


Abstract
Therapy-related myeloid neoplasms (t-MNs) include diseases onsetting in patients treated with chemo- and/or radiotherapy for a primary cancer, or an autoimmune disorder. Genomic variants, in particular, in familial cancer genes, may play a predisposing role. Recent advances in deep sequencing techniques have shed light on the pathogenesis of t-MNs, identifying clonal hematopoiesis of indeterminate potential (CHIP) as a frequent first step in the multihit model of t-MNs. CHIP is often detectable prior to any cytotoxic treatment, probably setting the fertile genomic background for secondary leukemogenesis. The evolution pattern toward t-MNs is then a complex process, shaped by the type of cancer therapy, the aging process, and the individual exposures, that favor additional hits, such as the acquisition of TP53 mutations and unfavorable karyotype abnormalities. The pathogenesis of t-MNs differs from MN associated with environmental exposure. Indeed, the genetic aberration patterns of MN developing in atomic bomb survivors show few mutations in classical DNA methylation genes, and a high prevalence of 11q and ATM alterations, together with TP53 mutations. Survival in t-MNs is poor. In addition to the biology of t-MNs, the patient’s previous disease history and the remission status at t-MN diagnosis are significant factors contributing to unfavorable outcome. New drugs active in secondary leukemias include CPX-351, or venetoclax in combination with hypomethylating agents, monoclonal antibodies as magrolimab, or targeted drugs against pathogenic mutations. Allogeneic stem cell transplantation remains the best currently available therapeutic option with curative intent for fit patients with unfavorable genetic profiles.
Introduction
Knowledge of therapy-related myeloid neoplasms (t-MNs) has significantlyimproved over the last 5 years, and some of the classical views are changing. t-MNs are classified as myelodysplasticsyndromes (MDS), acute myeloid leukemias (AMLs), and MDS/myeloproliferative neoplasms, diagnosed in patients treated with cytotoxic therapy for a primary neoplasm or an autoimmune disease.1,2
t-MNs were traditionally subgrouped according to the previous exposure to alkylating agents, topoisomerase II inhibitors, or radiotherapy (RT). More recently, new agents belonging to different classes of chemotherapy (CHT)drugs, such as poly(ADP-ribose) polymerase inhibitors, and purine analogshave been associated with t-MN development.2,3
On the contrary, the availability of new, targeted treatment options has reduced the prevalence of t-MNs in some settings, as in the case of acute promyelocytic leukemia (APL), where the prevalence of t-MNs in patients treated with the combination of all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) is significantly lower than that following the classical AIDA (ATRA-idarubicine) CHT protocol. Indeed, at a follow-up of 6 years, t-MNs occurred in none of the patients treated with ATRA/ATO in the Italian-German APL0406 study, vs 1.5% of those treated with the AIDA regimen.4 Similar results have been reported for the National Cancer Research Institute AML17 trial at a follow-up of 5.7 years, with 1% t-MNs in the AIDA arm, vs no t-MNs in the ATRA/ATO arm.5
In this review, we will update on the pathogenesis of t-MNs, including the evolution in the type of exposures, the predisposing factors, and the emerging treatment options, which are changing the clinical perspectives. For this purpose, we will concentrate our literature revision on data emerging from papers published in the last 5 years.
Genetic abnormalities in t-MNs
Karyotype
Karyotype abnormalities have been largely described in patients with t-MNs, who are characterized by a decreased prevalence of normal karyotype, and a predominance of complex or unbalanced karyotypes with chromosomal deletions, when compared with de novo (dn) AML or MDS.2,6-9 The widespread use of CHT combinations makes it difficult to ascribe the mutagenic potential to a single drug. With these limitations in mind, recurrent translocations as t(15;17), t(8;21), inv(16), and 11q23 abnormalities have been traditionally associated with topoisomerase II inhibitors, and t-MNs usually develop after a latency time of 1 to 3 years. Prognosis of t-AML with a recurrent translocation is similar to the dn counterpart in most case series, and “fit” patients should be treated according to standard protocols for these leukemia subtypes.10-12 Complete or partial deletions of chromosome 5 and 7, and very complex karyotypes, with >5 simultaneous chromosomal abnormalities,13,14 have been reported in the majority of t-MNs following alkylating agents or RT, occurring at a latency of 5 to 7 years.2
Normal karyotype is present in ∼33% of t-MNs (range, 23-37).2,9,14-17 In these cases, disease biology is similar to that of the dn counterpart, although the outcome is inferior in t-AML, because of a higher rate of deaths in remission.18 t-MNs with normal karyotype following RT have a very long latency and clinical characteristics that resemble dn diseases.19 This raises the issue of the definition of therapy-related MNs, solely based on the previous exposure to a previous cytotoxic treatment, vs a biology-based definition of these diseases, that will be discussed in the following paragraphs.
In t-MDS, very complex and deleted karyotypes are very frequent, leading to an overrepresentation of poor and very-poor cytogenetic risks.14 This results in a high prevalence of high- and very-high-risk patients following the revised international prognostic scoring system (IPSS-R) subgroups, and a significantly reduced very-low and low IPSS-R patients. The IPSS-R system is applicable to t-MDS as dn-MDS and reliably predicts AML transformation and overall survival (OS). However, patients with t-MDS in the low to intermediate IPSS-R subgroups had an inferior outcome, compared with the dn counterparts, partly because of patient-related factors, including activity of the primary disease, age, and cumulative toxicity of primary therapy.14 Overrepresentation of prognostically unfavorable mutations in lower-risk t-MDS, such as TP53, and reduced prevalence of favorable SF3B1 mutations also play a significant prognostic role.13,20-22
Somatic mutations
In recent years, the molecular characteristics of MNs have been deeply analyzed, and somatic mutations have been described in >95% of AML and MDS, without significant differences in the overall number of mutations in secondary vs dn subtypes, and none of the genes exclusively mutated in t-MNs.13,20,21 However, the proportion of cases mutated in specific genes differs in the 3 AML subtypes, dn, secondary to MDS or to myeloproliferative neoplasms (s-AML), and t-MNs. Indeed, mutations in SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, or STAG2 genes were >95% specific for the diagnosis of s-AML and are common in MDS.13,23 These same mutations were present in only 30% of t-AML. From a clinical point of view, t-AML with “de novo” alterations, including NPM1 mutations, MLL/11q23, or core binding factor rearrangements, are chemosensitive, similar to the corresponding dn-AML.13 The molecular classification may help to distinguish MN biologically dn from actual “therapy-related” forms, with implications for the treatment choice, as discussed below.
Mutations of TP53 are the single most frequent molecular abnormality in t-MNs, reported in 30% to 47% of cases in independent series, and associated with complex karyotype in ∼80% of cases.13,20-22 In clinically defined t-AML, Lindsley et al showed that TP53 mutations define a specific subgroup, with significant differences from s-AML, including younger age at diagnosis, lower number of recurrent driver mutations, higher number of cytogenetic abnormalities, and a reduced probability of achieving response after induction CHT.13 These data were recently confirmed in 229 cases of TP53mutt-MDS.22 Moreover, in t-MDS, TP53 mutations frequently occurred in a multihit state, including multiple TP53 mutations, associations with TP53 deletions, or copy-neutral loss of heterozygosity, as compared with TP53mut dn-MDS.22 TP53 mutations in single-, and even more in multihit state, were the major drivers of negative prognosis in MDS, independent of the dn or therapy-related subtype.21,22 TP53mut MN also display an immune-suppressive BM microenvironment that potentially is a synergistic driver of the poor prognosis.24 Mechanistically, mutant TP53 represses miR-34a expression, and that in turn results in MYC overexpression and upregulation of PDL1 in hematopoietic stem cells and BM blasts,24 providing these cells with an immunological privilege.
Among other recurrent molecular abnormalities in t-MDS are mutations of the PPMD1 gene, which have been detected in 15% of cases, at a significantly higher frequency than in dn-MDS (3%, P < .001).21 PPM1D encodes a protein phosphatase that regulates the cellular response to environmental stress, in part by inhibiting TP53 activity.25 PPM1D mutations may emerge or expand after CHT, and this provides evidence for their pathogenic role.26,27 TP53 and PPM1D genes are comutated in t-MNs more frequently than expected by chance, and because PPM1D mutations are frequently gain of function, the resulting increase in phosphatase activity may further inhibit the DNA-damage response (DDR) process leading to leukemic transformation.21,28,29
t-MNs’ predisposition
Germ-line susceptibility
In 10% to 15% of cases, MN occurs as a second neoplasm in patients who underwent surgery alone to treat the primary tumor, and a familial and/or personal history of multiple neoplasms is present in 5% to 10% of patients.9 Close relatives with breast, ovarian, or pancreatic cancer were present in the family history of 57% of patients with t-MNs and a previous breast cancer.30 These data indicate that individual predisposition to cancer development may play a role in t-MNs. Variants in detoxification and DNA-repair enzymes have been described in the past, but associations with t-MNs have not been confirmed, probably also because of the lack of adequate controls matched for age, primary disease, and therapy.31-37
Deleterious mutations typical of familial predisposition syndromes have been found in ∼20% of patients with a t-MN and a previous breast cancer or a lymphoproliferative disease. In particular, variants of the Fanconi Anemia pathway (including BRCA1, FANCA, FANCD2, FANCJ, and PALB2), TP53, and CHEK2 genes have been reported.30,38,39 Because many of these mutations were novel, with an unclear functional significance, these data will need further investigation. Hematologic malignancies are also a frequent event in individuals with a Li-Fraumeni syndrome.40,41 TP53 germline pathogenic or likely pathogenic variants were identified in 13 of 84 children with a t-MN, including 5 patients with mosaicism in the nontumor tissue.41
Familial recurrence of cancer is of particular relevance for the appropriate selection of allogeneic stem cell donors in younger patients with t-MNs, indicating the need for testing of family donors for germ-line mutations, and specific tumor surveillance programs after successful treatment of a primary tumor.42,43
Acquired susceptibility
One of the major discoveries in the last several years has been the confirmation of the presence of age-related clonal hematopoiesis of indeterminate potential (CHIP) in the peripheral blood of healthy individuals, associated with the presence of mutations in myeloid genes in ∼1% of the general population. This rate increases to >10% after the age of 70 years.44,45 These individuals are at increased risk of a myeloid neoplasm, at a rate of 1% per year.44-46
CHIP has been found at the time of the primary tumor diagnosis, prior to receiving any type of cytotoxic treatment in a high proportion of patients who later develop a t-MN, accounting for 20% to 60% of the cases.47-52 The mutated genes in these cases include classical CHIP genes, as DNMT3A, TET2, ASXL1, but also TP53 and PPM1D, and likely set the fertile genomic background favoring secondary leukemogenesis (Table 1; Figure 1). The presence of mosaic chromosomal alterations, such as amplifications, deletions, and copy-neutral loss of heterozygosity, synergizes with somatic mutations in increasing the t-MN risk.53
Role of CHIP as predisposing factor in t-MNs
| Primary disease (PD) | n | Age at t-MNs (y, median, range) | CHIP at PD diagnosis (%) | Frequently mutated genes* | Reference |
|---|---|---|---|---|---|
| NHL/HL MM Solid tumors | 3/1 2 1 | 56 (42-68) | 4 of 7 (57) | TP53 | 47 |
| NHL/HL APL/AML ALL | 7/2 4 1 | 62 (30-81) | 3 of 8 (21) | ASXL1 | 48 |
| NHL MM Solid tumors | 5 1 7 | 74 (70-82) | 8/13 (62) vs 15/56 (27) controls | TP53, TET2 | 49 |
| NHL/HL Solid tumors | 2/1 11 | 65 (28-77) | 10/14 (71) vs 17/54 (31) controls | TP53, TET2, DNMT3A, RUNX1, SRSF2 | 50 |
| NHL/HL MM/amyloidosis | 11/2 5 | 61 (41-71) | 7/10 (70) in PBSC grafts | TP53, DNMT3A | 51 |
| Hematological neoplasms Solid tumors | 35 | NA | 19 (54) | TP53 | 26 |
| Primary disease (PD) | n | Age at t-MNs (y, median, range) | CHIP at PD diagnosis (%) | Frequently mutated genes* | Reference |
|---|---|---|---|---|---|
| NHL/HL MM Solid tumors | 3/1 2 1 | 56 (42-68) | 4 of 7 (57) | TP53 | 47 |
| NHL/HL APL/AML ALL | 7/2 4 1 | 62 (30-81) | 3 of 8 (21) | ASXL1 | 48 |
| NHL MM Solid tumors | 5 1 7 | 74 (70-82) | 8/13 (62) vs 15/56 (27) controls | TP53, TET2 | 49 |
| NHL/HL Solid tumors | 2/1 11 | 65 (28-77) | 10/14 (71) vs 17/54 (31) controls | TP53, TET2, DNMT3A, RUNX1, SRSF2 | 50 |
| NHL/HL MM/amyloidosis | 11/2 5 | 61 (41-71) | 7/10 (70) in PBSC grafts | TP53, DNMT3A | 51 |
| Hematological neoplasms Solid tumors | 35 | NA | 19 (54) | TP53 | 26 |
HSCT, allogeneic stem cell transplantation; MM, multiple myeloma; NA, not applicable; NHL, non-Hodgkin lymphoma; PBSC, peripheral blood stem cell.
More than 1 patient.
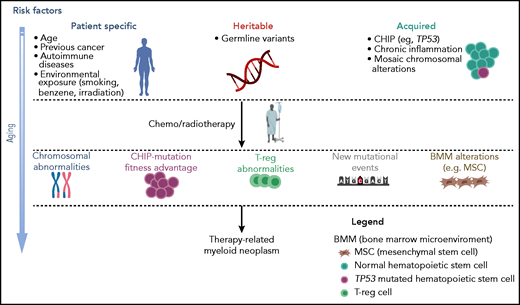
Multistep pathogenesis of t-MNs. Patient-related factors, including age, type, and treatment of primary disease, in the presence of germ-line variants, together with acquired factors, such as CHIP and inflammation, may all contribute to lay the groundwork for the development of myeloid diseases.40-47 The subsequent cytotoxic therapy may later induce further genetic changes, the selection of abnormal hematopoietic clones, and changes in the microenvironment, resulting in the onset of a t-MN. Professional illustration created with BioRender.com.
Multistep pathogenesis of t-MNs. Patient-related factors, including age, type, and treatment of primary disease, in the presence of germ-line variants, together with acquired factors, such as CHIP and inflammation, may all contribute to lay the groundwork for the development of myeloid diseases.40-47 The subsequent cytotoxic therapy may later induce further genetic changes, the selection of abnormal hematopoietic clones, and changes in the microenvironment, resulting in the onset of a t-MN. Professional illustration created with BioRender.com.
CHIP may play a role also in the context of recurrent chromosome translocations, which are traditionally regarded as primary leukemia-initiating events in AML. Dillon et al recently reported that at the time of complete remission (CR), driver mutations in DNMT3A, PPM1D, and MYCN were found in t-APL, but not in dn-APL.54 These mutations persisted in molecular CR, in some cases were present prior to induction CHT for APL, and affected multiple hematopoietic lineages (Figure 2). These data underline the relationship between CHIP and t-AML and may challenge the assumption that fusion genes are bona fide primary transforming events.
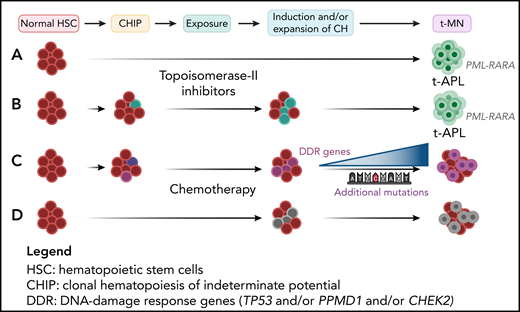
The cytotoxic treatment shapes disease evolution in t-MNs. (A) The type of cytotoxic treatment influences the genetic changes leading to t-MNs. In particular, in t-APL developing after treatment with topoisomerase II inhibitors, breakpoints are clustered within hotspot regions on chromosomes 15 and/or 17, in the PML and RARA genes, corresponding to preferential sites of topoisomerase II–mediated DNA cleavage.26 (B) However, in some cases, CHIP-related mutations were detectable in t-APL, but not in dn-APL and persisted at the time of CR. (C) Elderly patients with a solid tumor, who received RT or CHT with platinum compounds or topoisomerase-I inhibitors, frequently presented CHIP and additional mutations in the DDR genes TP53, PPMD1, and CHEK2, as compared with those who received surgery, immunotherapy, or targeted therapy.26 (D) Unlike adults, the majority of pediatric t-MNs were characterized by mutations arising as a consequence of cytotoxic therapy, without preexisting CHIP. Professional illustration created with BioRender.com.
The cytotoxic treatment shapes disease evolution in t-MNs. (A) The type of cytotoxic treatment influences the genetic changes leading to t-MNs. In particular, in t-APL developing after treatment with topoisomerase II inhibitors, breakpoints are clustered within hotspot regions on chromosomes 15 and/or 17, in the PML and RARA genes, corresponding to preferential sites of topoisomerase II–mediated DNA cleavage.26 (B) However, in some cases, CHIP-related mutations were detectable in t-APL, but not in dn-APL and persisted at the time of CR. (C) Elderly patients with a solid tumor, who received RT or CHT with platinum compounds or topoisomerase-I inhibitors, frequently presented CHIP and additional mutations in the DDR genes TP53, PPMD1, and CHEK2, as compared with those who received surgery, immunotherapy, or targeted therapy.26 (D) Unlike adults, the majority of pediatric t-MNs were characterized by mutations arising as a consequence of cytotoxic therapy, without preexisting CHIP. Professional illustration created with BioRender.com.
In the near future, molecular profiling of patients with a solid tumor or a lymphoproliferative disease may be one of the critical factors influencing treatment choice toward less-toxic, targeted agents in CHIP+ patients, and/or carriers of mosaic chromosomal alterations.53 Along this line, in women aged 50 to 75 years with breast cancer, CHIP, and abnormal blood parameters, the risk of t-MNs has been shown to be substantially increased up to 9% at long term.26 This would outweigh the potential benefits of adjuvant CHT in early-stage disease and should be taken into consideration in the treatment planning. Prospective studies are needed to determine the potential benefit of adjusting adjuvant CHT based on presence of CHIP.
Role of cytotoxic therapy in t-MNs development
Shaping disease evolution by the cytotoxic treatment
Although inborn or acquired genetic susceptibility has been shown, it is undeniable that the cytotoxic treatment plays a relevant role in the pathogenesis of t-MNs (Figure 2). This has been elegantly shown for t-APL, where clustering of breakpoints has been described within hotspot regions on chromosomes 15 and/or 17, in the PML and RARA genes.12,55-57 Functional analysis confirmed that the breakpoints were preferential sites of topoisomerase II–mediated DNA cleavage in the presence of mitoxantrone or epirubicine. This shows the direct genotoxic impact of topoisomerase II inhibitors on HSC, leading to the t(15;17) translocation in t-APL.12,55-57
CHT also influences the occurrence of myeloid gene mutations. Indeed, mutations in the DDR genes TP53, PPMD1, and CHEK2 were more frequently detectable in the peripheral blood of older patients with a solid tumor who had received RT or CHT with platinum compounds or topoisomerase II inhibitors, as compared with those who received surgery, immunotherapy, or targeted therapy.26 The same was true for smoking, which was associated with an increased mutation number, particularly of the ASXL1 gene, and a wider clone size. On the contrary, splicing factor (SF3B1, SRSF2, U2AF1) or epigenetic modifier (DNMT3A, TET2) genes were more frequently mutated in elderly untreated patients, compatible with their classical CHIP-related role (Figure 2). Mutations were also associated with specific primary cancers, with PPM1D being more frequently mutated in patients with ovarian or endometrial cancer vs other subtypes, after adjusting for age.26
The “fitness” advantage may endow small preexisting clones with a sufficient degree of chemoresistance that allows their survival and expansion post-therapy. Longitudinal analysis in 525 patients showed that DDR-mutated clones tended to expand in 61% of patients who received a cytotoxic CHT or RT, in particular, in patients with multiple mutations.26 Of 35 patients who later developed a t-MN, CHIP was detectable at the time of primary cancer diagnosis in 19 cases, with TP53 mutations in 10 cases. Secondary leukemogenesis was not only associated with expansion of the TP53mut clone, but also associated with acquisition of chromosomal aneuploidies or mutations in genes known to drive progression to AML as FLT3, KRAS, and NRAS.26
The effects of previous therapy on the induction of t-MNs seem very relevant in pediatric t-MNs. Using very deep sequencing technologies, a recent report showed that the majority of pediatric t-MNs was derived from mutated clones arising at a mean of 405 days (range, 118 to 748) prior to t-MN diagnosis.41 These variants were not detectable at the time of the primary malignancy diagnosis in 23 of 26 cases with available material.41
The cytotoxic treatment may also alter the BM microenvironment. Stoddart et al recently showed that treatment with the alkylating agent N-ethyl-N-nitrosourea induced senescence of mesenchymal stromal cells, decreased DNA synthesis and the growth of fibroblast colony-forming units in a TP53mut, 5qdel mouse model.58 Senescence was further confirmed by elevated levels of messenger RNA encoding p21 (Cdkn1a), p16INK4a (Cdkn2a), and IL-6. These mechanisms may be also active in vivo and contribute to the development of a proinflammatory environment, which may favor leukemogenesis. It will be interesting to study whether these alterations cooperate with the expansion of immunosuppressive T cells and myeloid-derived suppressor cells shown in TP53mut AML.24
Effects of evolution of cancer treatment on t-MN incidence
The beginning of the 21st century has seen substantial changes in the agents and clinical approaches to cancer CHT, with corresponding improvements in the prognosis of many cancers, and a consequently increasing number of long-term survivors, at risk of developing late treatment-related complications, including t-MNs. Recently, Morton et al, using US population-based registry data from the National Cancer Institute’s Surveillance, Epidemiology, and End Results Program, showed that the t-MN risks after CHT have been increasing in the years 2000 to 2014 for all solid tumors, except colon cancer, particularly in association with the use of platinum compounds.59 In this registry, treatment of primary cancers of the thyroid with radioiodine, of stage I to III prostate or of stage I to II uterine cancers with RT was associated with a three- to fivefold increased risk of t-AML with abnormal karyotype, with myelodysplasia-related changes, or of chronic myelogenous leukemia.60
An emerging subtype of primary malignancy associated with t-MNs is chronic lymphocytic leukemia, because of the prolonged survival of treated patients and the use of newer immunochemotherapy combinations, as the fludarabine-cyclophosphamide-rituximab regimen. This CHT protocol has been associated with up to 5% of t-MNs at a median latency of ∼4 years.61,62
Prolonged disease-free survival in multiple myeloma has led to increases in the onset of t-MNs, mainly because of the cumulative effect of cytotoxic therapies, which also include autologous stem cell transplantation (SCT). A recent meta-analysis showed that lenalidomide, used as maintenance treatment after autologous SCT, although associated with improved relapse-free survival, may increase the risk of t-MNs, particularly when preceded by oral melphalan treatment.63
Targeted, peptide receptor radionuclide therapy (PRRT) has been in use in the last 20 years in patients with inoperable gastroenteropancreatic neuroendocrine tumors, and prostatic cancers. Despite the targeted approach, this treatment has been associated with a 1.4% to 4.8% incidence of t-MNs.64-66 In particular, 30 of 1631 patients (1.8%) treated with different PRRT strategies between 1999 and 2019 in Germany developed a t-MN (23 MDS and 7 AML), at a median latency of 49 and 39 months from first PRRT dose, respectively.67 This incidence of t-MNs after PRRT is definitely lower than that previously reported by Brieau et al in a small cohort of 20 neuroendocrine tumors patients treated with 177Lu-DOTATATE as salvage treatment.68 Indeed, most of these patients had been previously exposed to alkylating agents, highlighting the synergistic leukemogenic role of CHT-PRRT combinations.
In some cases, the evolution in cancer treatment has reduced the t-MN risk. An emblematic example is Hodgkin lymphoma (HL), initially considered at “high risk” of t-MNs because of the use of “MOPP” (mecloretamine, vincristine, procarbazine, and prednisone), and later of “escalated-BEACOPP” (bleomicin, etoposide, doxorubine, cyclophosphamide, vincristine, procarbazine, and prednisone) regimens.69 In this context, medium- to long-term follow-up studies have favored the use of schemes such as ABVD (adriamycin, bleomycin, vinblastine, and dacarbazine) and restricted the indications and fields of RT, leading to a reduction of t-MN risk.
Granulocyte colony-stimulating factor (G-CSF) is widely used in solid tumors and hematologic malignancies to maintain optimal dose intensity and prevent treatment-induced febrile neutropenia. No significant association was found between the use of G-CSF and t-MN prevalence in a large cohort of women treated with CHT for primary invasive breast cancer, although the risk of t-MNs increased with the number of G-CSF doses.70 These findings call for further investigation into associations between G-CSF use and t-MNs.
MN after environmental exposure
MNs owing to environmental exposure are also a model of in vivo leukemogenesis, although not classified as t-MNs. They include MN onsetting after exposure to poisons because of professional reasons, as reported in workers exposed to benzene in the shoe or oil industries.71 However, the best model to document the role of environmental factors is probably MDS occurring in atomic bomb survivors, where the amount and type of exposure are measurable. In this setting, aberration profiles are different from those observed in t-MNs. Indeed, comparing genetic profiles of MDS in proximally exposed (PE; <1.5 km from the hypocenter) to that of distally exposed patients, a higher frequency of abnormal karyotypes was observed in PE cases, with higher prevalence of intermediate to very poor IPSS‐R cytogenetic categories.72,73 As in dn-MDS, IPSS‐R cytogenetic categories were independently associated with poor survival and cumulative incidence of leukemic transformation, but exposure distance did not play a significant role.74 Among frequent aberrations, chromosomal translocations and inversions in particular in chromosomes 3, 8, and 11 were frequent in PE patients.73 Furthermore, significantly fewer mutations in genes of the DNA methylation pathway were observed in patients with PE, who also had a significantly higher rate of 11q deletions, as compared with distally exposed patients.72 Biallelic alterations of the ATM gene, localized in the 11q region, were also very frequent. The rate of TP53 and of splicing gene mutations was similar in the 2 groups, whereas transcription and chromatin modification genes were more frequently mutated in patients with PE-MDS.72 The different mutation profile of MDS in PE patients highlights the direct effects of γ irradiation on the DDR pathway, and particularly on the ATM gene, on hematopoietic stem cells.
Radiation exposure from imaging tests, as from computed tomographic scans in patients with cancer, may also increase the t-MN risk. Recently, a 1.5-fold increased risk for leukemia associated with computed tomographic scans has been reported in a population-based case-control study, including 13 040 leukemia patients and 130 400 matched controls. The dose-response relationship was mostly evident in patients aged 45 years or younger.75
Emerging treatment options in t-MNs
Survival in t-MNs is poor, when compared with other MDS and AML subtypes. In addition to the unfavorable biologic characteristics of t-MNs described above, patient-related factors, such as age, comorbidities, the previous disease history, its treatment, and the remission status at t-MN diagnosis, are significant factors contributing to patient frailty, and to unfavorable outcome.
Until recent years, patients with t-MNs have been conventionally excluded from many clinical trials. Nowadays, because of improvements in supportive care and treatments, t-MN should be stratified according to standard prognostication, better if integrated with mutational profiling, and treated accordingly.
New drugs with specific activity on “secondary” leukemias, targeting pathogenic mutations or interfering with immune mechanisms, as the anti-CD47 antibody magrolimab, are or will be available in the future. CPX-351 (Vyxeos; Jazz Pharmaceuticals) is a liposomal formulation of a 5:1 combination of cytosine-arabinoside/daunoblastine, with a specific indication for newly diagnosed elderly s-AML, including t-AML.76 Originally tested in a wider population of AML patients, the phase 2 trial showed that clinical benefit was highest among patients with s-AML.77 These data were confirmed by the subsequent phase 3 study performed in s-AML, where CPX-351 significantly improved median OS (9.56 vs 5.95 months) and overall remission rate (47.7% vs 33.3%) vs the 7 + 3 regimen, in the age group between 65 and 70 years, particularly in therapy-related forms.76 This drug, although well tolerated, should be considered in the setting of conventional CHT and used in patients eligible for this treatment modality.
As TP53mut is very frequent in t-MNs, these mutations represent one of the major challenges to improving outcome in t-MNs. The hypomethylating agent (HMA) azacitidine (AZA) and decitabine have been used in t-MNs, with results similar to the dn counterpart.78,79 Also, the 10-day extended decitabine dosing has been shown to significantly reduce the TP53mut burden, reaching levels of undetectability in most patients.80 Efficacy of HMA in AML increases when combined with the BCL2-inhibitor venetoclax (Venclyxto; Abbvie). Indeed, in the phase 3 VIALE-A trial, secondary disease subtype did not influence response or survival.81 Looking at mutation profiling, TP53mut cases had a 55% overall probability of achieving CR, when treated with the venetoclax/AZA combination, vs 0% with AZA alone. This translated into a significantly longer survival for the combination in all patient subgroups.81
APR-246 is a novel small molecule anticancer compound that reactivates mutated and nonfunctional p53, shifting it toward the wild-type p53 conformation. The results of the phase 1b/2 study of APR-246 and AZA in HMA-naive, TP53mut MDS, and AML with 20% to 30% blasts showed that the combination was well tolerated with no dose-limiting toxicities.82 In patients evaluable for response, the overall response rate was 87%, with a 53% CR rate. The median OS of the cohort was 10.8 months, with improved OS in responders vs resistant patients (14.6 vs 7.5 months). Notably, the AZA/APR-246 was a successful bridge to HSCT in 40% of patients. Serial high-sensitivity next-generation sequencing using a 5% variant allele frequency cutoff identified deep molecular remission in 21 patients (38%), with a significant association between TP53 variant allele frequency clearance and CR.82 The phase 3 study of APR-246 in combination with AZA vs AZA alone is ongoing (#NCT03745716).
Short duration of response is still a major issue that must be tackled, in particular, for TP53mutt-MNs. For these reasons, the best available treatment option in t-MNs with poor prognostic features remains HSCT. Data on 228 patients with s-AML/t-AML compared with 416 dn-AML undergoing HSCT have been recently published.83 The s-AML/t-AML subtype was associated with older age and adverse European Leukemia Net (ELN) risk, and, although the cumulative incidence of relapse was similar, nonrelapse mortality was higher and OS was shorter in s-AML/t-AML as compared with dn-AML. However, in multivariate analyses, after adjustment for ELN risk and pre-HSCT measurable residual disease (MRD) status, disease subtype did not impact outcome.
These data show the importance to reduce the disease burden prior to HSCT to ideally achieve an MRD− status (CRMRD−), as dynamic surrogate marker of outcome. Future studies will address the use of targeted treatment combinations prior to transplant, and as posttransplant maintenance, as strategies with curative intent for patients with t-MNs.
Conclusions
The pathogenesis of t-MNs has been unraveled in the last years because of the availability of deep sequencing techniques, which have confirmed a germline cancer predisposition in 15% to 20% of t-MNs, and acquired susceptibility consisting of CHIP in a large proportion of adult cases. The mutagenic effect of cytotoxic therapy and the interactions with individual factors have also been better defined. It is hoped that in the near future, the improved genomic characterization of the patient background will be the basis for treatment decisions and individualized treatment approaches, at the time of both primary cancer and t-MN diagnosis.
Acknowledgments
The authors acknowledge their mentors, G. Leone and F. Lo-Coco, for inspiring discussions on t-MN and generous support, and their colleagues, M. Criscuolo, M. Divona, L. Fianchi, S. Hohaus, C. Gurnari, S. Lavorgna, L. Pagano, and T. Ottone, for sharing their scientific interests and for their constant and kind collaboration.
This work was supported by Italian Association for Cancer Research (AIRC) 5 × 1000 call “Metastatic disease: the key unmet need in oncology” to MYNERVA project 21267 (MYeloid NEoplasms Research Venture AIRC; a detailed description of the MYNERVA project is available at http://www.progettoagimm.it).
Authorship
Contribution: M.T.V., G.F., and E.F. revised the literature and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Maria Teresa Voso, Department of Biomedicine and Prevention, University of Rome Tor Vergata, Viale Oxford 81, 00133 Rome, Italy; e-mail: voso@med.uniroma2.it.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal