

Abstract
Cytogenetics has long represented a critical component in the clinical evaluation of hematologic malignancies. Chromosome banding studies provide a simultaneous snapshot of genome-wide copy number and structural variation, which have been shown to drive tumorigenesis, define diseases, and guide treatment. Technological innovations in sequencing have ushered in our present-day clinical genomics era. With recent publications highlighting novel sequencing technologies as alternatives to conventional cytogenetic approaches, we, an international consortium of laboratory geneticists, pathologists, and oncologists, describe herein the advantages and limitations of both conventional chromosome banding and novel sequencing technologies and share our considerations on crucial next steps to implement these novel technologies in the global clinical setting for a more accurate cytogenetic evaluation, which may provide improved diagnosis and treatment management. Considering the clinical, logistic, technical, and financial implications, we provide points to consider for the global evolution of cytogenetic testing.
Introduction
Genomic characterization has become an essential component in the clinical management of hematologic malignancies, supporting diagnosis and prognostication as well as informing therapeutic decisions.1-7 Cytogenetics, the study of chromosome number and structure, represents a central facet of diagnostic genomic analysis with a longstanding history. For the past 50 years, chromosome banding has permitted single-cell and genome-wide analysis of chromosomal alterations, generating a foundational knowledge base of chromosome abnormalities and their clinical associations.7-13 Presently, for most hematologic malignancies, chromosome banding is an essential component of the diagnostic workup that guides clinical care.1,2,14-16 Chromosome banding has long remained the gold standard for cytogenetic studies globally, primarily for its high success rate in detecting critical copy number variation (CNV) and structural variation (SV) known to initiate and drive disease progression.7,17-21
However, an inherent limitation of chromosome banding is the dependency on cells dividing in vitro. In some hematologic malignancies, a sufficient number of analyzable metaphase cells may not be obtained in culture to achieve a complete chromosome banding study.22 Moreover, even when cells readily divide and render ample metaphases, chromosome banding has limited resolution with challenges in detecting alterations <10 Mb. Supplementary technologies, namely fluorescence in situ hybridization (FISH) and chromosomal microarray (CMA), were subsequently developed and overcame many of the inherent limitations of chromosome banding analysis.23 These platforms do not necessarily require dividing cells and can detect abnormalities <10 Mb, effectively expanding the resolution from large chromosome bands down to gene-level imbalances. FISH is a targeted assay that can be used to detect CNVs and SVs in targeted regions. While widely adopted, there exists great variability in the clinical use of FISH testing, which is often predicated on a priori knowledge of a specific gene or region of interest.24 Moreover, clinical laboratories are also largely reliant on the development of commercially available FISH probes.25 CMA is a powerful whole-genome technology with the ability to detect CNV and copy-neutral loss-of-heterozygosity (CN-LOH) in the kilobase range using an unbiased approach.25-32 However, as CMA cannot detect balanced chromosomal rearrangements, which are hallmarks of many hematologic malignancies, its utility remains limited to diseases primarily driven by CNVs or unbalanced SVs. CMA adoption worldwide has been hampered by financial constraints and reimbursement practices. While several evidence-based reviews have highlighted the clinical utility of CMA for a diverse spectrum of hematologic malignancies, to date, CMA has not been universally incorporated into current clinical guidelines.32-38 Given the rapid advancements of genomic technologies that have the potential to comprehensively detect clinically significant cytogenetic alterations, the international diagnostic community must carefully evaluate the implementation of these next-generation cytogenetic methodologies and their implications for clinical care by considering the following questions.
Are our current traditional cytogenetic diagnostic assays sufficient, or could newer technologies improve clinical management?
For myeloid malignancies, chromosome banding and FISH can be used to detect the majority of the CNVs and SVs needed for appropriate clinical management.14,39-41 The high success rate at identifying acute myeloid leukemia (AML)-specific abnormalities reflects the ability of myeloid cells to divide readily in culture.42,43 Of 250 adult AML cases with concurrent chromosome banding and FISH testing, a successful karyotype result (defined as having ≥20 sufficiently analyzable metaphases) was attained in 88% of cases, 10% of cases had between 1 and 19 metaphases, and only 2% of cases had no metaphases for analysis.44 Supplementation by AML-directed FISH yielded a 98% concordance between chromosome and FISH analysis (in cases with an adequate chromosome study).44 Such evidence-based studies provided the foundation for the clinical guidelines supported by the European LeukemiaNet (ELN) (www.leukemia-net.org/home), the National Comprehensive Cancer Network (NCCN) (www.nccn.org), and the Revised International Prognostic Scoring System (IPSS-R) (www.mds-foundation.org).5,20,21,44-47
The success of chromosome analysis is, however, not uniform across all hematologic malignancies.48 A successful chromosome analysis would provide ≥20 metaphases of sufficient resolution to identify chromosomal abnormalities representative of the hematologic disease being evaluated. In contrast to myeloid malignancies, the success rate of chromosome analysis in acute lymphoblastic leukemia (ALL) is approximately 70% to 75%; failures are primarily attributed to substandard chromosome morphology rendering the cells unanalyzable, and/or poor in vitro cell growth of the ALL clone with an inadequate number of analyzable metaphases.48 When chromosome banding studies are successful, an ALL-specific clonal abnormality can be identified in about 60% to 80% of ALL cases.22,48 Similar to ALL, the frequency of detecting an abnormal clone in chronic lymphocytic leukemia (CLL) using a CpG oligodeoxynucleotide-stimulated culture is 50% to 70%, as reported in multilaboratory studies.49,50 In contrast, the frequency of identifying an abnormal plasma cell clone in the case of a plasma cell neoplasm (PCN) such as multiple myeloma is only 10% to 20%51 since malignant plasma cells rarely undergo cell division, except in cases of advanced disease.52
Ongoing cancer research studies utilizing novel technologies continue to expose inherent limitations of current cytogenetic diagnostic testing. In hematologic malignancies, SVs involving enhancer hijackers can reposition promoter and/or enhancer elements of the genome (such as immunoglobulin enhancer sequences) and drive the overexpression of a nearby oncogene without producing in-frame chimeric gene fusions.53-55 These types of abnormalities are relatively common in lymphoid malignancies and have been shown to lead to false-negative56-58 or, in rare instances, false-positive FISH results.59 For example, the MYC break-apart FISH probe demonstrates an approximately 4% false-negative rate in diffuse large B-cell lymphoma56 and a 50% to 70% false-negative rate in PCNs.58,60 Novel molecular approaches have also identified new aberrations that play a role in cancer initiation and progression, some of which can be targeted using novel therapeutics.61 Understanding the limitations of current “gold standard” diagnostic tools and identifying technologies that may be more effective in addressing these vulnerabilities will be crucial to the advancement of clinical care.
Innovative molecular approaches designed to overcome present-day limitations include mate pair sequencing (MPseq) and optical genome mapping (OGM). MPseq is a variation of whole-genome sequencing (WGS) utilizing a specialized library preparation of long input DNA followed by circularization, fragmentation, and sequencing of smaller paired-end fragments.58,62 OGM electrophoreses high molecular weight DNA into nanochannels, linearizes them for imaging, and creates a consensus genome map from processed images.63-65 Both MPseq and OGM can identify CNVs and SVs, including balanced rearrangements that escape detection by CMA. Recent studies have highlighted the advantages of OGM as an unbiased whole-genome technology in hematologic malignancies, demonstrating concordance with chromosome banding while revealing additional abnormalities missed by routine workups and refining complex karyotypes such as derivative and marker chromosomes.64,66-68 In addition, whole transcriptome sequencing (WTS) and targeted RNA-seq assays have been developed to enable the identification of expressed gene fusions. These approaches have shown promising results in pediatric ALL,69 some lymphomas,70 multiple myeloma,71,72 T-cell lymphoblastic lymphoma,73 and AML.74 Generally, RNA-based assays detect transcribed genomic regions and highly expressed fusion products, which preclude their detection of SVs involving enhancer hijackers that do not result in chimeric gene fusions. In contrast, DNA-based WGS provides a comprehensive and unbiased characterization of gene rearrangements. Importantly, formalin-fixed, paraffin-embedded material, commonly available for extramedullary hematologic malignancies such as lymphoma, generally only allows DNA-based assays such as WGS for fusion detection in these malignancies. Although targeted RNA-seq assays are commonly used in the context of hematologic malignancies, WTS has not been widely adopted in the clinical setting.
The technologies described above provide viable alternatives to traditional cytogenetic approaches (chromosome banding, FISH, and CMA) with improved resolution. However, all the aforementioned techniques examine the genome through a singular lens, limited to cytogenetic aberrations. Today, diagnostic evaluation of hematologic disorders requires the evaluation of other types of genomic alterations, including single nucleotide variants (SNVs) (eg, TP53, RUNX1, IDH, NPM1, and FLT3-ITD).9,15,75-77 Detection of these alterations can be achieved using multiple molecular methods, including reverse transcription polymerase chain reaction (RT-PCR), Sanger sequencing, or next-generation sequencing (NGS) approaches. Thus, for many hematologic malignancies, detection of clinically relevant genomic variants requires a battery of individual assays. Therefore, a single, comprehensive, and affordable assay that captures SNVs, CNVs, and SVs is highly desirable. Please see Table 1 for the list of currently available genomic technologies, their advantages, and limitations.
Advantages, limitations, and clinical utility of currently available genomic technologies in hematologic malignancies
| G-banding | FISH | CMA | WGS | Targeted sequencing panels | RT-PCR | MPSeq | WTS | OGM | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Analyte | Chromosome in dividing cells | DNA in interphase nuclei and metaphase | DNA | DNA | DNA | RNA | RNA | DNA | RNA | DNA |
| Coverage | Whole | Targeted | Whole | Whole | Targeted | Targeted | Targeted | Whole | Whole | Whole |
| Distinction of individual cell clones | Yes | Yes | No | No | No | No | No | No | No | No |
| Analysis bias | Yes | Yes (if cultured) | No | No | Yes | Yes | No | No | No | No |
| Turnaround time (d) | 3-7 | 4 h to 2 d | 3-7 | 7-14 | 7-14 | 7-14 | 4 h to 5 d | 7-14 | 14-21 | 7-10 |
| Unmapped region detection | Yes | No | No | No | No | No | No | No | No | Some Alu and LINE elements |
| Ability to multiplex | Low | Low | High | High | High | High | High | High | High | Low to medium |
| Analytical sensitivity (%) | 1*-3 out of 20 metaphases | 1-10 | 10-20 | 20-30 | 1-10 | 5-10 | ~0.01 | 10 | 1-10 | 5-20 |
| SVs | Yes | Yes | No | Yes (long-read or short-read deep sequencing) | No | Gene fusion | Limited | Yes | Yes | Yes |
| CNVs | Yes | Yes | Yes | Yes | Limited | Limited | Limited | Yes | Limited | Yes |
| SNVs | No | No | No | Yes | Yes | Yes | No | Limited | Yes | No |
| Disease status | Diagnosis, disease monitoring, relapse | Diagnosis, disease monitoring, relapse | Diagnosis, relapse | Diagnosis, relapse | Diagnosis, disease monitoring, relapse, MRD (if deep coverage) | Diagnosis, disease monitoring, relapse, MRD (if deep coverage) | Diagnosis, disease monitoring, MRD, relapse | Diagnosis, relapse | Diagnosis, relapse | Diagnosis, relapse |
| Well-established | High | High | High | Low | High | High | High | Low | Low | Low |
| Cost | ++ | ++ | +++ | +++++ | +++ | +++ | ++ | ++++ | ++++ | +++ |
| G-banding | FISH | CMA | WGS | Targeted sequencing panels | RT-PCR | MPSeq | WTS | OGM | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Analyte | Chromosome in dividing cells | DNA in interphase nuclei and metaphase | DNA | DNA | DNA | RNA | RNA | DNA | RNA | DNA |
| Coverage | Whole | Targeted | Whole | Whole | Targeted | Targeted | Targeted | Whole | Whole | Whole |
| Distinction of individual cell clones | Yes | Yes | No | No | No | No | No | No | No | No |
| Analysis bias | Yes | Yes (if cultured) | No | No | Yes | Yes | No | No | No | No |
| Turnaround time (d) | 3-7 | 4 h to 2 d | 3-7 | 7-14 | 7-14 | 7-14 | 4 h to 5 d | 7-14 | 14-21 | 7-10 |
| Unmapped region detection | Yes | No | No | No | No | No | No | No | No | Some Alu and LINE elements |
| Ability to multiplex | Low | Low | High | High | High | High | High | High | High | Low to medium |
| Analytical sensitivity (%) | 1*-3 out of 20 metaphases | 1-10 | 10-20 | 20-30 | 1-10 | 5-10 | ~0.01 | 10 | 1-10 | 5-20 |
| SVs | Yes | Yes | No | Yes (long-read or short-read deep sequencing) | No | Gene fusion | Limited | Yes | Yes | Yes |
| CNVs | Yes | Yes | Yes | Yes | Limited | Limited | Limited | Yes | Limited | Yes |
| SNVs | No | No | No | Yes | Yes | Yes | No | Limited | Yes | No |
| Disease status | Diagnosis, disease monitoring, relapse | Diagnosis, disease monitoring, relapse | Diagnosis, relapse | Diagnosis, relapse | Diagnosis, disease monitoring, relapse, MRD (if deep coverage) | Diagnosis, disease monitoring, relapse, MRD (if deep coverage) | Diagnosis, disease monitoring, MRD, relapse | Diagnosis, relapse | Diagnosis, relapse | Diagnosis, relapse |
| Well-established | High | High | High | Low | High | High | High | Low | Low | Low |
| Cost | ++ | ++ | +++ | +++++ | +++ | +++ | ++ | ++++ | ++++ | +++ |
Depending on the clinical situation, 1 metaphase with a recurring abnormality may be considered evidence for an abnormal clone.
Can a single assay provide comprehensive genomic characterization?
The evolution of Sanger sequencing to NGS, coupled with the decreasing costs of DNA resequencing, ushered in an unprecedented genomics era.78,79 In the current clinical diagnostics setting, NGS assays are frequently limited to targeted gene-specific panels that require regular updates as new clinically relevant genes are identified.80-82 However, there is considerable variability in content, analysis, and interpretation between different genomics laboratories.
Whole exome sequencing (WES) covers all exonic sequences and has the potential to eliminate much of the variance, at least relating to content. While WES costs have dramatically decreased over the past 5 years, they nonetheless remain prohibitive in many parts of the world. With each evolutionary advance, we see a benefit but also recognize other deficiencies.83-86 Improved bioinformatics algorithms have enabled WES to detect most CNVs, albeit with variable performance, depending on the quality and depth of the sequencing data and normalization parameters. In addition, since WES excludes intronic and regulatory genomic regions, it is unable to detect a large subset of SVs. This shortfall is significant, as detection of some intronic SVs is essential in the evaluation of certain hematologic malignancies. Thus, WES has the potential to rapidly adapt to new information stemming from translational research, while it also suffers from the inability to detect all pertinent genomic aberrations.84
WGS, particularly involving long-read or long-insert DNA fragments, is an attractive technology that can detect all 3 genomic variants (SNVs, CNVs, and SVs) in an unbiased manner.87-89 Despite these advantages, WGS is currently not adopted for clinical use worldwide because of the complexity of WGS compared with other technologies in relation to the processes, workflows, and resources necessary for its implementation. At present, WGS is primarily used in large academic centers, mostly for translational research purposes. A recent publication by Duncavage et al proposed a paradigm shift in clinical laboratory genomic testing by suggesting WGS as an alternative to cytogenetic analysis in myeloid cancers.90 The authors performed WGS in a cohort of 263 patients with myeloid malignancies and confirmed 40 recurrent translocations and 91 copy-number alterations previously identified by cytogenetic studies. In addition, WGS identified additional genomic events (CNVs and/or SVs) in 40 of 235 patients (17.0%). When used prospectively in 117 consecutive patients, WGS provided new genetic information in 29 (24.8%) and changed the risk category in 19 (16.2%). This publication was met with both enthusiasm and skepticism,5,91-95 and subsequently sparked a discussion among clinical laboratorians globally, dividing those who were actively promoting an evolution of testing and clinical adoption of WGS vs those who debated the real world and current feasibility of this transition.
Implementing new technologies in a clinical diagnostic laboratory
To justify the introduction of a new clinical genomic assay as a replacement for the current gold standard cytogenetic testing (ie, chromosome banding coupled with FISH), multiple factors should be carefully considered. These include the following interconnected considerations: clinical, logistic, technical, and financial.
Clinical considerations
The utilization of new comprehensive molecular methodologies has the potential to improve clinical care by leading to a more accurate diagnosis and enhancing treatment selection based on higher resolution and more precise genomic data. In these instances, a genome-wide assessment may reveal uncommon but known entities that are not routinely surveyed. Some example scenarios include, but are not limited to:
- 1.
Hematologic malignancies with a broad spectrum of genomic driver alterations of various classes that would require multiple assays to effectively evaluate, such as Ph-like B-ALL, PCN, CLL, and non-Hodgkin lymphomas.
- 2.
Hematologic malignancies with well-documented enhancer hijacking alterations associated with risks of false-negative or false-positive findings by conventional diagnostic approaches (eg, IGH and/or MYC rearrangements).
- 3.
Cases in which chromosome studies are not successful or are known to demonstrate an unacceptable failure rate (especially in follow-up studies of posttreatment specimens).
- 4.
Atypical clinical presentations warranting the identification of genomic drivers for appropriate management (eg, mixed lineage leukemias).
- 5.
Detection of cytogenetic abnormalities that require further workup to completely characterize their origin and genomic makeup (eg, derivative and marker chromosomes).
- 6.
Investigation of possible false-negative findings when a normal karyotype/FISH result is obtained, but clinical presentation and pathology support the evidence of a neoplasm.
Technological advances have uncovered additional genomic aberrations and complexities often unappreciated using conventional diagnostic approaches. However, interpretation of these novel findings with unclear clinical significance remains a challenge and will likely require additional translational research initiatives to understand their clinical significance. In the interim, the question remains as to how to handle these new findings that are revealed when employing novel technologies. While most laboratories are likely to only report abnormalities of known clinical significance, consideration should be given to possibly reporting novel abnormalities to potentially facilitate future translational research studies involving large patient cohorts. Such studies would focus on correlating novel genomic abnormalities with patient outcomes and their response to therapies. Indeed, this would require a clear description/disclaimer of the unknown significance of the novel findings, which could potentially be reported under a special heading on the clinical report.
The value of this approach was recently demonstrated by Tyner et al.96 In this study, the authors reported the integration of whole-exome and transcriptome sequencing in the analysis of 672 patients with AML while examining recurrent mutations (and combinations thereof) to determine drug sensitivity. This study summarized functional, genomic, and transcriptomic data from this large, clinically characterized AML cohort to date and paved the way for establishing the clinical utility of novel technologies in myeloid disorders. Similar reports have been published for other hematologic malignancies such as pediatric ALL and PCN. Nordlund et al used linked-read WGS to generate haplotype information and successfully detected CNVs and SVs in ALL patients, including cryptic and key abnormalities such as DUX4, ZNF384, and PAX5 gene rearrangements.97 Moreover, Hollein et al recently reported the use of WGS in combination with RNA-seq in 211 diagnostic PCN samples and observed 92% concordance of FISH and WGS results for CNVs and SVs evaluation.72 However, some abnormalities, such as MYC translocations in PCN, are more reliably identified using WGS rather than FISH approaches.60 Such translational studies afford the opportunity to correlate conventional diagnostic approaches with novel genomic studies. In this instance, a comparative nomogram study could correlate the clinical significance of low-level abnormal clones detected by WGS with the number of abnormal metaphase cells detected by chromosome banding. This would be critical to our future understanding of small clonal populations identified by WGS. As the clinical significance of most submicroscopic CNVs below the resolution of current cytogenetic profiling is not known, sufficiently powered studies are needed to delineate their clinical, prognostic, and therapeutic relevance. Publicly available databases are also critically needed to facilitate data sharing (both genomic and clinical information) to enhance our understanding of these genomic findings within the clinical setting. This is by no means an easily accomplished task, as it will require the interpretation and integration of large amounts of data for each patient while holding the promise of improved clinical management. To this end, population reference databases such as the Genome Aggregation Database98 and Database of Genomic Variants,99 among others,100,101 have provided reference maps allowing improved interpretation of SNVs and CNVs, including some SVs. Recently, a population reference map of SVs has been made available,102 providing a reference of SVs from nearly 15 000 genomes from a diverse population. This tool will be instrumental in the interpretation of incidental and benign SVs.
One commonly encountered clinical challenge in oncology testing is the identification of incidental findings of germline variants of clinical significance. To this end, the American College of Medical Genetics and Genomics has published guidance for the reporting of secondary findings in the context of clinical exome and genome sequencing.103 The secondary findings maintenance working group has recommended a list of genes that should be evaluated in individuals undergoing clinical-grade sequencing. Based on these recommendations, these secondary findings should also be reported when encountered in the analysis of SVs and CNVs using NGS. To simplify workflows, however, filters can be applied to allow a focused analysis on clinically significant regions related to the reason for testing. Informed consent and genetic counseling should also be recommended.103
In some circumstances, particularly when the tumor represents nearly 100% of the tested tissue, it is unclear if the abnormality identified by NGS is germline or somatic. In such instances, similar to other clinical testing scenarios, a nontumor sample can be requested (blood or skin) to evaluate the germline status. This can be a desirable follow-up approach that minimizes the need to perform side-by-side germline and somatic testing. Currently, as there are no guidelines to address all the above possibilities in oncology testing, each laboratory should establish its own policies to address incidental findings, including the option to only analyze targeted regions.
We suggest that for early adopters of new technologies without a full understanding of many of the novel variants that will be encountered, a tiered reporting strategy is necessary to clearly delineate the well-established genomic alterations from other variants of unknown significance. Such a tiered approach has been recommended by numerous professional organizations in the analysis of CNVs, SNVs, and CN-LOH in cancer.26,82,104-106 Moreover, as translational and clinical research studies continue to deliver evidence-based data supporting the significance of novel genomic findings, reanalysis of existing WGS data can be achieved to update the patient’s diagnosis/prognosis/management if novel, clinically significant variants are identified, potentially minimizing additional expenses associated with repeat genomic testing.
Logistic considerations
The impact of adopting new technologies such as WGS must be considered at a system level. On a global scale, the replacement of conventional testing may be impractical at this time. Multiple factors must be considered and carefully navigated, including the availability of sequencing equipment, trained personnel, infrastructure to support reagent transportation and distribution, impacts of turnaround time, complexity of analysis, development of an integrated workflow between the molecular and cytogenetics laboratories, computing and data storage infrastructure, and cost (including reimbursement).107 Given that evaluation of novel technologies may require a significant upfront investment in the forms of capital equipment, bioinformatics means, data storage capabilities, and human resources, the transition to any disruptive technology must be well-planned in order to avoid disruption to patient care and confusion to the clinical stakeholders. The gradual expansion of testing should enable harmonization of laboratory processes, maintaining quality control and reporting guidelines, and facilitate the adoption of those technologies with the greatest improvements over present standards.
Technical considerations
The early adoption of WGS as an alternative to conventional cytogenetic testing has brought about questions regarding the need to replace current cytogenetic approaches. Indeed, this “replacement” is much more likely to be an “evolution” with the gradual adoption of innovative technologies following the establishment of carefully considered disease lists and/or clinical scenarios where a given technology shows the greatest promise. Presently, many clinical situations optimally require the combinatorial use of multiple technologies to appropriately balance time, cost, and clinical needs. Targeted assays may be supplemented if new technologies cannot return critical results in a timely fashion, potentially delaying appropriate clinical management. In the context of AML, WGS will likely take a longer time to complete than traditional approaches. Therefore, coupling WGS with more rapid, targeted assays, such as FISH for PML::RARA or BCR::ABL1 fusion, may permit results to be returned in a timely fashion to determine initial therapy at diagnosis/presentation. Subsequently, WGS results can be delivered before the end of the initial induction therapy for a more comprehensive genomic profiling that may provide novel but clinically important information for subsequent therapeutic/risk stratification decisions. This approach requires built-in redundancies in testing parameters until turnaround times can match present standards.
Despite the comprehensive nature of genome-wide assays such as WGS, there remain regions in the genome or critical aberrations that are not amenable to WGS. For example, a fraction of the genome, such as subtelomeric and centromeric regions, is refractory to sequencing.108 Although some of these limitations could be overcome with the use of ultralong read sequencing,109 this approach is not widely accessible. In addition, internal tandem repeats (eg, FLT3-ITD) may escape detection by amplicon-based WGS and will likely require orthogonal approaches to uncover these types of genomic lesions.110
Current published guidelines for clinical germline WGS specifically note that laboratories are not required to validate all classes of variation that include all potential iterations of genomic lesions but can instead adopt a phased approach to validations. However, laboratories must establish operational familiarity with all quality control parameters and identify any factors that would adversely affect the detection of certain variants (eg, PCR amplification for library preparation and CNV detection). In certain clinical situations, orthogonal confirmation of clinically actionable pathogenic calls may be warranted, although this additional effort would further increase the cost and turnaround time of clinical testing.105
Another challenge in the universal use of WGS is the inconsistency in the variant components of the bioinformatics pipelines used by each laboratory. These include alignment and variant calling in addition to the transfer of data to new human genomic builds. In addition, the depth of sequencing coverage would optimally be adapted according to the reason for testing (diagnosis, response to treatment, or minimal residual disease [MRD] assessment). In general, cancer NGS tests with the goal to guide targeted therapy have a limit of detection (LOD) of around 5%. However, in the context of MRD detection, cancer NGS tests may need to reliably detect alterations down to ≤1%.111,112 The sensitivity of WGS is typically lower than other testing methodologies such as targeted RT-PCR. Thus, for MRD purposes, private and novel variants identified by WGS at diagnosis could be used to customize an RT-PCR assay that would allow higher sensitivity. While Duncavage and colleagues suggested that WGS could be a viable alternative to cytogenetic analysis in myeloid cancers with a target coverage of 60x for their entire cohort, this level of coverage is predicted to provide a LOD of 10% using 3 reads as the minimum number to call SNVs with 95% confidence interval based on binomial probability distribution. This would be insufficient for either guiding targeted therapy or tracking MRD.112-114 In addition, a small subset (4.3%) of their cohort had less than 25x genome coverage, corresponding to a LOD of 23%. To reach a LOD of 5% and 1% for appropriate NGS tests to guide targeted therapy or to detect MRD, the sequence coverage needs to be at 124x and 625x, respectively, which would increase sequencing cost significantly (Figure 1). The LOD to detect CNV, on the other hand, varies based on the size and genomic location of these alterations. For CNVs that include junction sequences, split reads can support breakpoint detection and improve the sensitivity compared with CNV detected using read-depth alone.115 In the Duncavage et al study, the following criteria were used to call CNVs >5Mb: (1) Having at least 2 paired and 2 split reads supporting the breakpoints, and (2) the ratio between coverage depth and normalized control being <0.8 for deletion or >1.3 for duplication. This entails that the tumor burden is ≥20% to call a deletion >5Mb or ≥30% to call a duplication >5Mb.
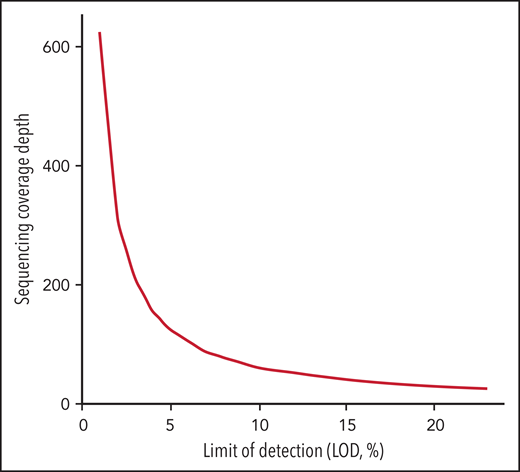
Sequencing coverage depth needed for LOD in the range of 1% to 25% calculated based on confidence interval at 0.95 with threshold of minimum 3 unique sequencing reads according to the binomial distribution.
Sequencing coverage depth needed for LOD in the range of 1% to 25% calculated based on confidence interval at 0.95 with threshold of minimum 3 unique sequencing reads according to the binomial distribution.
In general, LOD increases for smaller size CNVs, thus making it challenging to identify gene-level or exon-level CNVs <5Mb with 20% to 30% tumor burden using ∼60x WGS. The LOD for CN-LOH detection is still not well understood, and further studies are needed. It is therefore critical that such issues be thoroughly investigated so that laboratories can identify both advantages and limitations and ensure that alternative tests are available in the laboratory’s testing menu if, and when, a diagnostic result cannot be achieved (eg, due to sequencing failures, gaps in coverage, complex repetitive sequences, appropriately tracking MRD, etc). Therefore, if WGS is not successful, the laboratory can use conventional chromosome studies from samples that have been already grown and stored as a fixed cell pellet. This illustrates the importance of maintaining some degree of redundancy, with competency in a more diversified testing portfolio to address the various testing deficiencies defined above. Put simply, FISH and chromosome banding may remain necessary in certain instances, particularly in instances of compromised WGS studies. All these regulatory processes are time-consuming and will slow down the speed of establishment of WGS for routine diagnostics.
Financial considerations
The financial considerations for performing WGS as a clinical test include both the cost to adopt and validate WGS and the cost to routinely perform it in a clinical laboratory setting. The major cost to adopt WGS is capital equipment, personnel cost for staff technologist/scientist with experience in NGS and bioinformaticians to build and maintain the pipeline, reagents for test validation, data storage, database generation, and qualified laboratory geneticists or molecular pathologists for data interpretation and reporting. As shown in Table 2, the total cost is estimated in the $0.9 to $1.5 million range in each laboratory.
Estimated cost for the adoption and validation of WGS in a clinical laboratory setting
| Item | Estimated cost range ($) | Comments |
|---|---|---|
| Capital equipment | 800 000-1 000 000 | Ex: Illumina NovaSeq 6000 |
| Bioinformatics pipeline | 10 000-30 000 | Ex: Illumina Dragon license |
| Test validation | 39 000-100 000 | 30-50 samples |
| Staff salary | 50 000-200 000 (varies per country) | Technologist and research and development scientist (3-6 mo), bioinformatician (3-6 mo), laboratory geneticist and/or pathologist (1-3 mo) |
| Total | 899 000-1 330 000 |
| Item | Estimated cost range ($) | Comments |
|---|---|---|
| Capital equipment | 800 000-1 000 000 | Ex: Illumina NovaSeq 6000 |
| Bioinformatics pipeline | 10 000-30 000 | Ex: Illumina Dragon license |
| Test validation | 39 000-100 000 | 30-50 samples |
| Staff salary | 50 000-200 000 (varies per country) | Technologist and research and development scientist (3-6 mo), bioinformatician (3-6 mo), laboratory geneticist and/or pathologist (1-3 mo) |
| Total | 899 000-1 330 000 |
The total cost to run WGS as a clinical test with ∼60x coverage is estimated to be in the $1300 to $2500 range,90,116,117 with the cost breakdown to routinely perform WGS in a clinical laboratory setting discussed in detail in Duncavage et al (∼$2 million for a laboratory running 1000 WGS tests per year). This is significantly higher compared with chromosome banding, which remains in the range of $150 to $750 (varies by country). Moreover, this cost is based on the estimate of batching ∼10 to 20 samples per sequencing flow cell, which may not be feasible for most laboratories, as batching will invariably interfere with maintaining an appropriate turn-around-time for hematologic malignancy testing. In addition, depending on the robustness of curated knowledge bases, the cost for employing scientists and laboratory geneticists and/or pathologists to interpret and report cases will vary and remain a significant financial consideration. Optimistically, for future adoption of WGS, laboratories must batch patient samples with different clinical indications, including hematologic malignancies, solid tumors, and congenital conditions, which could help maintain a clinically appropriate turn-around-time and decrease cost.118
Based on these cost considerations, currently, only large institutions in developed countries with substantial financial support can afford the implementation of WGS. The financial and other logistical barriers remain prohibitive for smaller laboratories, especially those in low-income countries, to adopt WGS.
In the United States, reimbursement by private and public payers only occurs if a test has an associated current procedural terminology code (CPT; a medical code used to report medical, surgical, and diagnostic procedures and services to entities such as physicians, health insurance companies, and accreditation organizations) (www.cms.gov).119,120 The CPT codes for a given test are linked to a specific technology (eg, chromosome banding/FISH/RT-PCR/CMA/WES/WGS); however, a technology-associated CPT code does not necessarily guarantee reimbursement. The newer the code, the more likely insurance companies are to deny payment, substantiating their actions by claiming the technology to be “experimental.” US Food and Drug Administration (FDA) approval of a specific technology-associated test essentially converts an experimental test to one accepted for use in clinical diagnosis, thus facilitating reimbursement. However, considerable resources are needed to navigate the FDA approval process, making this pathway more likely to be initiated by commercial laboratories or large medical institutions. Reimbursement can be linked to FDA approval, particularly when a therapeutic drug is associated with a specific diagnosis supported using an FDA-approved test (www.fda.gov).121 In such instances, access to therapy may be tied to the specific testing dictated by pharmaceutical companies. Therefore, even if a laboratory offers WGS, a specific targeted assay such as FISH may instead be required to assure reimbursement for the disease-associated therapy. In countries with public health systems (eg, Canada, Germany, Italy, Spain, and the United Kingdom), government bodies work with scientific associations or physician organizations to determine which tests will be financially reimbursed and often have fixed annual budgets that make adding expensive testing difficult.116 In many instances, centralized clinical laboratories then provide these approved tests. The same holds true for many Latin American countries, such as Argentina, that have a mix of public and private health systems. Lagging reimbursement by public and private payers remains the primary obstacle to the widespread adoption of novel technologies that offer more sensitive and comprehensive testing modalities. Unfortunately, this factor often discourages the adoption of new tests despite a body of literature that clearly demonstrates their clinical utility and validity.
Financial considerations remain key factors for clinical laboratories to consider adopting clinical WGS. The cost to adopt and validate WGS, followed by routinely performing WGS while maintaining a reasonable turn-around-time, may not be currently applicable for most laboratories globally. In addition, without clear pathways for reimbursement, creating a business plan to justify adopting the new technologies may be impractical at this time.116
Toward real-time changes in clinical guidelines
Chromosome banding is an integral part of current clinical guidelines and prognostic systems (NCCN, ELN, and IPSS-R). The value of novel technologies has been highlighted in this review, and the frequent lag in their adoption constitutes an obstacle to improved diagnostics and patient outcomes. This is especially significant as access to the newest therapies through enrollment in clinical trials relies on current guidelines. It is therefore important for our laboratory and clinical communities to formulate a more dynamic response to the evidence emerging from novel technologies and more rapidly and continuously update the guidelines. We argue that clinical guidelines should adopt technology-agnostic standards so long as a laboratory appropriately validates a given technology. Constraining diagnostic genomic laboratories to a particular testing methodology is counterproductive and decreases patient access to local laboratories that could potentially offer the same test by another validated method in a more affordable manner.
Alternative approaches to improving patient outcomes
As improvement in patient outcomes remains the ultimate objective of clinical genomic testing, it is important to consider alternative and innovative approaches that may reduce the necessity for comprehensive genome-wide testing strategies. Clinical management is often guided by the optimal therapy choice based on biomarker status: as more targeted therapies for specific biomarkers are developed, clinical management via personalized medicine becomes paramount and may eliminate reliance on WGS altogether. While we remain in the early days of single-cell sequencing, a deeper understanding of clonal heterogeneity and a change in therapeutic strategies based on these minor but potentially aggressive clones may become more widely used. In addition, the significance of novel therapies such as chimeric antigen receptor T-cell or tumor mRNA vaccines may be agnostic to tumor biomarkers and become a “magic bullet” for some types of cancer therapy.122 Finally, it is unclear whether a combined omics approach (genomic, transcriptomic, proteomic, methylomic) may be a better and more comprehensive approach not only to understand but also to treat human disease.
Concluding remarks
From a global health perspective, with the goal to provide equitable care for all patients worldwide, access to novel and sophisticated genomic testing tools may not be immediately feasible across all clinical laboratories. Presently, there are significant impediments to the immediate adoption of WGS by clinical laboratories worldwide. While the clinical utility of WGS is undeniable, chromosome banding and FISH with SNP array integration will likely continue to serve as the gold standard of cytogenetic testing for hematologic malignancies. We remain optimistic that by establishing data-generating initiatives through translational research, expediting the adoption of published guidelines (such as NCCN and ELN), validating novel technologies for clinical utility, and ensuring appropriate turnaround times and reimbursement policies, we can embrace the change and drive the evolution of genomic technologies. Looking ahead, and as our understanding of the genome and the molecular pathogenesis of disease continues to advance, we will undoubtedly find ourselves looking into (epi)genomic, transcriptomic, and proteomic tools for the full characterization of human diseases. While one single test may not replace the totality of tests that currently exist, the active exploration and adoption of novel approaches in defined clinical settings may indeed allow for a continued and controlled evolution resulting in the successful clinical implementation of WGS in the near future.
Acknowledgments
The authors thank Jill Kappers from the Mayo Clinic for providing administrative assistance in the preparation of this document.
Supported by National Cancer Institute grant P01 CA108671 11 (R.L.L.).
Authorship
Contribution: X.X., Y.M.N.A., L.B.B., A.M.D., and B.L. researched data for the article, provided substantial contribution to discussion and content, writing, and review/editing of the manuscript before submission; M.M., C.M., R.L.L., R.P.H., and F.S. researched data for the article, provided substantial contribution to discussion and content and review/editing of the manuscript before submission; and A.C.S., P.D.C., M.D.C., M.S.G., I.G.F., D.T.H., B.S., and I.S. reviewed and edited the manuscript before submission.
Conflict-of-interest disclosure: R.L.L. is on the supervisory board of Qiagen and is a scientific advisor to Imago, Mission Bio, Zentalis, Ajax, Auron, Prelude, C4 Therapeutics, and Isoplexis; receives research support from and consulted for Celgene and Roche and has consulted for Incyte, Janssen, Astellas, Morphosys, and Novartis; has received honoraria from Astra Zeneca, Roche, Lilly, and Amgen for invited lectures and Gilead for grant reviews. A.C.S. reports personal financial interests in Bionano Genomics. The remaining authors declare no competing financial interests.
Correspondence: Xinjie Xu, Division of Hematopathology, Division of Laboratory Genetics and Genomics, Department of Laboratory Medicine and Pathology, Mayo Clinic, 200 First Street SW, Rochester, MN 55905; e-mail: xu.xinjie@mayo.edu; Brynn Levy, College of Physicians and Surgeons, Columbia University Medical Center and the New York Presbyterian Hospital, 3959 Broadway CHC – Room 406b, New York, NY 10032; e-mail: bl2185@cumc.columbia.edu; and Francesc Solé, MDS Group, Institut de Recerca Contra la Leucèmia Josep Carreras, Edifici IJC, Campus ICO-Germans Trias i Pujol, Ctra de Can Ruti, Camí de les Escoles s/n, 08916 Badalona, Barcelona, Spain; e-mail: fsole@carrerasresearch.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal