

Key Points
Blinatumomab is safe and feasible for use in B-ALL after allogeneic HCT.
The composition of a patient’s T-cell subsets at the time of treatment is indicative of whether they will respond to blinatumomab.

Abstract
Patients with B-lineage acute lymphoblastic leukemia (ALL) are at high-risk for relapse after allogeneic hematopoietic cell transplantation (HCT). We conducted a single-center phase 2 study evaluating the feasibility of 4 cycles of blinatumomab administered every 3 months during the first year after HCT in an effort to mitigate relapse in high-risk ALL patients. Twenty-one of 23 enrolled patients received at least 1 cycle of blinatumomab and were included in the analysis. The median time from HCT to the first cycle of blinatumomab was 78 days (range, 44 to 105). Twelve patients (57%) completed all 4 treatment cycles. Neutropenia was the only grade 4 adverse event (19%). Rates of cytokine release (5% G1) and neurotoxicity (5% G2) were minimal. The cumulative incidence of acute graft-versus-host disease (GVHD) grades 2 to 4 and 3 to 4 were 33% and 5%, respectively; 2 cases of mild (10%) and 1 case of moderate (5%) chronic GVHD were noted. With a median follow-up of 14.3 months, the 1-year overall survival (OS), progression-free survival (PFS), and nonrelapse mortality (NRM) rates were 85%, 71%, and 0%, respectively. In a matched analysis with a contemporary cohort of 57 patients, we found no significant difference between groups regarding blinatumomab’s efficacy. Correlative studies of baseline and posttreatment samples identified patients with specific T-cell profiles as “responders” or “nonresponders” to therapy. Responders had higher proportions of effector memory CD8 T-cell subsets. Nonresponders were T-cell deficient and expressed more inhibitory checkpoint molecules, including T-cell immunoglobulin and mucin domain 3 (TIM3). We found that blinatumomab postallogeneic HCT is feasible, and its benefit is dependent on the immune milieu at time of treatment. This paper is posted on ClinicalTrials.gov, study ID: NCT02807883.
Introduction
The therapeutic landscape of acute lymphoblastic leukemia (ALL) is rapidly evolving. Allogeneic hematopoietic cell transplantation (HCT) is a potentially curative option for patients with high-risk ALL, with overall survival (OS) ranging from 30% to 60%, depending on disease characteristics and risk profile.1,2 Measurable residual disease (MRD) and high-risk cytogenetics/molecular features are key determinants of relapse risk and outcomes in patients.3-9 The management of ALL patients who relapse after allogeneic HCT is challenging with poor survival outcomes, regardless of the treatment modality used.2,10 Thus, it is critical to employ strategies to minimize the risk for relapse, even after transplantation. However, other than tyrosine kinase inhibitor (TKI) maintenance post-HCT in patients with Philadephia chromosome-positive (Ph+) ALL, there are limited maintenance strategies. Donor lymphocyte infusion (DLI) has been used preemptively in patients with signs of early relapse with some success,11-15 but is generally associated with remission rates below 10% and an elevated risk of graft-versus-host disease (GVHD).16 Hence, there continues to be an unmet need for mitigation strategies to reduce the risk of relapse in high-risk ALL patients.
Blinatumomab is a bispecific T-cell engager (BiTE), with one arm targeting CD19 on B-ALL blasts and the other arm binding to CD3ζ on T cells. Upon binding, T cells become activated and exert perforin-dependent cytotoxicity against target cells expressing CD19. Studies have shown a clinical benefit of blinatumomab in MRD-positive ALL patients in morphologic complete response (CR), where 80% successfully converted into MRD negativity.17 Its clinical benefit was also investigated in the relapsed and refractory (R/R) setting. A phase 2 study of 189 patients with R/R ALL treated with blinatumomab showed an overall response rate (ORR) of 43%, and with a median follow-up of 8.9 to 9.8 months, the median progression-free survival (PFS) and OS were 5.9 and 6.1 months, respectively.18 Similar findings were noted in children with R/R ALL.19 Its activity was confirmed in the phase 3 clinical trial (TOWER) by Kantarjian and colleagues with 405 patients with CD19-positive R/R B-ALL who were randomized to either blinatumomab (n = 271) or standard chemotherapy (n = 134).20 Compared with the chemotherapy-treated cohort, the blinatumomab-treated cohort had superior outcomes, as shown by higher rates of ORR (44% vs 25%, P < .001), superior 6-month EFS (31% vs 12%, HR 0.55; 95% CI 0.43-0.71, P < .001), and superior median OS (7.7 months vs 4 months, HR 0.71; 95% CI 0.55-0.93, P = .01).20 Finally, the feasibility of blinatumomab following allogeneic HCT was illustrated in a study by Stein and colleagues; 64 patients received blinatumomab after relapse following prior allogeneic HCT.21 Grade 3 to 4 cytokine release syndrome (CRS) was noted in 3% of patients, and grade 3 to 4 neurotoxicity was noted in 16% of patients. GVHD occurred in 11% of patients and did not require blinatumomab discontinuation or hospitalization. Efficacy was similar to what was observed in patients without prior HCT, where 45% had CR/CRh within 2 cycles of blinatumomab, and 34% had MRD response.21
Many mechanisms of immune evasion exist for relapse following allogeneic HCT, including low levels of cytotoxic effector T cells and upregulation of inhibitory checkpoint molecules soon after transplant.22 Based on the safety profile of blinatumomab, specifically its lack of hematopoietic cell cytotoxicity, and its mechanism of action of specifically directing cytotoxic T cells to leukemic blasts during the immediate posttransplant time period when there may be low levels of mature T cells present, we postulated that blinatumomab would be an ideal agent to test in this setting. We conducted a phase 2 study to evaluate the feasibility and clinical benefit of blinatumomab administered for 1 year after allogeneic HCT in high-risk B-ALL patients. While the benefit of blinatumomab use may be highest in MRD-positive patients prior to allogeneic HCT, we included in our study other high-risk populations to see if they may benefit from this strategy.
Methods
Study design
This was a prospective, open-label, single-arm, single-center clinical trial evaluating the use of blinatumomab postallogeneic HCT in B-ALL patients at high risk of relapse after transplant. The primary endpoint was feasibility, defined by the rate of treatment-related toxicities attributable to blinatumomab, acute GVHD (aGVHD), and secondary graft failure. Secondary endpoints were PFS, OS, impact of MRD status, and nonrelapse mortality (NRM). Continuous monitoring of toxicity was conducted for all patients starting with the first cohort of 5 patients. The study followed a Bayesian model, and dose-limiting toxicities were defined as grades 3 to 4 aGVHD >30%, secondary graft failure >30%, or NRM within 1 cycle of blinatumomab. The study was conducted after the protocol was reviewed and approved by MD Anderson Cancer Center’s Institutional Review Board. Patients provided informed consent prior to enrollment in the clinical study in accordance with the Declaration of Helsinki. This phase 2 clinical trial was registered at ClinicalTrials.gov (NCT02807883).
Patient eligibility
The study was initially limited to adults only, but after safety was confirmed following treatment in 8 adults, it was subsequently amended to include children. The final eligibility age at time of enrollment was 1 to 70 years. Other key eligibility criteria included receiving allogeneic HCT within the last 100 days for B-ALL at high risk of relapse as determined by: (1) complete hematologic remission beyond CR1 at the time of allogeneic HCT, (2) primary induction failure requiring more than 1 line of therapy, (3) positive MRD by flow cytometry (threshold >0.01%) or molecular assays by PCR (sensitivity 1/10 000) before allogeneic HCT, and/or (4) high-risk cytogenetic or molecular profile defined as Ph+ ALL, Ph-like ALL, KMT2A gene rearrangement, complex cytogenetics, or hypodiploid cytogenetics at diagnosis. Additionally, patients with MRD positivity after HCT were eligible for the study.
Enrollment occurred within 30 to 100 days after allogeneic HCT and after adequate hematologic recovery, defined as an absolute neutrophil count ≥0.5 × 109/L and platelet count >20 × 109/L. Patients needed to have adequate performance status (ECOG ≤2 or Karnofsky ≥50) and have adequate organ function, defined as creatinine clearance greater than 30 mL/min, ALT/AST <5 × upper limit of normal (ULN) and serum bilirubin <3 × ULN. Patients were excluded from the study if they had relapsed disease, defined as >5% blasts in bone marrow (BM) or peripheral blood (PB) and/or active involvement of the central nervous system (CNS) by ALL (defined as ≥5 leukocytes/μL with the presence of blast cells in the CNS or cranial-nerve palsy), active GVHD requiring systemic steroids, systemic steroids beyond physiologic replacement, and/or uncontrolled infection.
Treatment
Patients received blinatumomab as continuous IV infusion on days 1 through 28 of each cycle. Dosing and administration followed the standard FDA guidelines for children and adults. Briefly, patients ≥45 kg received 28 μg of blinatumomab daily administered as a continuous infusion on days 1 through 28, and patients <45 kg (dose based on BSA) received 15 μg/m2 per day (maximum: 28 μg/day) as a continuous infusion on days 1 through 28.23 Patients were premedicated with dexamethasone 16 to 20 mg IV prior to the start of each cycle. Patients were hospitalized for observation during the first 3 days of cycle 1 and the first 2 days of cycle 2. The first cycle was given within the first 3 months after allogeneic HCT and then at approximately 6, 9, and 12 months following transplant. The study did not require patients to be off immunosuppression prior to initiating therapy with blinatumomab. The transplant preparative regimen, GVHD prophylaxis, graft source, and graft allotype were determined by the treating physician and not prescribed by this protocol. For patients with Ph+ ALL, the use of TKI therapy posttransplant was permitted.
Safety and evaluations
Disease assessments with PB studies (eg, flow cytometry and chimerisms) and BM examinations were done prior to each cycle and at study completion. Patients who remained in remission after blinatumomab therapy were labeled “Responders,” and patients with disease progression defined by recurrence of MRD by flow cytometry or morphologic leukemic blasts were labeled “Nonresponders.” The Common Terminology Criteria for Adverse Events (CTCAE) version 4 was used to grade toxicities.
Flow cytometry data analysis
Samples were collected following informed consent from 15 consecutive patients (4 nonresponders, 11 responders), separated using Ficoll density separation (Lymphoprep, STEMCELL Technologies), and cryopreserved. PB mononuclear cells (PBMC) were then thawed and immunostained with CD127 (BD Biosciences: Clone HIL7RM21), CD25 (BioLegend: Clone BC96), LAG-3 (BD Biosciences: Clone T47-530), PD-1 (BioLegend: Clone EH12.2H7), CCR7 (BD Biosciences: Clone 3D12), CD3 (BD Biosciences: Clone UCHT1), CD45RO (BD BioSciences: Clone UCHL1), CD8 (BioLegend: Clone SK1), 2B4 (BioLegend: Clone C1.7), CTLA-4 (BioLegend: Clone L3D10), CD160 (BioLegend: Clone 341204), CD4 (BioLegend: Clone RPA-T4), PDL1 (BD Biosciences: Clone MH1), CD19 (BD Biosciences: Clone HIB19), TIM-3 (BD Biosciences: Clone 7D3), and TIGIT (eBioscience: Clone MBSA43). Cells were acquired using an X-20 Fortessa (BD Biosciences). The analysis was implemented with the cyt3 package in MATLAB (https://github.com/dpeerlab/cyt3).24 Raw data were first transformed using hyperbolic arcsin with a cofactor of 150. Bh-SNE25 version of SNE was run against the collection of all samples, with 8000 data points subsampled for each; viSNE reduced the high-dimensional flow cytometry data into 2 dimensions, and the result was visualized in the viSNE map (2D scatterplot). PhenoGraph clustering26 was then used to identify subpopulations in the viSNE map. The number of nearest neighbors was set at 100. The annotation of subpopulations by PhenoGraph was directly shown in the viSNE map, where cells that belong to the same subpopulation were shown in the same color.
Response definitions and outcome
CR was defined as having ≤5% malignant blasts in the BM, normal blood counts with ANC ≥0.5 × 109/L and platelet >20 × 109/L, normal karyotype, and absence of extramedullary disease. MRD was assessed using multiparameter flow cytometry with a threshold of >0.01%. Myeloablative and reduced-intensity conditioning regimens were defined according to the Center for International Blood and Marrow Transplant Research (CIBMTR) criteria.27 aGVHD was staged and graded according to the criteria published by Przepiorka and colleagues.28 Chronic GVHD (cGVHD) was reported based on the 2014 NIH cGVHD Consensus Conference.29
Statistical methods
OS time and NRM were computed from date of allogeneic HCT to last known vital sign. Patients alive at the last follow-up date were censored. PFS time was computed from date of allogeneic HCT to date of disease progression or death (if he or she died without disease progression) or the last evaluation date. Patients who were alive and did not experience progression of disease at the last follow-up date were censored. In addition, relapse and GVHD were computed from date of allogeneic HCT to date of event; patients who did not experience the event were censored at their last follow-up date. Patients who remained in remission were grouped as “responders,” and those that progressed during therapy with blinatumomab were grouped as “nonresponders.” OS and PFS were estimated using the Kaplan-Meier method. The cumulative incidences of relapse, NRM, and GVHD were evaluated using the competing risks method. The competing risk for relapse was death and for NRM was relapse, while the competing risks for GVHD were relapse and death.
A contemporary cohort of patients with high-risk B-ALL who received allogeneic HCT was identified retrospectively. To correct for potential bias in the HCT comparisons, treatment patients were matched to controls (1:2 where possible) using the following steps: 1) patients were divided into groups based on disease status at allogeneic HCT, cytogenetic risk, and MRD prior to allogeneic HCT; we did not match for conditioning regimen intensity as a separate analysis comparing the 2 treatment groups by regimen intensity did not yield different results (data not shown); 2) within each subgroup of completely matched patients, a random number generator was employed from the uniform distribution on the interval (0,1) using a prime modulus multiplicative generator; and 3) each treated patient was matched with 1 or 2 (where possible) control patient(s) starting from the lowest generated random number. Group differences in OS and PFS were evaluated using a stratified log-rank test while group differences in cumulative incidences were assessed by stratified Gray’s test.30
All statistical analyses were performed using SAS 9.4 for Windows (SAS Institute Inc., Cary, NC). All statistical tests used a significance level of 5%. No adjustments for multiple testing were made.
Results
Patient and disease characteristics
Twenty-three patients signed consent, and 21 patients who received at least 1 dose of blinatumomab therapy postallogeneic HCT were included in the analysis. Two patients never received therapy due to GVHD that required treatment. Table 1 summarizes patient and treatment characteristics for the study cohort. Eighty-one percent of the patients were male, 62% were White, with a median age of 30 (17 to 66) years, and 90% (19 of 21) had a high-risk cytogenetic or molecular profile at diagnosis. Two patients had >1 HCT prior to blinatumomab therapy. Seventy-six percent of patients were exposed to blinatumomab prior to allogeneic HCT. The median days from transplant to the first day of cycle 1 of blinatumomab was 78 (range, 44 to 105), and MRD was detected prior to the start of blinatumomab in 2 patients. Fifty-seven percent of patients (12 of 21) completed all 4 cycles of therapy (Table 1). Three patients could not complete treatment due to GVHD, and the remaining patients (n = 6) relapsed before they could complete all 4 intended cycles. All patients were on tacrolimus during cycle 1 of blinatumomab, with a mean tacrolimus level of 7.4 ng/mL (range, 4.3 to 10.3 ng/mL).
Patient and treatment characteristics
| Measure | All patients, n = 21, (%) |
|---|---|
| Gender | |
| Male | 17 (81) |
| Female | 4 (19) |
| Age | |
| Mean (SD) | 37.9 (16.2) |
| Median (range) | 29.9 (16.9-65.6) |
| Race/ethnicity | |
| White | 13 (62) |
| Hispanic | 8 (38) |
| High-risk cytogenetic/molecular risk at diagnosis | 18 (90) |
| Ph+ | 2 |
| Ph-like | 8 |
| KMT2A | 4 |
| Complex* | 2 |
| Hypodiploid | 2 |
| Days from HCT to blinatumomab start | |
| Median (range) | 78.0 (44.0-105.0) |
| MRD prior to blinatumomab treatment | |
| Detected | 2 (10) |
| Not detected | 19 (90) |
| Number of cycles | |
| 1 | 21 |
| 2 | 13 |
| 3 | 12 |
| 4 | 12 |
| Measure | All patients, n = 21, (%) |
|---|---|
| Gender | |
| Male | 17 (81) |
| Female | 4 (19) |
| Age | |
| Mean (SD) | 37.9 (16.2) |
| Median (range) | 29.9 (16.9-65.6) |
| Race/ethnicity | |
| White | 13 (62) |
| Hispanic | 8 (38) |
| High-risk cytogenetic/molecular risk at diagnosis | 18 (90) |
| Ph+ | 2 |
| Ph-like | 8 |
| KMT2A | 4 |
| Complex* | 2 |
| Hypodiploid | 2 |
| Days from HCT to blinatumomab start | |
| Median (range) | 78.0 (44.0-105.0) |
| MRD prior to blinatumomab treatment | |
| Detected | 2 (10) |
| Not detected | 19 (90) |
| Number of cycles | |
| 1 | 21 |
| 2 | 13 |
| 3 | 12 |
| 4 | 12 |
Complex karyotype defined as 5 or more cytogenetic abnormalities.
Transplant characteristics for the study group and the matched cohort are listed in Table 2. In the study group, about half of the patients were in CR1, and 80% had no detectable MRD at the time of HCT. The Karnosky performance score was ≥80% in 94% of patients, and the median comorbidity index was 3 (range, 0 to 8). Regarding the source of allogeneic HCT graft, 33% were matched siblings, 48% matched unrelated donors, and 19% had haploidentical family donors. Approximately half of the patients received a reduced-intensity conditioning regimen. The median time from diagnosis to HCT was 8.8 months (range, 3.2 to 107.7 months). The median number of blinatumomab cycles received was 4 (range, 1 to 4). Except for gender, all key characteristics were similar between the study and control groups (Table 2).
Patient and transplant characteristics for study group and matched cohort
| HCT patients | ||||
|---|---|---|---|---|
| Measure | All, n = 57 (%) | Blinatumomab n = 21 (%) | Controls n = 36 (%) | P value* |
| Gender | ||||
| Male | 33 (58) | 17 (81) | 16 (44) | .012 |
| Female | 24 (42) | 4 (19) | 20 (56) | |
| Age in years, median (range) | 38.0 (16.0-66.0) | 29.0 (16.0-65.0) | 41.0 (19.0-66.0) | .26† |
| Race/ethnicity | ||||
| White | 36 (65) | 13 (62) | 23 (68) | .13 |
| Hispanic | 15 (27) | 8 (38) | 7 (21) | |
| Other | 4 (7) | 0 (0) | 4 (12) | |
| High cytogenetic/molecular risk | 49 (89) | 18 (90) | 31 (89) | 1.00 |
| Months from diagnosis to HCT | ||||
| Median (range) | 7.8 (2.9-107.7) | 8.8 (3.2-107.7) | 7.0 (2.9-99.8) | .25† |
| Disease status at HCT | ||||
| CR 1 | 32 (56) | 11 (52) | 21 (58) | .86 |
| CR 2 | 18 (32) | 7 (33) | 11 (31) | |
| CR 3+ | 7 (12) | 3 (14) | 4 (11) | |
| MRD at HCT | ||||
| Detected | 10 (18) | 4 (20) | 6 (17) | .73 |
| Not detected | 46 (82) | 16 (80) | 30 (83) | |
| Unknown | 1 | 1 | 0 | |
| Karnofsky performance status | ||||
| ≤80 | 5 (11) | 1 (6) | 4 (13) | .65 |
| ≥80 | 42 (89) | 15 (94) | 27 (87) | |
| HCT-CI | ||||
| Median (Range) | 3.0 (0.0-8.0) | 3.0 (0.0-8.0) | 3.0 (0.0-8.0) | .91† |
| Donor type | ||||
| MRD | 22 (39) | 7 (33) | 15 (42) | .48 |
| MUD | 21 (37) | 10 (48) | 11 (31) | |
| Haplo | 14 (25) | 4 (19) | 10 (28) | |
| Conditioning regimen | ||||
| MAC | 25 (44) | 4 (19) | 21 (58) | .010 |
| MAC/TBI | 8 (14) | 5 (24) | 3 (8) | |
| RIC | 24 (42) | 12 (57) | 12 (33) | |
| HCT patients | ||||
|---|---|---|---|---|
| Measure | All, n = 57 (%) | Blinatumomab n = 21 (%) | Controls n = 36 (%) | P value* |
| Gender | ||||
| Male | 33 (58) | 17 (81) | 16 (44) | .012 |
| Female | 24 (42) | 4 (19) | 20 (56) | |
| Age in years, median (range) | 38.0 (16.0-66.0) | 29.0 (16.0-65.0) | 41.0 (19.0-66.0) | .26† |
| Race/ethnicity | ||||
| White | 36 (65) | 13 (62) | 23 (68) | .13 |
| Hispanic | 15 (27) | 8 (38) | 7 (21) | |
| Other | 4 (7) | 0 (0) | 4 (12) | |
| High cytogenetic/molecular risk | 49 (89) | 18 (90) | 31 (89) | 1.00 |
| Months from diagnosis to HCT | ||||
| Median (range) | 7.8 (2.9-107.7) | 8.8 (3.2-107.7) | 7.0 (2.9-99.8) | .25† |
| Disease status at HCT | ||||
| CR 1 | 32 (56) | 11 (52) | 21 (58) | .86 |
| CR 2 | 18 (32) | 7 (33) | 11 (31) | |
| CR 3+ | 7 (12) | 3 (14) | 4 (11) | |
| MRD at HCT | ||||
| Detected | 10 (18) | 4 (20) | 6 (17) | .73 |
| Not detected | 46 (82) | 16 (80) | 30 (83) | |
| Unknown | 1 | 1 | 0 | |
| Karnofsky performance status | ||||
| ≤80 | 5 (11) | 1 (6) | 4 (13) | .65 |
| ≥80 | 42 (89) | 15 (94) | 27 (87) | |
| HCT-CI | ||||
| Median (Range) | 3.0 (0.0-8.0) | 3.0 (0.0-8.0) | 3.0 (0.0-8.0) | .91† |
| Donor type | ||||
| MRD | 22 (39) | 7 (33) | 15 (42) | .48 |
| MUD | 21 (37) | 10 (48) | 11 (31) | |
| Haplo | 14 (25) | 4 (19) | 10 (28) | |
| Conditioning regimen | ||||
| MAC | 25 (44) | 4 (19) | 21 (58) | .010 |
| MAC/TBI | 8 (14) | 5 (24) | 3 (8) | |
| RIC | 24 (42) | 12 (57) | 12 (33) | |
Fisher’s exact test.
Wilcoxon rank-sum test.
Safety and feasibility
Blinatumomab was well-tolerated posttransplantation, with the most common severe adverse events being limited to hematologic cytopenias, including leukopenia (19% G3) and neutropenia (19% G4), as noted in Table 3. Diarrhea occurred in 7 patients (33%) and was mostly grade 1 (5 patients) and not GVHD-related. Importantly, only 1 patient developed grade 1 CRS, and 1 patient developed grade 2 neurotoxicity in the form of confusion that resolved with a temporary hold of the blinatumomab infusion (Table 3). Furthermore, rates of GVHD were acceptable, with cumulative rates of aGVHD grades 2 to 4 and 3 to 4 noted at 33% and 5%, respectively. Two patients (10%) were noted to have NIH mild cGVHD (oral 1/3; oral 1/3, liver 1/3), and 1 patient (5%) had moderate cGVHD (skin 3/3, liver 1/3). Finally, none of the patients developed secondary graft failure. Study accrual was slow, and consequently, the study was stopped early due to the sponsor’s decision. Since none of the 21 patients enrolled met the toxicity criteria for stopping the trial, the feasibility of blinatumomab postallogeneic HCT as consolidation therapy in patients with B-lineage ALL was met.
Incidence of toxicities graded by CTCAE, V4
| Toxicity | Maximum grade | |||
|---|---|---|---|---|
| Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
| n (%) | n (%) | n (%) | n (%) | |
| Constitutional | ||||
| Fatigue | 3 (14) | 0 (0) | 1 (5) | 0 (0) |
| Fever | 1 (5) | 2 (10) | 0 (0) | 0 (0) |
| Flu-like syndrome | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Hematologic | ||||
| Anemia | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Leukopenia | 2 (10) | 6 (29) | 4 (19) | 0 (0) |
| Neutropenia | 2 (10) | 0 (0) | 0 (0) | 4 (19) |
| Thrombocytopenia | 3 (14) | 0 (0) | 1 (5) | 0 (0) |
| Cardiovascular | ||||
| Chest pain | 0 (0) | 1 (5) | 0 (0) | 0 (0) |
| Hypotension | 0 (0) | 1 (5) | 0 (0) | 0 (0) |
| Arrythmias | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Thromboembolic event | 2 (10) | 0 (0) | 0 (0) | 0 (0) |
| Pulmonary | ||||
| Dyspnea | 0 (0) | 1 (5) | 0 (0) | 0 (0) |
| Cough | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Gastrointestinal | ||||
| Abdominal pain | 0 (0) | 1 (5) | 0 (0) | 0 (0) |
| Nausea | 2 (10) | 1 (5) | 0 (0) | 0 (0) |
| Vomiting | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Oral mucositis | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Diarrhea | 5 (24) | 1 (5) | 1 (5) | 0 (0) |
| Elevated liver enzymes | 4 (19) | 5 (24) | 1 (5) | 0 (0) |
| Infections | ||||
| Viral | 2 (10) | 0 (0) | 0 (0) | 0 (0) |
| Bacterial | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Electrolyte abnormalities | ||||
| Hypokalemia | 0 (0) | 0 (0) | 1 (5) | 0 (0) |
| Hypomagnesemia | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Neurologic | ||||
| Headache | 3 (14) | 0 (0) | 0 (0) | 0 (0) |
| Dizziness | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Confusion | 0 (0) | 1 (5) | 0 (0) | 0 (0) |
| Transient dysphasia | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Tremors | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Skin | ||||
| Rash | 4 (19) | 1 (5) | 2 (10) | 0 (0) |
| Renal | ||||
| Elevated creatinine | 1 (5) | 1 (5) | 0 (0) | 0 (0) |
| Others | ||||
| Dry eye | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Alopecia | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Anxiety | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Arthralgia | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Hiccups | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Insomnia | 3 (14) | 0 (0) | 0 (0) | 0 (0) |
| Toxicity | Maximum grade | |||
|---|---|---|---|---|
| Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
| n (%) | n (%) | n (%) | n (%) | |
| Constitutional | ||||
| Fatigue | 3 (14) | 0 (0) | 1 (5) | 0 (0) |
| Fever | 1 (5) | 2 (10) | 0 (0) | 0 (0) |
| Flu-like syndrome | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Hematologic | ||||
| Anemia | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Leukopenia | 2 (10) | 6 (29) | 4 (19) | 0 (0) |
| Neutropenia | 2 (10) | 0 (0) | 0 (0) | 4 (19) |
| Thrombocytopenia | 3 (14) | 0 (0) | 1 (5) | 0 (0) |
| Cardiovascular | ||||
| Chest pain | 0 (0) | 1 (5) | 0 (0) | 0 (0) |
| Hypotension | 0 (0) | 1 (5) | 0 (0) | 0 (0) |
| Arrythmias | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Thromboembolic event | 2 (10) | 0 (0) | 0 (0) | 0 (0) |
| Pulmonary | ||||
| Dyspnea | 0 (0) | 1 (5) | 0 (0) | 0 (0) |
| Cough | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Gastrointestinal | ||||
| Abdominal pain | 0 (0) | 1 (5) | 0 (0) | 0 (0) |
| Nausea | 2 (10) | 1 (5) | 0 (0) | 0 (0) |
| Vomiting | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Oral mucositis | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Diarrhea | 5 (24) | 1 (5) | 1 (5) | 0 (0) |
| Elevated liver enzymes | 4 (19) | 5 (24) | 1 (5) | 0 (0) |
| Infections | ||||
| Viral | 2 (10) | 0 (0) | 0 (0) | 0 (0) |
| Bacterial | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Electrolyte abnormalities | ||||
| Hypokalemia | 0 (0) | 0 (0) | 1 (5) | 0 (0) |
| Hypomagnesemia | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Neurologic | ||||
| Headache | 3 (14) | 0 (0) | 0 (0) | 0 (0) |
| Dizziness | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Confusion | 0 (0) | 1 (5) | 0 (0) | 0 (0) |
| Transient dysphasia | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Tremors | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Skin | ||||
| Rash | 4 (19) | 1 (5) | 2 (10) | 0 (0) |
| Renal | ||||
| Elevated creatinine | 1 (5) | 1 (5) | 0 (0) | 0 (0) |
| Others | ||||
| Dry eye | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Alopecia | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Anxiety | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Arthralgia | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Hiccups | 1 (5) | 0 (0) | 0 (0) | 0 (0) |
| Insomnia | 3 (14) | 0 (0) | 0 (0) | 0 (0) |
Laboratory correlates
To study the kinetics of T-cell response after blinatumomab therapy, we studied the T-cell populations and expression of checkpoint molecules in serial PBMCs for 15 patients with available samples (11 responders and 4 nonresponders). Responders had greater numbers of CD3, CD4, and CD160 T cells compared with nonresponders, both at baseline and posttreatment (Figure 1). In addition, responders had higher levels of CD8 T cells after therapy (Figure 1). Detailed quantitative values for the heat map findings are supplied in a box-plot diagram (supplemental Figure 1). viSNE analysis confirmed increased numbers of effector memory and terminally differentiated effector memory cells both within the CD8 and CD4 T-cell compartments in responders compared with nonresponders, pointing to their critical role in mediating cytotoxicity (Figure 2). Detailed quantitative values for the viSNE analysis are supplied in a box-plot diagram (supplemental Figure 2).
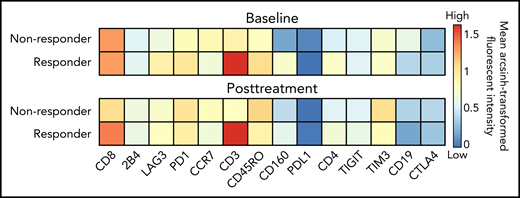
Heatmap of surface marker expression of T cells in nonresponders and responders. Hyperbolic arcsin transformed fluorochrome expression for 14 markers were averaged for baseline samples taken from nonresponders (n = 3) and responders (n = 10), and posttreatment samples from nonresponders (n = 4) and responders (n = 11).
Heatmap of surface marker expression of T cells in nonresponders and responders. Hyperbolic arcsin transformed fluorochrome expression for 14 markers were averaged for baseline samples taken from nonresponders (n = 3) and responders (n = 10), and posttreatment samples from nonresponders (n = 4) and responders (n = 11).
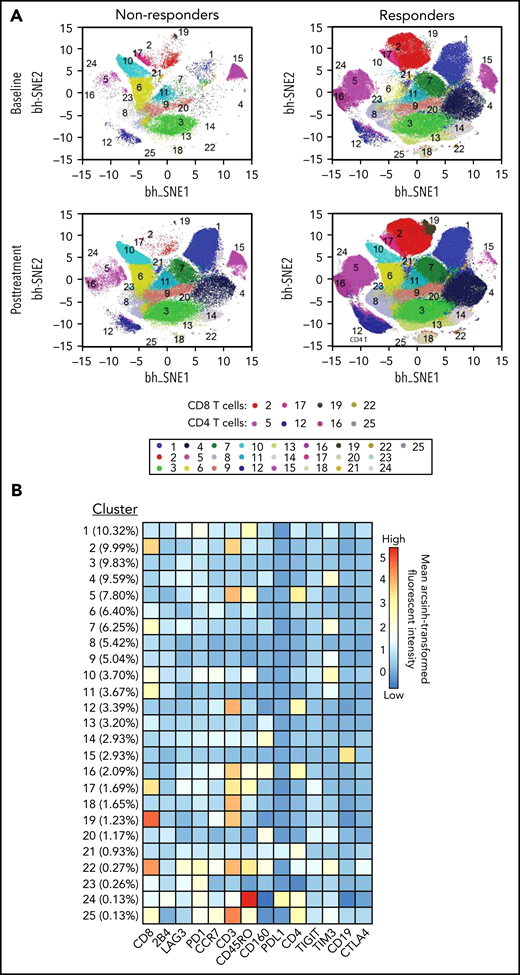
Subpopulations identified via viSNE analysis of 14 surface markers in all 56 samples. (A) viSNE map for nonresponders and responders color-coded according to PhenoGraph cluster annotation. viSNE maps were separated to baseline and posttreatment in both nonresponders and responders groups. (B) Heatmap of mean surface marker expression in each cluster. Percentage in parentheses denotes the size of each cluster.
Subpopulations identified via viSNE analysis of 14 surface markers in all 56 samples. (A) viSNE map for nonresponders and responders color-coded according to PhenoGraph cluster annotation. viSNE maps were separated to baseline and posttreatment in both nonresponders and responders groups. (B) Heatmap of mean surface marker expression in each cluster. Percentage in parentheses denotes the size of each cluster.
We also examined the expression of checkpoint molecules on T cells, including LAG3, PD1, PDL1, TIGIT, TIM3 (T-cell immunoglobulin and mucin domain 3), and CTLA4. Checkpoint molecules LAG3, PD1, TIGIT, and TIM3 were expressed at high levels both at baseline and after treatment in all patient samples (Figure 1); however, TIM3 was the only checkpoint that was expressed at statistically higher levels in nonresponders compared with responders after blinatumomab treatment (P = .04) (supplemental Figure 1). Finally, CD19 expression was lowest after 1 cycle of blinatumomab in responders compared with nonresponders (Figure 1), and the difference was statistically significant after treatment (supplemental Figure 1).
Efficacy
Seventeen of the 21 (81%) patients were alive at the end of the study, and the median follow-up time for all patients was 14.3 months (range, 7.5 to 52.4 months). Six patients progressed, including the 2 patients who had MRD positivity prior to the start of blinatumomab therapy, for a cumulative incidence of relapse of 29% (95% CI 11%-49%). The 1-year OS and PFS for patients were 85% (95% CI 61%-95%) and 71% (95% CI 47%-86%), respectively (Figure 3). There were no regimen-related deaths. We compared our results to a contemporary cohort control that included information for 128 patients (Table 2). Using a 2:1 (control:treated) ratio, the matched analysis dataset included information for 57 (36:21) patients. The median follow-up time for the control group was 24.6 months (range, 3.4 to 67.4). No statistically significant differences in PFS and OS were observed between groups (Figure 4).
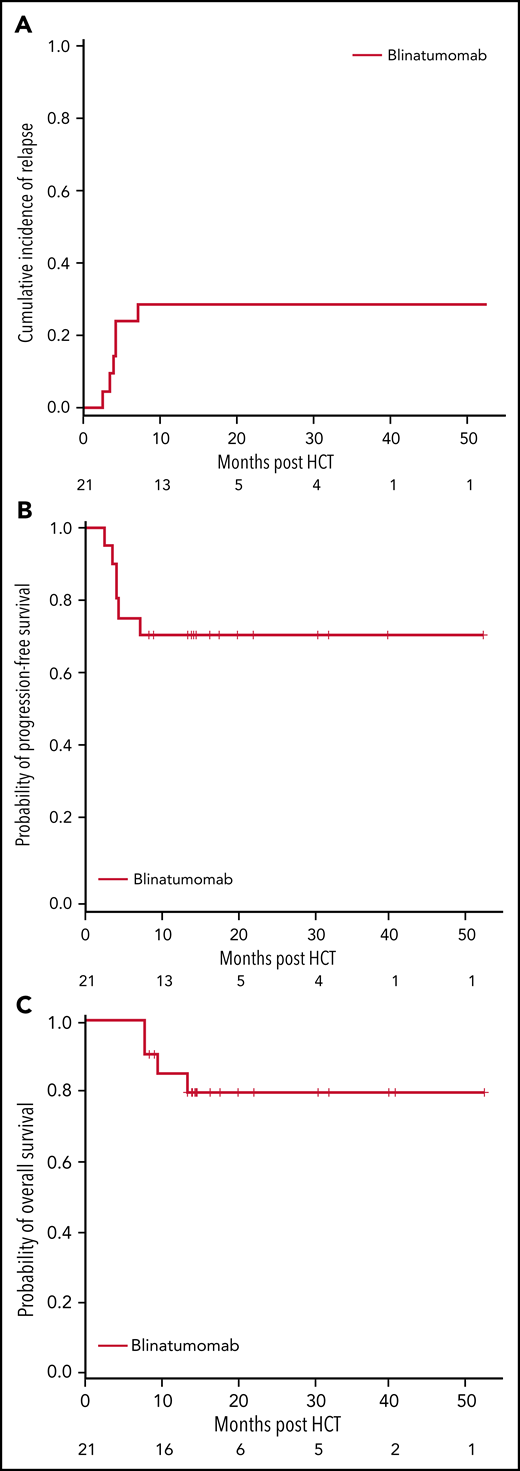
Study outcomes for patients treated with blinatumomab. At 1 year, the rate of relapse was 29% (95% CI, 11%-49%) (A), progression-free survival (PFS) 71% (95% CI, 47%-86%) (B), and overall survival (OS) 85% (95% CI, 61%-95%) (C).
Study outcomes for patients treated with blinatumomab. At 1 year, the rate of relapse was 29% (95% CI, 11%-49%) (A), progression-free survival (PFS) 71% (95% CI, 47%-86%) (B), and overall survival (OS) 85% (95% CI, 61%-95%) (C).

Comparison of PFS and OS between patients treated with blinatumomab maintenance and no posttransplant maintenance (matched-case cohort). At 1 year, the rates of PFS for the blinatumomab vs the control group were 71% vs 68%, P = .44 (A), and the rates for OS for the blinatumomab vs the control group were 85% vs 76%, P = .23 (B).
Comparison of PFS and OS between patients treated with blinatumomab maintenance and no posttransplant maintenance (matched-case cohort). At 1 year, the rates of PFS for the blinatumomab vs the control group were 71% vs 68%, P = .44 (A), and the rates for OS for the blinatumomab vs the control group were 85% vs 76%, P = .23 (B).
Discussion
To our knowledge, this is the first study to investigate the use of prophylactic blinatumomab in the posttransplant setting to mitigate the risk of relapse. Our study established the feasibility of this approach, with 91% of enrolled patients able to receive at least 1 cycle of blinatumomab at a median of 78 days following the day of HCT and 57% completing all 4 intended cycles of treatment. As expected, based on the previously published toxicity profile of blinatumomab, the drug was well tolerated, with no significant toxicity noted. Importantly, treatment did not need to be stopped secondary to cytopenias, and there were no cases of secondary graft failure. Furthermore, despite the expectant hypogammaglobulinemia with blinatumomab, there were no excess infections, as shown in Table 3, and the NRM rate was 0. Finally, GVHD rates were not in excess of what would be expected, and in fact, grades 3 to 4 aGVHD were quite low at 5%.
Notably, we were able to glean important mechanistic insights into why this type of cellular therapy had benefits in only a subset of patients. Broadly, cluster analysis clearly identifies responders as having higher frequencies of CD4 and CD8 T cells with an effector memory phenotype compared with nonresponders (Figure 2; supplemental Figure 2). Furthermore, at baseline, prior to the initiation of blinatumomab, the responder group had relatively higher levels of CD160 which, while shown to be inhibitory to CD4 T-cell function in some studies,31 has been associated with CD8+ T-cell effector function in other studies (Figure 1; supplemental Figure 2).32 Interestingly, however, we did not find a difference in the CD4 Treg frequencies at baseline33 between responders and nonresponders (P = .378, data not shown). Our findings are limited by both the small sample size included in the correlative analysis and the imbalance of a greater number of responders (73%) vs nonresponders. Thus, our exploratory findings require further investigation in larger, prospective studies.
The in vivo modulation of blinatumomab has been recently reported by Puzzolo and colleagues.34 Extensive in vivo monitoring was performed in 43 patients treated on the GIMEMA LAL2216 study of upfront induction with dasatinib followed by 2 to 5 cycles of consolidation with blinatumomab in adult patients with Ph+ ALL. They noted a progressive increase in CD3+ T cells after each cycle of blinatumomab that became significant after cycle 3, specifically with an increase in the CD3/CD8 T-cell subset. Furthermore, they noted increases in CD4/CD45RO+ T-NK and NK lymphocyte populations, while they noted a progressive reduction in T-regulatory (Tregs) CD4+ T cells, which have been shown in some studies to drive tumor evasion and limit the efficacy of blinatumomab.33,35,36 In contrast to our study, they did not find a correlation between immune modulation and the degree of molecular response that was reached in about 80% of patients.34
The association of higher memory CD4 and CD8 T-cell subsets in blinatumomab-responding patients has been noted before.37 One approach to address low levels of T cells would be to combine DLI with blinatumomab. In fact, this strategy was investigated by Ueda and colleagues in 4 patients with B-ALL who relapsed following allogeneic HCT and subsequently received DLI concurrently during the second or later cycles of blinatumomab. Prolonged remission was noted in 2 of the patients.38 Variable efficacy, along with the risk for GVHD, has limited the enthusiasm for further investigating this approach. Another strategy may be to use cytokines to restore T-cell activity. Interleukin-7 (IL-7) is a common γ-chain cytokine required for lymphocyte survival and expansion, specifically preventing lymphocyte apoptosis and restoring CD4+ and CD8+ T-cell function.39 Although not currently approved for clinical use, IL-7 is under investigation in numerous clinical trials, including one in which it was used to promote T-cell recovery after T-cell-deplete allogeneic HCT.40
Upregulation of exhaustion markers PD1, TIM3, and LAG3 have been previously noted on ALL blast cells,41 and at lower levels of TIGIT.42 Furthermore, treatment with blinatumomab resulted in an increase in CTLA-4, PD1, TIM3, and LAG3 expression in cell assays.41 While we observed mildly elevated levels of these checkpoint molecules in our patient samples (Figure 1), critically, only TIM3 expression was found to be significantly higher in nonresponders after treatment with blinatumomab (Figure 1; supplemental Figure 1). TIM3 has been shown to regulate both innate and adaptive immune responses, potentially acting as a positive or negative regulator.43 Checkpoint blockade, including against TIM3, is most actively investigated in solid tumors. However, there are several trials addressing this route of resistance in ALL by combining blinatumomab with checkpoint inhibitors. Inhibitors of PD1 in combination with blinatumomab are actively being investigated in the following ongoing clinical trials for adults with relapsed B-ALL (ClinicalTrials.gov Identifier: NCT03512405, NCT03168079, NCT04524455). Another ongoing trial is investigating the combination of blinatumomab with checkpoint inhibitors of PD1 and CTLA4 in patients with relapsed B-ALL (ClinicalTrials.gov identifier: NCT02879695). Five patients have been treated with blinatumomab combined with nivolumab, and 4 have achieved CR with MRD negativity.44
Efficacy for blinatumomab maintenance is difficult to determine since this was not a prospectively randomized study. In efforts to estimate possible activity, we compared outcomes with a matched 2:1 cohort, and based on this analysis, saw no significant benefit for blinatumomab maintenance (Figure 4). In addition to the inherent limitations of this type of analysis, we acknowledge the short follow-up duration for the blinatumomab group, and we will need to monitor outcomes with longer follow-up. In the nontransplant setting, Rambaldi and colleagues recently reported on the outcomes for the subset of patients who received continued blinatumomab for maintenance and consolidation45 from the original randomized, phase 3 study of blinatumomab vs standard chemotherapy in patients with Ph-negative, refractory, or relapsed B-ALL20; 267 patients received blinatumomab induction, and 36 (13%) received continued maintenance, defined as 6 or more cycles of blinatumomab. The maintenance cohort had longer OS (median unreached vs 15.5 months, OR 0.37, 95% CI 0.16-0.88) and PFS (14.5 months [95% CI 7.1-21.9] vs 9.8 months [95% CI 8.5-11.1], OR 0.48, 95% CI 0.22-1.03) compared with those who didn’t receive maintenance.45
In conclusion, we demonstrated that blinatumomab maintenance therapy following transplant is feasible and has a very safe toxicity profile. While our study did not demonstrate a clear clinical advantage of this approach in the entire cohort, we showed that response to blinatumomab therapy posttransplantation is dependent on the immune profile of the patient posttransplantation, with a distinct immune phenotype-predicting response. This may inform which patients will most likely benefit from blinatumomab therapy posttransplantation. Larger-prospective studies are needed to confirm these findings. In addition, we also found that overexpression of checkpoint molecules, specifically TIM3, may be implicated as a mechanism of resistance, and therefore combination therapies with blinatumomab and immune checkpoint inhibitors may result in improved efficacy.
Acknowledgments
This work was supported by grants (1 R01 CA211044-01, 5 P01CA148600-03, and P50CA100632-16) from the National Institutes of Health (NIH), the Specialized Program of Research Excellence (SPORE) in Leukemia grant (P50CA100632), grant (CA016672) to the Anderson Cancer Center from the NIH, the Cancer Center Support Grant (NCI Grant P30 CA016672), and the clinical trial was supported by Amgen Pharmaceutical.
Authorship
Contribution: M.R.G., P.B., X.J., K.R., and P.K. were involved in writing the manuscript, study design, data collection, data analysis, data interpretation, and reviewing and editing of manuscript; V.N., S.I., M.K., and R.B. were involved in running the laboratory studies, data analysis, and reviewing and editing of the manuscript; D.R.M. was involved in data analysis, data interpretation, literature search, writing the manuscript, and creating the tables and figures; and C.G., S.K., M.D., A.A., R.M., G.A., I.K., B.O., D.M., U.P., A.O., P.T., N.J., E.J., F.R., H.K., K.C., R.C., and E.S. were involved in data collection, data interpretation, and reviewing and editing of the manuscript.
Conflict-of-interest disclosure: K.R., P.B., M.D., R.B., and The University of Texas MD Anderson Cancer Center (MDACC) have an institutional financial conflict of interest with Takeda Pharmaceutical for the licensing of the technology related to CAR-NK cells. K.R., R.B., and The University of Texas MD Anderson Cancer Center have an institutional financial conflict of interest with Affimed GmbH. K.R. participates on scientific advisory boards for GemoAb, Avenge Bio, Kiadis, GSK, and Bayer. All other authors declare no competing financial interests.
Correspondence: Partow Kebriaei, Department of Stem Cell Transplantation & Cellular Therapy, The University of Texas, M.D. Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: pkebriae@mdanderson.org.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal