Cells were cultured with 50 ng/mL of TPO only from days 6 to 13. On day 13, megakaryocytes were harvested, washed, and treated with 50 ng/mL of TPO together with either IFN-γ at 250 U/mL or lipopolysaccharide (LPS) at 100 ng/mL, and LPS-binding protein and CD14 were added to the cell culture to facilitate LPS binding, both at a final concentration of 100 ng/mL. After 20-hour stimulation, HLA-A, -B, and -C expression on megakaryocytes was measured by flow cytometry.
CLP
CLP was used to induce polymicrobial sepsis in mice. The mice were first anesthetized with ketamine and xylazine. The lower abdominal area of anesthetized mice was sterilized using iodine swabs. A small incision was made in the lower abdominal area to expose the cecum. The cecum was ligated with a 3.0 silk suture and then carefully punctured a single time with a 21-gauge needle to expose fecal contents into the abdominal cavity. The abdominal cavity was then closed with nonabsorbable nylon sutures and the area resterilized with iodine. The mice were then given a subcutaneous injection of 600 μL of saline and returned to clean cages on a heating pad during recovery. Mice received buprenex immediately after the surgery and again a few hours postsurgery subcutaneously. For sham control conditions, all these steps were followed, except the cecum was not ligated or punctured. Mice were monitored daily for disease severity and euthanized 4 days after CLP unless otherwise stated. Platelets were harvested 4 days after CLP induction.
OVA immunization
To elicit antigen-specific CD8+ T-cell responses in vivo, chicken ovalbumin (OVA) at 2 mg/mL was mixed with equal volume of either complete Freund adjuvant or incomplete Freund adjuvant and injected subcutaneously at 50 μL per site at the left and right hock (100 μL per mouse). For in vivo OVA treatment (pulse) experiments, mice were administered OVA without adjuvants intraperitoneally at 3 mg per mouse per day for 3 consecutive days.
DQ-OVA and OVA pulse assay
Platelets (2 × 107/mL) in Medium 199 were treated (pulsed) with DQ-OVA at 100 μg/mL at 37°C for 1 hour, followed by CD41 surface staining at room temperature in the dark for 15 to 30 minutes and acquired on flow cytometry immediately. For negative controls, platelets were stained for CD41, fixed with equal volume of 4% paraformaldehyde (PFA), and then pulsed with DQ-OVA for 1 hour. Proteasome inhibitors MG-132, bortezomib, and lactacystin and corresponding vehicles were added to platelet suspension at a final concentration of 40 μM for 30 minutes, followed by DQ-OVA pulse for 1 hour; CD41 staining was added during the last 30 minutes of DQ-OVA pulse. Samples were acquired on a flow cytometer immediately. To confirm the successful inhibition of proteasome activity in platelets, the 20S Proteasome Activity Assay Kit was used according to the manufacturer’s recommendations (MilliporeSigma). Briefly, platelets isolated from control mice were used for the indicated treatment and prepared in the cell lysis buffer following the manufacturer’s manual and measured for their 20S proteasome activity. For OVA pulse, platelets (2 × 107/mL) in Medium 199 were pulsed with OVA at 1 mg/mL at 37°C with gentle rotation for 6 hours, washed, and then stained for CD41, H-2Kb, and SIINFEKL (SIIN):H2Kb surface molecules. For SIIN peptide pulse, platelets were pulsed with SIIN peptide (OVA 257-264 [chicken]) at 25 μM for 2 hours and washed 2 times.
AST activity assay
To biochemically evaluate the disease severity in our CLP sepsis model, AST blood levels were measured using the commercial AST Activity Assay Kit from MilliporeSigma following the manufacturer’s protocol. Briefly, mouse blood was collected by cardiac puncture without anticoagulants. After clotting for 30 minutes undisturbed at room temperature, blood samples were centrifuged at 2000g for 10 minutes at 4°C. Serum was collected and centrifuged at 12 000g for 2 minutes to further remove debris and saved for the AST activity assay.
Murine CD8+ T-cell isolation and in vitro labeling
Single-cell suspensions of splenocytes from Rag2−/− OT-I mice were lysed with ACK red blood cell lysis buffer and then washed with phosphate-buffered saline (PBS). CD8+ T cells were isolated by negative selection using a CD8+ T-cell Isolation Kit (mouse) from Miltenyi Biotec following the manufacturer’s instructions. The purity and viability of isolated CD8+ T cells were measured by flow cytometry (all isolations were >95% purity and >95% viability). For proliferation assays, CD8+ T cells were washed with PBS twice and then stained with Violet Proliferation Dye 450 (V450) following the manufacturer’s instructions.
CD8+ T-cell proliferation assay
The isolated V450-stained CD8+ T cells from Rag2−/− OT-I mice mentioned above were used for the proliferation assay. To induce CD8+ T-cell activation and proliferation in the absence of stimulation or an exogenous cytokine like interleukin-2, we added fixed APCs, which stimulate CD8+ T-cell proliferation weakly.32 As to the preparation of fixed APCs, we pretreated CD45.1 splenocytes with SIIN at 25 μM in the presence of LPS, CD14, and LPS-binding protein, all at 100 ng/mL for 6 hours, and then fixed the splenocytes (0.8% PFA for 5 minutes) for a consistent stimulatory signal for CD8+ activation.
Platelets from indicated conditions were used at a ratio of platelet:CD8 of 50:1 (comparable to physiological ratios). Fixed CD45.1 was used at a number equal to CD8+ T cells. For CD3/CD28 antibody–stimulated conditions, we coated the cell culture plate with a CD3 antibody at 1 μg/mL overnight at 4°C, washed with PBS before the experiment, and then added a CD28 antibody at 2 μg/mL at the time of the experiment.
Quantitative real-time PCR
Total RNA was isolated from platelets lysed in TRIZOL reagent. Quantitative real-time polymerase chain reaction (PCR) analysis was then performed with the Quanta Perfecta SYBR Supermix Kit. HLA-A messenger RNA expression was normalized to human TUBB1. Thermal cycling conditions were as follows: 2 minutes at 95°C, 40 cycles of 30 seconds at 94°C, 30 seconds at 54°C, 45 seconds at 72°C, and 10 minutes at 72°C. Primers used for HLA-A and TUBB1 were: HLA-A forward 5′-GATTACATCGCCTTGAACGAGG-3′, HLA-A reverse 5′-GCAGGGTAGA AGCTCAGGG-3′; TUBB1 forward 5′-GCGTGTACTACAACGAAGCCTAC-3′, and TUBB reverse 5′-AAAACTGTCGGGTTGAAAGAGAG-3′.
Flow cytometry
For the MHC-I expression assay, platelets were incubated with anti-human HLA-A, -B, or -C, antimouse H-2D or H-2K, or antimouse SIIN:H-2Kb antibodies (depending on the experiment) at room temperature in the dark (20-30 minutes, depending on the antibody). Platelets were then fixed with equal volume of 4% PFA for 15 minutes. Samples were acquired on a flow cytometer immediately or, in some experiments, washed with platelet binding buffer after fixation and acquired on a flow cytometer within 2 hours. For CD8+ T cells, dendritic cells, and monocytes, single-cell suspensions of peripheral blood mononuclear cells, lymph nodes, and splenocytes were Fc blocked using rat anti-mouse CD16/32 (clone2.4G2, BD 553141). Cells were then incubated with the appropriate antibody (depending on the experiment) or viability dye at room temperature for 30 minutes and then fixed and washed. For tetramer staining, cells were incubated with either isotype tetramer control or a specific tetramer at 4°C for 30 minutes after Fc blocking. Cells were then washed and stained for CD45, TCRβ, and CD8, followed by fixation. Supplemental Table 2 lists antibody information. Intracellular IFN-γ staining was carried out following the BD Fixation/Permeabilization Solution Kit protocol. Briefly, cells were stimulated with eBioscience Cell Stimulation Cocktail for 4 to 6 hours in vitro, followed by surface staining including TCRβ, CD8, CD44, and CD62L, fixation/permeabilization, and intracellular staining for IFN-γ.
Western blotting
Washed platelets were saved in lysis buffer, separated by gel electrophoresis, electrotransferred onto polyvinylidene fluoride membranes, and then blocked in Tris-buffered saline, 0.1% Tween 20, and 5% dry milk. Western blots were probed with antibodies against B2M (Abcam), human MHC-I (Santa Cruz Biotechnology), or mouse MHC-I (Bio-Rad Laboratories). Beta-actin (Santa Cruz Biotechnology) was used as a control for protein loading and densitometric quantification.
Immunofluorescence microscopy
For platelet MHC-I staining, platelets (0.5 × 108 to 1 × 108/mL) suspended in Medium 199 were fixed with equal volume of 4% PFA and cytospun onto a cover glass. Platelets were then washed, permeabilized with 0.1% Triton-X 100 in Hanks balanced salt solution, blocked with 10% goat serum, and incubated with primary antibodies at 4°C overnight, followed by incubation with secondary antibodies at room temperature for 2 hours. For platelet and CD8+ T-cell coculture and staining, chamber wells were precoated with either fibrinogen or poly-L-lysine. For fibrinogen coating, chamber wells were coated with fibrinogen at 100 μg/mL at 37°C for 30 minutes or at 4°C overnight and blocked with 2% serum albumin. For poly-L-lysine coating, chamber wells were coated with poly-L-lysine at 100 μg/mL overnight at 4°C. OVA- or SIIN-pulsed platelets were then washed and seeded onto the fibrinogen-coated chamber wells at a density of 4 × 107platelets per mL for 30 to 40 minutes. The wells were then carefully washed to remove nonadherent platelets. CD8+ T cells isolated from Rag2−/− OT-I mice were next added to the chamber wells at a density of 4 × 106/mL CD8+ T cells per well (ratio of platelets/CD8+ T cells, 10:1). After 30 minutes of coincubation, unbound cells were washed off. The remaining cells were fixed with 2% PFA, followed by permeabilization with 0.1% Triton and blocking with 2% serum albumin, and then stained with primary and secondary antibodies. Alternatively, SIIN-pulsed platelets were seeded onto poly-L-lysine–coated chamber wells. Because platelets remain quiescent on poly-L-lysine, platelets were seeded at a higher concentration of 6.6 × 107platelets per mL for 1 hour. The nonadherent platelets were carefully removed. CD8+ T cells from Rag2−/− OT-I mice were added at a platelet/CD8+ T-cell ratio of 10:1 for 1 hour. Samples were fixed with 2% PFA and then washed. Cells were allowed to settled overnight at 4°C, washed again the following day, treated with 50 mM of ammonium chloride for antigen retrieval (15 minutes), and continued for further permeabilization with 0.1% Triton-X 100 in Hanks balanced salt solution, blocking with 10% goat serum, incubation with primary antibodies at 4°C overnight, and incubation with immunofluorescent secondary antibodies at room temperature for 2 hours. Antibodies used included anti-B2M antibody, anti-CD8a antibody, anti–integrin αIIb antibody, anti-GPIbβ derivative DyLight649, 4′,6-diamidino-2-phenylindole, and secondary antibodies (supplemental Table 2). Two-dimensional and z-stack images were acquired using an Olympus IX81 FV300 (Olympus, Melville, NY) confocal scanning microscope equipped with 60×/1.42 NA and 40×/1.35 NA oil objectives. Four laser excitation wavelengths were used (405, 488, 568, and 640 nm). Z-stack images were used for the reconstructed 3-dimensional structure for 3-dimensional videos and max projection images.
Statistical analyses
Statistical analyses were performed using Prism 8 software (GraphPad Software). The 2-tailed Student t test or analysis of variance with post hoc test was used to compare continuous variables when data were normally distributed. The Mann-Whitney test was used to compare nonnormally distributed variables. The log-rank (eg, Mantel-Cox) survival test was used for survival. A 2-tailed P value of <.05 was considered statistically significant.
Results
Platelets actively internalize, process, and cross-present exogenous antigens through MHC-I during sepsis
Platelets have been shown to be capable of cross-presenting exogenous antigens in vitro,21 suggesting that platelets can both internalize and proteolyze exogenous antigens similarly to professional APCs. However, the efficacy of this process has not been evaluated. To determine this, we pulsed platelets with DQ-OVA, a self-quenched conjugate of OVA that, when proteolyzed, emits fluorescence (BODIPY; Figure 1A, schematic). Platelets fixed with 2% PFA were used as negative controls. We found that a majority of murine platelets were able to internalize and proteolyze exogenous antigens as indicated by the BODIPY signal (Figure 1B-C). To evaluate antigen processing in platelets during sepsis, we compared the platelets isolated from sham and CLP mice 4 days after CLP. Antigen internalization and processing by platelets were similar under sham and CLP conditions, suggesting that platelets retain their robust capacity for antigen uptake and processing during sepsis. As shown in Figure 1D, antigen internalization and processing by human platelets from healthy donors and patients with sepsis were consistent with the murine findings. Because protein degradation in the proteasome is the most common pathway for proteolysis, we also examined whether platelet proteasome activity was necessary for DQ-OVA processing. Platelets were treated with 3 different proteasome inhibitors: MG-132, bortezomib, and lactacystin. All 3 inhibitors successfully inhibited the 20S proteasome activity (supplemental Figure 1A-B). However, they did not inhibit DQ-OVA processing (supplemental Figure 1C-D), suggesting that platelets internalize and process exogenous antigens in a proteasome-independent manner.

Platelets actively phagocytose, immunoproteolyze, and present exogenous antigens through MHC-I. (A) To determine if murine and human platelets are capable of internalization and proteolysis of exogenous antigens, isolated mouse platelets (day 4 after CLP) or human platelets (day 0 upon recruitment) were pulsed with DQ-OVA for 1 hour at 37°C. BODIPY signal indicates successful internalization and processing of the DQ-OVA, which can be detected by flow cytometry. (B) Representative flow plots demonstrating BODIPY signal in murine platelets labeled with CD41. A majority of platelets were positive for BODIPY after DQ-OVA pulse (but not when platelets were fixed), showing active antigen internalization and processing by platelets. (C) Platelets isolated from mice after sham or CLP-induced sepsis actively internalized and proteolyzed DQ-OVA (n = 8-14). Fixed platelets pulsed with DQ-OVA were used as negative control. (D) Human platelets from both healthy donors and patients with sepsis actively internalized and proteolyzed DQ-OVA (n = 6-8). (E) Platelets isolated from sham or CLP mice were pulsed with OVA in vitro for 6 hours, and the OVA antigen peptide–MHC-I complex (SIIN:H-2Kb) was detected. As the arrows demonstrate, we observed significantly increased SIIN:H-2Kb expression in OVA-pulsed platelets from CLP mice compared with sham mice, suggesting increased antigen cross-presentation by platelets during sepsis. Summarized percentages of SIIN:H-2Kb platelets are shown on the right. Platelets from CLP mice showed significantly increased antigen cross-presentation (n = 9-12). (F) Mice received OVA without adjuvant (3 mg per day) for 3 consecutive days, and on day 4, SIIN:H-2Kb expression on the surface of platelets was measured by flow cytometry (n = 4-5; representative of 3 independent experiments). (G) SIIN:H-2Kb expression on the surface of platelets after OVA pulse positively correlated with total H-2Kb expression (n = 4-5; representative of 3 independent experiments). One-way analysis of variance with Tukey post hoc analysis, unpaired Student t test, and simple linear regression test were used. ****P < .0001. MFI, mean fluorescence intensity; plts, platelets; WGA, wheat germ agglutinin.
Platelets actively phagocytose, immunoproteolyze, and present exogenous antigens through MHC-I. (A) To determine if murine and human platelets are capable of internalization and proteolysis of exogenous antigens, isolated mouse platelets (day 4 after CLP) or human platelets (day 0 upon recruitment) were pulsed with DQ-OVA for 1 hour at 37°C. BODIPY signal indicates successful internalization and processing of the DQ-OVA, which can be detected by flow cytometry. (B) Representative flow plots demonstrating BODIPY signal in murine platelets labeled with CD41. A majority of platelets were positive for BODIPY after DQ-OVA pulse (but not when platelets were fixed), showing active antigen internalization and processing by platelets. (C) Platelets isolated from mice after sham or CLP-induced sepsis actively internalized and proteolyzed DQ-OVA (n = 8-14). Fixed platelets pulsed with DQ-OVA were used as negative control. (D) Human platelets from both healthy donors and patients with sepsis actively internalized and proteolyzed DQ-OVA (n = 6-8). (E) Platelets isolated from sham or CLP mice were pulsed with OVA in vitro for 6 hours, and the OVA antigen peptide–MHC-I complex (SIIN:H-2Kb) was detected. As the arrows demonstrate, we observed significantly increased SIIN:H-2Kb expression in OVA-pulsed platelets from CLP mice compared with sham mice, suggesting increased antigen cross-presentation by platelets during sepsis. Summarized percentages of SIIN:H-2Kb platelets are shown on the right. Platelets from CLP mice showed significantly increased antigen cross-presentation (n = 9-12). (F) Mice received OVA without adjuvant (3 mg per day) for 3 consecutive days, and on day 4, SIIN:H-2Kb expression on the surface of platelets was measured by flow cytometry (n = 4-5; representative of 3 independent experiments). (G) SIIN:H-2Kb expression on the surface of platelets after OVA pulse positively correlated with total H-2Kb expression (n = 4-5; representative of 3 independent experiments). One-way analysis of variance with Tukey post hoc analysis, unpaired Student t test, and simple linear regression test were used. ****P < .0001. MFI, mean fluorescence intensity; plts, platelets; WGA, wheat germ agglutinin.
We then assessed whether platelets are able to load processed antigen peptides onto their MHC-I complexes and, if so, whether peptide loading onto MHC-I was altered during sepsis. Using an antibody specific for the immunogenic OVA peptide SIIN (amino acid residues 257-264) bound to the MHC-I H-2Kb allele,29 antigen cross-presentation by OVA-pulsed platelets from sham and CLP mice was examined by immunofluorescent microscopy. Platelets were counterstained with the membrane marker wheat germ agglutinin Alexa Fluor 555. Only a few OVA-pulsed platelets from sham control mice were positive for SIIN:H-2Kb (Figure 1E, top panel). However, this antigen cross-presentation was significantly increased in platelets from CLP mice with sepsis (Figure 1E, bottom panel; quantification on the right). As an alternative method, we also measured the mean fluorescence intensity of SIIN:H-2Kb on the surface of platelets by flow cytometry as an indicator of antigen cross-presentation by MHC-I. We observed a modest but significant increase in antigen cross-presentation by platelets from CLP mice as compared with sham controls, which correlated with H-2Kb expression (Figure 1F-G). Increased SIIN:H-2Kb expression on platelets from CLP mice was detected by 2 hours, peaked at 6 hours, and remained elevated for up to 24 hours (data not shown). This suggests that although platelets actively internalize and process exogenous antigens under both healthy and septic conditions, cross-presentation of exogenous antigen peptides is increased during sepsis.
MHC-I on human and murine platelets is increased during sepsis
We and others have reported that the platelet transcriptome and proteome are dynamically altered during sepsis.13,30 We therefore evaluated whether increased antigen cross-presentation by platelets during sepsis is associated with upregulation of MHC-I genes.
We first interrogated a platelet RNA sequencing data set from clinical and experimental sepsis that we recently published.13 The messenger RNA expression of MHC-I alleles HLA-A, HLA-B, and HLA-C were significantly upregulated in human platelets during clinical sepsis (Figure 2A; supplemental Figure 2A). Genes mediating other components of the MHC-I pathway, including complex assembly, antigen loading, and transportation, such as calreticulin (CALR), calnexin (CANX), ERp57 (PDIA3), and TAPBP, were also increased in platelets during clinical sepsis (supplemental Figure 2B). Increased MHC-I expression on platelets was independently confirmed in a larger cohort of patients with sepsis by real-time PCR, flow cytometry, immunofluorescent microscopy, and western blotting (Figure 2B-D; supplemental Figure 2D). MHC-I expression was not significantly associated with age (R2 = 0.00002; P = .98) or sex (P = .63) in either healthy donors or patients with sepsis. The expression of MHC-II alleles was not increased in human platelets during sepsis (supplemental Figure 2C), suggesting a degree of selectivity for upregulation of the MHC-I pathway. Because platelets are anucleate, we also evaluated whether agonists generated in sepsis (eg, IFN-γ and LPS) upregulate MHC-I expression in megakaryocytes. Both IFN-γ and LPS significantly increased MHC-I expression on human CD34+–derived megakaryocytes (supplemental Figure 2E).
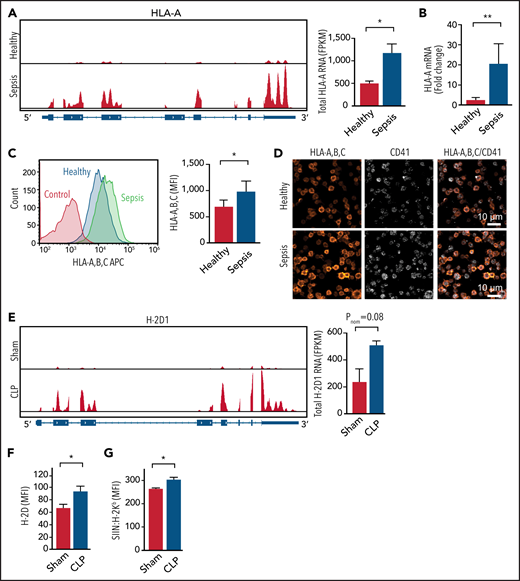
MHC-I expression increases in both human and mouse platelets during sepsis. (A) IGV snapshots on the left show representative reads of 1 MHC-I allele HLA-A by RNA sequencing (RNA-seq) in platelets from a healthy donor and a patient with sepsis. The summarized HLA-A RNA expression in platelets from healthy donors and septic patients is shown at the right (n = 5). (B) Fold increase of HLA-A messenger RNA (mRNA) expression detected by quantitative real-time PCR in platelets from patients with sepsis compared with platelets from healthy donors normalized to human TUBB1 (n = 8-9). (C) Representative flow plots on the left showing increased MHC-I on the surface of platelets from a patient with sepsis compared with platelets from a healthy donor and summarized quantification on the right (n = 20). (D) Representative immunofluorescent images showing increased MHC-I in platelets from a patient with sepsis compared with platelets from a healthy donor. (E) IGV snapshots on the left show representative reads of H-2D1 from control and CLP mice by RNA-seq (n = 3-4). (F) Increased expression of H-2D on platelet plasma membrane as detected by flow cytometry (n = 7-14). (G) Increased antigen cross-presentation in vivo as measured by SIIN:H-2Kb expression on platelets from mice after OVA treatment (n = 3-4). An unpaired Student t test and Mann-Whitney test were used. *P < .05, **P < .01. FPKM, fragments per kilobase; MFI, mean fluorescence intensity.
MHC-I expression increases in both human and mouse platelets during sepsis. (A) IGV snapshots on the left show representative reads of 1 MHC-I allele HLA-A by RNA sequencing (RNA-seq) in platelets from a healthy donor and a patient with sepsis. The summarized HLA-A RNA expression in platelets from healthy donors and septic patients is shown at the right (n = 5). (B) Fold increase of HLA-A messenger RNA (mRNA) expression detected by quantitative real-time PCR in platelets from patients with sepsis compared with platelets from healthy donors normalized to human TUBB1 (n = 8-9). (C) Representative flow plots on the left showing increased MHC-I on the surface of platelets from a patient with sepsis compared with platelets from a healthy donor and summarized quantification on the right (n = 20). (D) Representative immunofluorescent images showing increased MHC-I in platelets from a patient with sepsis compared with platelets from a healthy donor. (E) IGV snapshots on the left show representative reads of H-2D1 from control and CLP mice by RNA-seq (n = 3-4). (F) Increased expression of H-2D on platelet plasma membrane as detected by flow cytometry (n = 7-14). (G) Increased antigen cross-presentation in vivo as measured by SIIN:H-2Kb expression on platelets from mice after OVA treatment (n = 3-4). An unpaired Student t test and Mann-Whitney test were used. *P < .05, **P < .01. FPKM, fragments per kilobase; MFI, mean fluorescence intensity.
We next examined the expression of MHC-I in murine platelets during CLP-induced experimental sepsis. Similar to changes in human septic platelets, the expression of the murine MHC-I allele H-2D was upregulated during CLP sepsis (Figure 2E). Other murine MHC-I alleles and genes involved in the antigen presentation pathway were also consistently increased in our RNA sequencing data set (supplemental Figure 3A-B). Similar to platelets from patients with sepsis, MHC-II alleles were not increased in murine platelets during experimental sepsis (supplemental Figure 3C). We confirmed in an independent set of experiments that H-2D surface protein expression was also significantly increased on murine platelets during CLP by flow cytometry and total MHC-I expression by western blotting (Figure 2F; supplemental Figure 3D). Increased MHC-I expression on platelets during sepsis was accompanied by increased antigen presentation in vitro (Figure 1E) and in vivo (Figure 2G). These data indicate that platelet MHC-I expression and activity are increased in platelets during clinical and experimental sepsis.
Platelets interact via MHC-I with antigen-specific CD8+ T cells during sepsis
A primary function of MHC-I is to present peptides to antigen-specific TCRs on CD8+ T cells. During sepsis, whether exogenous antigen cross-presentation by platelets increases engagement of antigen-specific CD8+ T cells is unknown. To establish this, we pulsed platelets with the OVA peptide SIIN. We then seeded platelets onto fibrinogen-coated chamber wells, cocultured platelets with CD8+ T cells from Rag2−/− OT-I mice at a ratio of 10:1, and measured CD8+ T-cell binding (Figure 3A, schematic). Rag2−/− OT-I is a transgenic mouse strain that expresses SIIN:H-2Kb-specific TCRs on CD8+ T cells.31-33 Therefore, CD8+ T cells from Rag2−/− OT-I mice are commonly used for studying antigen-specific interactions between MHC-I and CD8+ T cells. As shown in Figure 3B and supplemental Videos 1 and 2, very few CD8+ T cells bound to unpulsed platelets from either CLP or sham control mice. In contrast, CD8+ T cells readily bound to SIIN-pulsed platelets, and the number of attached CD8+ T cells was significantly increased during sepsis (Figure 3C; supplemental Videos 3 and 4). Because fibrinogen can activate platelets, we also seeded platelets onto poly-L-lysine–coated chambers and cocultured platelets with CD8+ T cells to examine interactions between quiescent platelets and CD8+ T cells. As shown in supplemental Figure 4, consistent with our previous findings, platelets from CLP mice induced significantly increased CD8+ T-cell attachment. These findings indicate that platelet MHC-I can present antigen peptides and engage antigen-specific CD8+ T cells and that this is increased during sepsis.
![Platelet MHC-I mediates increased interactions between platelets and antigen-specific CD8+ T cells during sepsis. (A) To study whether the antigen–MHC-I complex on platelets can directly engage antigen-specific CD8+ T cells, platelets were isolated from either sham or CLP mice, pulsed with SIIN for 2 hours, washed, seeded onto fibrinogen-coated chamber wells, and cocultured with SIIN:H-2Kb-specific CD8+ T cells from Rag2−/− OT-I mice. Unbound cells were washed off. Attached CD8+ T cells were fixed and stained for confocal immunofluorescent imaging. (B) Representative Z stack maximum projection images showing an increased number of CD8+ T cells (labeled with 4′,6-diamidino-2-phenylindole [DAPI] and CD8) attached to platelets (labeled with GPIb) from CLP mice compared with sham mice. Yellow arrows illustrate CD8+ T cells that are attached to platelets. (C) Quantification of CD8+ T cells per image shows the interaction between platelets and CD8+ T cells in an antigen-specific manner (n = 15). One-way analysis of variance with Tukey post hoc analysis was used. **P < .01, ****P < .0001. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/5/10.1182_blood.2020008958/4/m_bloodbld2020008958f3.png?Expires=1769092730&Signature=ajDKKVsg5tBjGTixGwARcXkCji1w5ios5u6X6xFgpwYS2SXM~KAhwfzccshMjBVya3BL0iXZJdR41osu4AijTW5KNrB487MkpFjoa-3QTDjpLUp8HtueAHENQahzebZC6Ts2uQa20blmUkL~xr3eZU6-y9IHmwg5-csB83cLh-dvrD4dRrQ~-pjzB0YhKC16sIW~0h-OvalcM0eM9ysysjLw4nLmXRSZNi9Kw41S~5L19E3htiIjUJpmCCHOeIm19mCaQYWikw2zqvN2RmRv40zsw~4VDkAlcLm1go7yBwih8GWktfJLaGRlEv~tZ7RqqM9rGGwn~jKRZoZkaxt49w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Platelet MHC-I mediates increased interactions between platelets and antigen-specific CD8+ T cells during sepsis. (A) To study whether the antigen–MHC-I complex on platelets can directly engage antigen-specific CD8+ T cells, platelets were isolated from either sham or CLP mice, pulsed with SIIN for 2 hours, washed, seeded onto fibrinogen-coated chamber wells, and cocultured with SIIN:H-2Kb-specific CD8+ T cells from Rag2−/− OT-I mice. Unbound cells were washed off. Attached CD8+ T cells were fixed and stained for confocal immunofluorescent imaging. (B) Representative Z stack maximum projection images showing an increased number of CD8+ T cells (labeled with 4′,6-diamidino-2-phenylindole [DAPI] and CD8) attached to platelets (labeled with GPIb) from CLP mice compared with sham mice. Yellow arrows illustrate CD8+ T cells that are attached to platelets. (C) Quantification of CD8+ T cells per image shows the interaction between platelets and CD8+ T cells in an antigen-specific manner (n = 15). One-way analysis of variance with Tukey post hoc analysis was used. **P < .01, ****P < .0001. ns, not significant.
Platelet MHC-I mediates increased interactions between platelets and antigen-specific CD8+ T cells during sepsis. (A) To study whether the antigen–MHC-I complex on platelets can directly engage antigen-specific CD8+ T cells, platelets were isolated from either sham or CLP mice, pulsed with SIIN for 2 hours, washed, seeded onto fibrinogen-coated chamber wells, and cocultured with SIIN:H-2Kb-specific CD8+ T cells from Rag2−/− OT-I mice. Unbound cells were washed off. Attached CD8+ T cells were fixed and stained for confocal immunofluorescent imaging. (B) Representative Z stack maximum projection images showing an increased number of CD8+ T cells (labeled with 4′,6-diamidino-2-phenylindole [DAPI] and CD8) attached to platelets (labeled with GPIb) from CLP mice compared with sham mice. Yellow arrows illustrate CD8+ T cells that are attached to platelets. (C) Quantification of CD8+ T cells per image shows the interaction between platelets and CD8+ T cells in an antigen-specific manner (n = 15). One-way analysis of variance with Tukey post hoc analysis was used. **P < .01, ****P < .0001. ns, not significant.
Platelet MHC-I suppresses CD8+ T-cell numbers and functions in vivo during sepsis
To evaluate the in vivo significance of the interactions between platelet MHC-I and CD8+ T cells in sepsis, we used a platelet lineage–specific B2M knockout mouse strain (B2Mf/f-Pf4Cre) and littermate control mice (B2Mf/f; referred to herein as B2M+/+; Figure 4A).34 B2M, which encodes β2-microglobulin, is required for MHC-I complex assembly and antigen cross-presentation. MHC-I expression was intact on platelets from B2m+/+ mice (Figure 4B). Consistent with our findings in C57BL/6J WT mice with sepsis (Figure 2E-F; supplemental Figure 3A,D), platelet MHC-I expression was upregulated in B2M+/+ mice after CLP sepsis (Figure 4B). In contrast, MHC-I expression on platelets from B2Mf/f-Pf4Cre mice was defective and did not increase after CLP (Figure 4B). As shown in supplemental Figure 5, MHC-I expression on total leukocytes, dendritic cells, and monocytes was intact in both B2Mf/f-Pf4Cre and littermate control B2M+/+ mice.

Platelet MHC-I mediates CD8+ T-cell suppression during sepsis in vivo. (A) To study the function of platelet MHC-I function in vivo, we used a platelet lineage–specific B2M knockout strain (B2Mf/f -Pf4Cre). A representative western blot probed for B2M in platelets from a B2Mf/f-Pf4Cre mouse and littermate B2M+/+ control mouse is shown. (B) B2M+/+ or B2Mf/f-Pf4Cre mice were subjected to sham or CLP and euthanized on day 4. H-2D expression on platelets was measured by flow cytometry. Only platelets from B2M+/+ mice had increased H-2D expression during sepsis. Platelets from B2Mf/f -Pf4Cre mice remained negative and unchanged (n = 7-13). (C) Representative flow plots of the CD8+ T-cell population from sham or CLP mouse mesenteric lymph nodes. B2M+/+ mice had decreased CD8+ T cells when subjected to CLP, whereas the B2Mf/f-Pf4Cre mice did not. (D-E) Summarized CD8+ T-cell frequencies and absolute numbers, CD44highCD8+ T-cell frequencies, and CD4+ T-cell frequencies in the mesenteric lymph nodes (D) and peripheral blood (E) (n = 10-24). Upper bracket compares the percentage of CD8+ T cells between littermate control sham (eg, baseline) and B2M knockout during CLP. This bracket is included to visually highlight that the absence of MHC-I results in near-normal levels of CD8+ T cells during sepsis. One-way analysis of variance with Tukey post hoc analysis was used. *P < .05, **P < .01, ***P < .001. ns, not significant.
Platelet MHC-I mediates CD8+ T-cell suppression during sepsis in vivo. (A) To study the function of platelet MHC-I function in vivo, we used a platelet lineage–specific B2M knockout strain (B2Mf/f -Pf4Cre). A representative western blot probed for B2M in platelets from a B2Mf/f-Pf4Cre mouse and littermate B2M+/+ control mouse is shown. (B) B2M+/+ or B2Mf/f-Pf4Cre mice were subjected to sham or CLP and euthanized on day 4. H-2D expression on platelets was measured by flow cytometry. Only platelets from B2M+/+ mice had increased H-2D expression during sepsis. Platelets from B2Mf/f -Pf4Cre mice remained negative and unchanged (n = 7-13). (C) Representative flow plots of the CD8+ T-cell population from sham or CLP mouse mesenteric lymph nodes. B2M+/+ mice had decreased CD8+ T cells when subjected to CLP, whereas the B2Mf/f-Pf4Cre mice did not. (D-E) Summarized CD8+ T-cell frequencies and absolute numbers, CD44highCD8+ T-cell frequencies, and CD4+ T-cell frequencies in the mesenteric lymph nodes (D) and peripheral blood (E) (n = 10-24). Upper bracket compares the percentage of CD8+ T cells between littermate control sham (eg, baseline) and B2M knockout during CLP. This bracket is included to visually highlight that the absence of MHC-I results in near-normal levels of CD8+ T cells during sepsis. One-way analysis of variance with Tukey post hoc analysis was used. *P < .05, **P < .01, ***P < .001. ns, not significant.
We next examined CD8+ T-cell numbers and frequency in the B2Mf/f-Pf4Cre and B2M+/+ mice subjected to CLP or sham conditions. Published studies have found that the number of CD8+ T cells are reduced after clinical and CLP-triggered sepsis.1,35-38 Consistent with this, we also found that the numbers of CD8+ T cells were significantly decreased in B2M+/+ mice after CLP sepsis when we measured the CD3+CD8+ population in the viable CD45+ leukocytes isolated from the mesenteric lymph nodes (the draining lymph nodes during CLP sepsis), peripheral blood, spleen, and nondraining lymph nodes (Figure 4C-E; supplemental Figure 6A). In contrast, CD8+ T-cell numbers from B2Mf/f-Pf4Cre mice (where platelet MHC-I was defective) did not significantly change during sepsis in mesenteric lymph nodes and were at levels comparable to those in nonseptic sham control conditions (Figure 4C-D). Consistent findings were observed in CD8+ T cells from the peripheral blood and spleen, although levels were incompletely restored in the spleen (Figure 4E; supplemental Figure 6A). We further characterized the CD8+ T-cell populations, including the CD62LhighCD44low naïve CD8+ T cells and CD44high effector/memory CD8+ T cells. During sepsis, CD44highCD8+ T cells were decreased in the mesenteric lymph nodes of B2M+/+ mice but did not change in B2Mf/f-Pf4Cre mice (Figure 4D). There was no difference in CD62LhighCD44low naïve CD8+ T cells between B2M+/+ and B2Mf/f-Pf4Cre mice (data not shown). IFN-γ production is a hallmark of CD8+ T-cell function, and we measured the intracellular IFN-γ production in CD8+ T cells. Splenic CD8+ T cells from B2M+/+ mice subjected to CLP sepsis showed suppressed IFN-γ intracellular expression (supplemental Figure 6B). In contrast, IFN-γ production by CD8+ T cells in B2Mf/f-Pf4Cre mice subjected to CLP sepsis was similar to that in sham control conditions (supplemental Figure 6B). We also examined the number of CD8+ T cells in nondraining lymph nodes during sepsis (including cervical and axillary lymph nodes) but did not see any differences between B2M+/+ and B2Mf/f-Pf4Cre mice (data not shown). In addition, we examined CD4+ T cells in mesenteric lymph nodes, peripheral blood, and spleen. As anticipated, CD4+ T-cell numbers and frequency were not affected by platelet MHC-I deficiency (Figure 4D-E; supplemental Figure 6A). These data suggest that both CD8+ T-cell numbers and functions (eg, IFN-γ production) are regulated by platelet MHC-I during sepsis.
To assess whether loss of B2M might alter platelet count or size during sepsis, we measured platelet count and mean platelet volume. There was no significant difference between B2M+/+ and B2Mf/f-Pf4Cre mice (supplemental Figure 7A-B). We also examined the monocytes in the spleen and mesenteric lymph nodes after CLP, because platelet-derived β2M has been shown to be able to regulate monocytes in vivo.34 However, there were no differences in the number of monocytes between B2M+/+ and B2Mf/f-Pf4Cre mice (data not shown). In addition, we measured the serum AST levels as an indicator of liver injury in sepsis. Both B2M+/+ and B2Mf/f-Pf4Cre mice showed similarly increased AST as compared with sham control mice (supplemental Figure 7C).
Platelet MHC-I mediates decreased antigen-specific CD8+ T-cell proliferation in vitro during sepsis
Our in vivo data suggest that platelet MHC-I mediates decreased numbers of CD8+ T cells in sepsis. This suppression could occur through direct interactions between platelets and CD8+ T cells or through indirect interactions with other cells or molecules. We therefore next evaluated whether platelet MHC-I could mediate direct suppression of antigen-specific CD8+ T cells in vitro. Because antigen-specific CD8+ T-cell proliferation was previously shown to be reduced in sepsis,39 we carried out CD8+ T-cell proliferation assays in the presence of platelets from WT and MHC-I–deficient mice under sham or CLP sepsis conditions. We stained CD8+ T cells from Rag2−/− OT-I mice with the proliferation dye V450, which was incorporated into live cells. With each cell division, the V450 dye transfers to a new generation of cells, and the fluorescent intensity in each cell is approximately half of the previous. SIIN-pulsed and LPS-stimulated CD45.1 splenocytes were fixed and added to the coculture to induce consistent baseline levels of CD8+ T-cell proliferation. Platelets were pulsed with SIIN, washed, and added to the culture at a platelet/CD8+ T-cell ratio of 50:1 to mimic the estimated ratio in peripheral blood (Figure 5A, schematic). Baseline CD8+ T cells proliferated at a rate of ∼8% to 9% (Figure 5B). The addition of platelets from sham-treated C57BL/6J WT mice, as an additional control, did not substantially alter the rate of baseline CD8+ T-cell proliferation (∼10%; Figure 5C, left panel). In contrast and consistent with our in vivo findings, CD8+ T-cell proliferation was suppressed by ∼30% by platelets from C57BL/6 WT mice with sepsis (Figure 5C, right panel; Figure 5D). We next tested the effect of platelets from either B2M+/+ control or B2Mf/f-Pf4Cre mice on CD8+ T-cell proliferation during sepsis. Platelets from B2Mf/f-Pf4Cre mice, where B2M and therefore MHC-I were absent, showed near-normal levels of CD8+ T-cell proliferation (∼8%; Figure 5E-F). These data suggest that platelet MHC-I directly mediates CD8+ T-cell proliferation during sepsis.

Platelets from mice with sepsis directly induce antigen-specific CD8+ T-cell suppression in vitro. (A) Platelets isolated from B2M+/+ or B2Mf/f-Pf4Cre mice on day 4 after CLP were pulsed with SIIN for 2 hours, washed, and cocultured with antigen-specific CD8+ T cells isolated from Rag2−/− OT-I mice for 60 hours at 37°C in uncoated cell culture plates. (B) Representative flow plots for CD8+ T-cell proliferation at baseline. CD8+ T cells were stained with the proliferation dye V450 before coincubation. (C-D) A representative flow plot and quantitation demonstrating decreased CD8+ T-cell proliferation when CD8+ T cells were incubated with platelets isolated from C57BL/6 mice after CLP compared with sham. Proliferation was normalized to CD8+ T cells incubated with platelets from sham mice. (E-F) A representative flow plot and quantitation demonstrating suppressed CD8+ T-cell proliferation during sepsis, when CD8+ T cells were incubated with platelets isolated from B2M+/+ mice compared with platelets from B2Mf/f-Pf4Cre mice. Proliferation was normalized to CD8+ T cells incubated with platelets from B2Mf/f-Pf4Cre mice after CLP. All plots are representative of 3 independent experiments. An unpaired Student t test and Mann-Whitney test were used. *P < .05. FSC, forward scatter; NIR, near infrared region; SSC, side scatter.
Platelets from mice with sepsis directly induce antigen-specific CD8+ T-cell suppression in vitro. (A) Platelets isolated from B2M+/+ or B2Mf/f-Pf4Cre mice on day 4 after CLP were pulsed with SIIN for 2 hours, washed, and cocultured with antigen-specific CD8+ T cells isolated from Rag2−/− OT-I mice for 60 hours at 37°C in uncoated cell culture plates. (B) Representative flow plots for CD8+ T-cell proliferation at baseline. CD8+ T cells were stained with the proliferation dye V450 before coincubation. (C-D) A representative flow plot and quantitation demonstrating decreased CD8+ T-cell proliferation when CD8+ T cells were incubated with platelets isolated from C57BL/6 mice after CLP compared with sham. Proliferation was normalized to CD8+ T cells incubated with platelets from sham mice. (E-F) A representative flow plot and quantitation demonstrating suppressed CD8+ T-cell proliferation during sepsis, when CD8+ T cells were incubated with platelets isolated from B2M+/+ mice compared with platelets from B2Mf/f-Pf4Cre mice. Proliferation was normalized to CD8+ T cells incubated with platelets from B2Mf/f-Pf4Cre mice after CLP. All plots are representative of 3 independent experiments. An unpaired Student t test and Mann-Whitney test were used. *P < .05. FSC, forward scatter; NIR, near infrared region; SSC, side scatter.
We next evaluated if platelets still suppress CD8+ T-cell activation and proliferation when CD8+ T cells are provided with additional costimulatory signals. Exogenous CD3/CD28 antibodies were added to platelets cocultured with CD8+ T cells to activate CD3 and CD28 on CD8+ T cells.40 As shown in supplemental Figure 8A-B, platelets also reduced CD8+ T-cell proliferation in sepsis, even in the presence of exogenous robustly activating CD3/CD28 antibodies.
Platelet MHC-I regulates antigen-specific CD8+ T-cell responses and outcomes during sepsis
We next sought to determine the impact of platelet MHC-I on antigen-specific CD8+ T-cell responses in vivo. Mice were first administered OVA–complete Freund adjuvant to induce OVA-specific CD8+ T-cell responses, followed by induction of sepsis with CLP and then vaccination with OVA–incomplete Freund adjuvant (Figure 6A, schematic; Figure 6B, gating strategy). Draining lymph nodes were harvested 3 days later, and antigen-specific CD8+ T cells were measured using SIIN:H-2Kb BV421 tetramers. Consistent with our other findings (Figures 4C-E and 5C-F), both the frequency and numbers of antigen-specific CD8+ T cells during CLP sepsis were higher in B2Mf/f-Pf4Cre mice compared with B2M+/+ littermate controls (Figure 6C-D).
![Antigen-specific CD8+ T cells are protected in platelet MHC-I–deficient mice during sepsis. (A) To induce OVA-specific CD8+ T-cell responses, mice were given OVA–complete Freund adjuvant (CFA) immunization 12 days before CLP surgery and OVA–incomplete Freund adjuvant (IFA) 2 days after CLP. On day 5 after CLP, draining lymph nodes (LNs) were collected, and SIIN:H-2Kb tetramers were used to measure the antigen-specific CD8+ T-cell population. (B) Representative flow plots showing the gating strategy for SIIN:H2Kb+ tetramers using splenocytes from Rag2−/− OT-I mice as positive control. (C-D) Representative flow plots (C) and summary of the percentage and absolute number (D) of SIIN:H-2Kb–specific CD8+ T-cell population in the draining LNs from B2M+/+ and B2Mf/f-Pf4Cre mice after CLP. The antigen-specific CD8+ T-cell population was significantly protected in the B2Mf/f-Pf4Cre mice (n = 5). (E) Comparable survival of the platelet MHC-I–deficient mice (B2Mf/f-Pf4Cre) and littermate controls (B2M+/+) when subjected CLP. (F) To evaluate the impact of the MHC-I–regulated antigen-specific CD8+ T-cell responses in vivo, B2M+/+ or B2Mf/f-Pf4Cre mice were crossed with OT-I mice to generate a platelet MHC-I–deficient CD8+ T-cell transgenic strain of mice (B2M+/+–OT-I [WT OT-I] and B2Mf/f–Pf4Cre–OT-I [KO–OT-I]). (G) WT–OT-I or KO–OT-I mice were immunized with OVA-CFA on day −12 and subjected to CLP on day 0 and OVA-IFA on day 2. Survival was monitored daily for 5 days. Whereas 7 of 10 WT–OT-I mice died by day 5, only 3 of 7 KO–OT-I died. Mann-Whitney and log-rank (eg, Mantel-Cox) survival tests were used. *P < .05, **P < .01. FSC, forward scatter; PE, phycoerythrin; SSC, side scatter; WBC, white blood cell.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/5/10.1182_blood.2020008958/4/m_bloodbld2020008958f6.png?Expires=1769092730&Signature=KztwPy6oZDIWue0mwWG85UXyS4pvBm9tHLuQN0XhDDxDPPjRFNHtlTyiawWMIU8YB4rMGIcNcHWocWAhK9rS49gkB4ejGf6STEIOBEjEKARaw3nsEcSKvZIUA-4wTkkwP2kl4rk1cYwLFkGtFjCi-jWehxZoRUtXiKiMLD4Kcsm~aMrpCtBFJDKSCz7Y6Pqc~SWQ-H~TIzQ74JYIw-82s06kFYpofk0~6j0qm4bzvwsQxRjRtqy-Hf02J7~bp0B2BuhsuZkwtB0qyjQnU01jWFL9APx3jy00CfeMlGIekmZy2r9xab9qEnCZ1z9Ir~5dKYi6la8l1YOg9tjNoMrQSw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Antigen-specific CD8+ T cells are protected in platelet MHC-I–deficient mice during sepsis. (A) To induce OVA-specific CD8+ T-cell responses, mice were given OVA–complete Freund adjuvant (CFA) immunization 12 days before CLP surgery and OVA–incomplete Freund adjuvant (IFA) 2 days after CLP. On day 5 after CLP, draining lymph nodes (LNs) were collected, and SIIN:H-2Kb tetramers were used to measure the antigen-specific CD8+ T-cell population. (B) Representative flow plots showing the gating strategy for SIIN:H2Kb+ tetramers using splenocytes from Rag2−/− OT-I mice as positive control. (C-D) Representative flow plots (C) and summary of the percentage and absolute number (D) of SIIN:H-2Kb–specific CD8+ T-cell population in the draining LNs from B2M+/+ and B2Mf/f-Pf4Cre mice after CLP. The antigen-specific CD8+ T-cell population was significantly protected in the B2Mf/f-Pf4Cre mice (n = 5). (E) Comparable survival of the platelet MHC-I–deficient mice (B2Mf/f-Pf4Cre) and littermate controls (B2M+/+) when subjected CLP. (F) To evaluate the impact of the MHC-I–regulated antigen-specific CD8+ T-cell responses in vivo, B2M+/+ or B2Mf/f-Pf4Cre mice were crossed with OT-I mice to generate a platelet MHC-I–deficient CD8+ T-cell transgenic strain of mice (B2M+/+–OT-I [WT OT-I] and B2Mf/f–Pf4Cre–OT-I [KO–OT-I]). (G) WT–OT-I or KO–OT-I mice were immunized with OVA-CFA on day −12 and subjected to CLP on day 0 and OVA-IFA on day 2. Survival was monitored daily for 5 days. Whereas 7 of 10 WT–OT-I mice died by day 5, only 3 of 7 KO–OT-I died. Mann-Whitney and log-rank (eg, Mantel-Cox) survival tests were used. *P < .05, **P < .01. FSC, forward scatter; PE, phycoerythrin; SSC, side scatter; WBC, white blood cell.
Antigen-specific CD8+ T cells are protected in platelet MHC-I–deficient mice during sepsis. (A) To induce OVA-specific CD8+ T-cell responses, mice were given OVA–complete Freund adjuvant (CFA) immunization 12 days before CLP surgery and OVA–incomplete Freund adjuvant (IFA) 2 days after CLP. On day 5 after CLP, draining lymph nodes (LNs) were collected, and SIIN:H-2Kb tetramers were used to measure the antigen-specific CD8+ T-cell population. (B) Representative flow plots showing the gating strategy for SIIN:H2Kb+ tetramers using splenocytes from Rag2−/− OT-I mice as positive control. (C-D) Representative flow plots (C) and summary of the percentage and absolute number (D) of SIIN:H-2Kb–specific CD8+ T-cell population in the draining LNs from B2M+/+ and B2Mf/f-Pf4Cre mice after CLP. The antigen-specific CD8+ T-cell population was significantly protected in the B2Mf/f-Pf4Cre mice (n = 5). (E) Comparable survival of the platelet MHC-I–deficient mice (B2Mf/f-Pf4Cre) and littermate controls (B2M+/+) when subjected CLP. (F) To evaluate the impact of the MHC-I–regulated antigen-specific CD8+ T-cell responses in vivo, B2M+/+ or B2Mf/f-Pf4Cre mice were crossed with OT-I mice to generate a platelet MHC-I–deficient CD8+ T-cell transgenic strain of mice (B2M+/+–OT-I [WT OT-I] and B2Mf/f–Pf4Cre–OT-I [KO–OT-I]). (G) WT–OT-I or KO–OT-I mice were immunized with OVA-CFA on day −12 and subjected to CLP on day 0 and OVA-IFA on day 2. Survival was monitored daily for 5 days. Whereas 7 of 10 WT–OT-I mice died by day 5, only 3 of 7 KO–OT-I died. Mann-Whitney and log-rank (eg, Mantel-Cox) survival tests were used. *P < .05, **P < .01. FSC, forward scatter; PE, phycoerythrin; SSC, side scatter; WBC, white blood cell.
Prior studies have demonstrated that reduced CD8+ T-cell numbers and functions in sepsis are associated with adverse outcomes.2,35,36,41 We therefore next assessed whether interactions between platelet MHC-I and CD8+ T cells regulated outcomes in sepsis. CLP is a polymicrobial sepsis model. As expected, mortality from CLP sepsis did not differ between B2M+/+ and B2Mf/f-Pf4Cre mice, because both these strains of mice have a diverse TCR repertoire in their CD8+ T-cell population (Figure 6E). We next evaluated the impact of antigen-specific interactions between platelet MHC-I and CD8+ T cells on sepsis outcomes. To do this, we crossed B2M+/+ and B2Mf/f-Pf4Cre mice with OT-I mice to generate a platelet MHC-I–deficient, CD8+ T-cell transgenic strain with a dominant OVA-specific CD8+ T-cell population. We have employed the following nomenclature for these 2 strains: B2M+/+–OT-I (WT–OT-I), because their platelet MHC-I expression is intact and CD8+ T cells are transgenic, and B2Mf/f–Pf4Cre–OT-I (KO–OT-I), because their platelet MHC-I expression is defective and the CD8+ T cells are transgenic (Figure 6F). When the mice were subjected to CLP, sepsis-associated mortality trended lower in KO–OT-I mice, compared with WT–OT-I littermate controls (survival, 60% vs 30%; P = .35; Figure 6G).
Discussion
Platelets are dynamic effector cells that orchestrate immune and inflammatory host responses during infectious and inflammatory diseases.8,42,43 Interactions between platelets and CD8+ T cells are generally less well understood. In our current study, we found that platelets actively internalize and process exogenous antigens and present antigen peptides to antigen-specific CD8+ T cells. In sepsis, platelet MHC-I is increased, leading to decreased CD8+ T-cell numbers, reduced CD8+ T cell proliferation, and impaired CD8+ T-cell intracellular IFN-γ production.
CD8+ T-cell responses during sepsis have unique features compared with other infectious settings. For example, during malaria, CD8+ T-cell numbers and functional responses are generally upregulated.21 In contrast, during sepsis, CD8+ T-cell numbers and functional responses are suppressed and associated with increased secondary infections and mortality.2,6,36,44 Reduced CD8+ T-cell numbers during sepsis involve all CD8+ T-cell compartments, including naïve, effector, and memory cells.2,5,6 Sepsis may also suppress CD8+ T-cell responses against heterologous pathogens by diminishing the diversity of naïve CD8+ T cells and causing loss of CD8+ T-cell effector functions.35,36,38 MHC-I is a key molecular complex regulating antigen presentation to CD8+ T cells and subsequent CD8+ T-cell responses. Although platelets are known to possess robust levels of MHC-I and signal to CD8+ T cells,21 whether platelet MHC-I contributes to CD8+ T-cell suppression during sepsis has not been examined.
Antigen presentation includes antigen internalization and processing into peptides, binding of antigen peptides to MHC-I, and transportation to the cell surface.45 In vitro activated platelets are capable of antigen cross-presentation, indicating that platelets can internalize and process OVA.21 Megakaryocytes are also capable of antigen cross-presentation.22,46 However, the dynamics and efficacy of the internalization and proteolysis processes have not been evaluated. When we pulsed platelets with an exogenous antigen (eg, DQ-OVA, commonly used to evaluate antigen internalization and processing by APCs47 ), human and murine platelets robustly internalized and processed exogenous antigen within an hour. Moreover, processed peptides were loaded onto platelet MHC-I through cross-presentation.
Platelets from mice with sepsis have significantly increased antigen cross-presentation. MHC-I complexes are often the limiting factor for the efficiency of antigen presentation and cross-presentation by APCs.45 We also observed that increased antigen cross-presentation by platelets during sepsis was associated with increased expression of MHC-I alleles in platelets. In addition to sepsis, increased platelet MHC-I was also previously demonstrated in mice infected with the malaria strain P berghei.21 Although we did not measure the processing of specific viral or bacterial pathogens, platelets have been shown to be capable of internalizing influenza virus.11 Recent work by Pariser et al46 has also shown that megakaryocytes (platelet precursor cells) internalize E coli. These findings indicate that platelets are able to internalize other antigens and/or pathogens besides SIIN. Although OVA cannot passively enter cells, OVA can be internalized through phagocytosis or receptor-mediated endocytosis.48,49 Proteasome-mediated proteolysis is a common mechanism in other cells.50,51 However, based on our data, platelets seem to process OVA in a proteasome-independent mechanism under the conditions of our experiments. It may be that autophagosomes and lysosomes, which generate antigen peptides in other cells,52,53 are responsible for processing exogenous antigen in platelets.
In our current study, increased platelet MHC-I expression during sepsis also resulted in greater platelet–CD8+ T-cell interactions in vitro. Our previous studies found increased platelet-leukocyte aggregates, including platelet–CD8+ T-cell aggregates, in the peripheral blood from patients with sepsis and COVID.14,19 Consistently, recent work by Albayati et al54 also showed that platelet–T-cell aggregates are increased after CLP sepsis and that the platelet P2Y12 receptor regulates direct platelet–T-cell aggregation formation as well as T regulatory cell numbers and functions. Published work has shown that dendritic cell–derived exosomes are able to regulate CD8+ T-cell numbers and functions.55 Platelets release soluble and exosome-packaged mediators upon activation. In preliminary experiments, we did not observe any evidence that platelet releasate suppresses CD8+ T-cell proliferation during CLP sepsis (data not shown). Whether direct platelet binding to CD8+ T cells is required for T-cell responses remains largely unknown. Although our data suggest that platelet MHC-I engages CD8+ T cells in an antigen-specific manner, we cannot completely exclude the possibility that soluble factors, exosomes, or microvesicles released by platelets during sepsis may contribute to altered CD8+ T-cell numbers or functions.
In WT mice where platelet MHC-I was intact and upregulated, CD8+ T cell numbers, proliferation, and IFN-γ production were significantly reduced during sepsis. This is consistent with prior studies showing decreased CD8+ T-cell numbers in lymphoid organs and peripheral blood during experimental and clinical sepsis.35,37,41,56,57 When we examined CD8+ T cells in mice with sepsis lacking platelet MHC-I, CD8+ T-cell numbers, proliferation, and levels of IFN-γ production were restored to normal or near-normal levels. We could not clearly attribute these findings to other cells, including classical APCs, because we did not identify any differences in the numbers of dendritic cells, monocytes, or leukocytes between platelet MHC-I–sufficient and MHC-I–deficient mice. Furthermore, by using MHC-I tetramers, we identified that the number of antigen specific CD8+ T cells is maintained during sepsis when platelet MHC-I is absent. In a small sample size, preservation of CD8+ T-cell numbers and functions in the absence of platelet MHC-I was associated with a trend toward reduced mortality that did not reach significance after CLP sepsis in a platelet MHC-I–deficient CD8+ T-cell transgenic strain (B2Mf/f-Pf4Cre/OT-I). Although this is consistent with studies where either pharmacological or genetic rescue of CD8+ T-cell numbers improved survival in mice after CLP sepsis,41,58 these findings will need to be examined further in future studies.
Strengths of our study include parallel investigations in human and murine platelets during sepsis and concordant findings that MHC-I is upregulated in both clinical and experimental sepsis. We also used mouse models where MHC-I alleles and CD8+ T-cell epitopes are known, and peptide–MHC-I tetramers to quantify antigen-specific CD8+ T-cell responses in sepsis. We recognize that our patients with sepsis and healthy donors were not matched on the variable of age. However, based on published platelet RNA data sets from healthy human donors, MHC-I expression does not significantly change with aging.59,60 Consistent with this, there was no association between MHC-I allele expression and age in our patient cohorts. We also appreciate the possibility that cosignaling ligands for CD8+ T cells may also mediate platelet MHC-I-CD8+ T-cell interactions.25 Finally, although dendritic cells may suppress CD8+ T-cell functions, we did not observe any differences in dendritic cell numbers during sepsis or between platelet MHC-I–sufficient and platelet MHC-I–deficient mice.39
In summary, the results from this study indicate that through platelet MHC-I, which is upregulated similarly during clinical and murine sepsis, platelets internalize, process, and present antigens to CD8+ T cells. Through this mechanism, increased MHC-I on platelets during sepsis results in suppressed CD8+ T-cell numbers, proliferation, and functional responses.
The RNA sequencing data was published and are publicly available (Bioproject PRJNA521077).13
Requests for materials can be directed to the lead contact Li Guo (amilyg@u2m2.utah.edu), or corresponding authors, Andrew S. Weyrich (andy.weyrich@utah.edu) and Matthew T. Rondina (matthew.rondina@hsc.utah.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors acknowledge figure preparation expertise from Diana Lim; experiment technical support from Claudia Araujo, Mark Cody, Zhonglou Sun, Hu Dai, and Xiaoming Wang; assistance with participant recruitment from Antoinette Blair, Macy Barrios, Amber Plante, Jordan Greer, Amy DeNardo, Amanda Bailey, and Lindsey Waddoups; and thoughtful discussions from Guy A. Zimmerman, Weiquan (Wendy) Zhu, and Matthew A. Williams.
This work was supported by grants K01AG059892 (National Institute on Aging [NIA]) (R.A.C.), R35HL145237 (National Heart, Lung, and Blood Institute [NHLBI]) (A.S.W.), R01HL142804 (NHLBI) (M.T.R., A.S.W.), and R01AG048022, R56AG059877 (both NIA), and R01HL130541 (NHLBI) (M.T.R.) from the National Institutes of Health (NIH); in part, by Merit Review Award I01 CX001696 from the US Department of Veterans Affairs Clinical Sciences Research and Development (M.T.R.); by research fellowship KR 4945/1-1 from the German Research Foundation (K.K.); by Fonds voor Wetenschappelijk Onderzoek Vlaanderen grant 12U7818N (F.D.); and by the Flow Cytometry Core at the University of Utah. This material is the result of work supported by resources from and use of facilities at the George E. Wahlen VA Medical Center (Salt Lake City, UT). Research reported in this publication was supported by the National Center for Research Resources, NIH, under award 1S10RR026802-01. Research reported in this publication used the High-Throughput Genomics and Bioinformatic Analysis Shared Resource at Huntsman Cancer Institute, University of Utah, and was supported by the NIH/National Cancer Institute under award P30CA042014.
The contents do not represent the views of the US Department of Veterans Affairs or the US Government. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the NIH.
Authorship
Contribution: L.G. designed and performed experiments, analyzed the data, and wrote the manuscript; S.S., J.W.R., N.D.T., W.J., B.K.M., K.N.M., B.B., Y.K., K.K., F.D., S.P.J., A.S.E., R.A.C., and E.A.M. performed experiments; J.W.R., R.A.C., X.H., A.S.W., and M.T.R. analyzed results; J.W.R., S.M.B, C.N.M, A.S.W., and M.T.R. wrote the manuscript; and all authors reviewed and critically edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Andrew S. Weyrich, University of Utah, University of Utah Health Sciences Center, Eccles Institute of Human Genetics, 15 North 2030 East, Room 4220A, Salt Lake City, UT 84112; e-mail: andy.weyrich@utah.edu; and Matthew T. Rondina, University of Utah, University of Utah Health Sciences Center, Eccles Institute of Human Genetics, 15 North 2030 East, Room 4220A, Salt Lake City, UT 84112; e-mail: matthew.rondina@hsc.utah.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal