

Key Points
GATA1s-generating mutations and trisomy 21 in either order results in a megakaryoblastic leukemia similar to myeloid leukemia of Down syndrome.
Germline GATA1s-generating mutations are associated with familial predisposition for leukemia and myelodysplastic syndrome.

Abstract
Individuals with Down syndrome are at increased risk of myeloid leukemia in early childhood, which is associated with acquisition of GATA1 mutations that generate a short GATA1 isoform called GATA1s. Germline GATA1s-generating mutations result in congenital anemia in males. We report on 2 unrelated families that harbor germline GATA1s-generating mutations in which several members developed acute megakaryoblastic leukemia in early childhood. All evaluable leukemias had acquired trisomy 21 or tetrasomy 21. The leukemia characteristics overlapped with those of myeloid leukemia associated with Down syndrome, including age of onset at younger than 4 years, unique immunophenotype, complex karyotype, gene expression patterns, and drug sensitivity. These findings demonstrate that the combination of trisomy 21 and GATA1s-generating mutations results in a unique myeloid leukemia independent of whether the GATA1 mutation or trisomy 21 is the primary or secondary event and suggest that there is a unique functional cooperation between GATA1s and trisomy 21 in leukemogenesis. The family histories also indicate that germline GATA1s-generating mutations should be included among those associated with familial predisposition for myelodysplastic syndrome and leukemia.
Introduction
Down syndrome (DS) is associated with transient abnormal myelopoiesis (DS-TAM) in newborns and a more than 100-fold increased risk of myeloid leukemia during the first 5 years of life.1 Myeloid leukemia of DS (ML-DS) is characterized by megakaryoblastic phenotype, early age of onset, and favorable response to chemotherapy. Virtually all patients with DS-TAM and ML-DS have somatic mutations in exon 2 (or rarely exon 3) of the X-linked gene GATA1, which encodes a key erythroid and megakaryocytic transcription factor. These mutations produce a short GATA1 isoform (called GATA1s) that lacks the amino terminal transactivation domain. Similar germline GATA1s-generating mutations result in congenital anemia in males,2 which is sometimes considered Diamond Blackfan anemia.3,4
Leukemias typically arise after the sequential acquisition of mutations in a single-cell lineage, suggesting that there is a requirement for cooperating events in leukemogenesis.5 The importance of the order of the mutations (initiating and secondary events) is uncertain, although studies in myeloproliferative neoplasms suggest different phenotypes according to the order in which the mutations were acquired.6
We describe 2 families with germline GATA1s-producing mutations in which several members developed acute megakaryoblastic leukemia (AMKL) in early childhood. All evaluable leukemias had acquired trisomy 21 or tetrasomy 21 and clinical and molecular characteristics similar to those of ML-DS.
Methods
All family members provided written informed consent to participate in this study. The study was approved by the institutional review boards of Boston Children’s Hospital and Massachusetts Institute of Technology and the ethical committee of the Health Region Midt (Denmark).
Targeted GATA1 sequencing
For family 1, genomic DNA was purified from Ficoll-separated blood or bone marrow (BM) or from paraffin-embedded material. Sanger sequencing of GATA1 exon 2 and adjacent regions were performed on genomic DNA using published polymerase chain reaction primers.7 Myeloid next-generation sequencing (NGS) in the index patient of family 1 was performed by Sophia Genetics.
Data processing and whole-exome sequencing
For family 2, genomic DNA was purified from peripheral blood. Whole-exome sequencing (WES) and data processing were performed by the Genomics Platform at the Broad Institute of Massachusetts Institute of Technology and Harvard (http://genomics.broadinstitute.org/) using Illumina Nextera exome capture (∼38 Mb target) and sequenced (150-bp paired reads) to cover >80% of targets at 20× and a mean target coverage of >100×. WES sequencing data were processed by using a pipeline consisting of Picard command-line tools with genomic alignment using the Burrows-Wheeler Alignment (BWA) aligner (BWA-MEM) to the human genome build 38. Single nucleotide variants and insertions/deletions (indels) were jointly called across all samples in the full Center for Mendelian Genomics cohort using Genome Analysis Toolkit HaplotypeCaller package version 3.5. Default filters were applied to single nucleotide variant and indel calls using the Genome Analysis Toolkit Variant Quality Score Recalibration approach.
Variant annotation and identification
Called variants were annotated as previously described.8 Two main inheritance models were explored: X-linked recessive for the anemia seen in the 2 affected males (III-3 and III-4) and dominant inheritance for the leukemias seen throughout the family. In both inheritance models, the GATA1 variant chrX:48791111:T>C was flagged as the likely causal variant. The genes ANKRD26, ETV6, GATA2, CEBPA, RUNX1, SAMD9, SAMD9L, and DDX41 were manually inspected for potentially relevant variants, and none were identified.
Results
Family 1
The index case (IV-2, Figure 1A; supplemental Table 1) presented at 27 months of age with AMKL, karyotype 48,XX,der(6)del(6)(q15q22),der(7)ins(6;7)(q23q25;q32),+21,+21[24]/46,XX[1]. Myeloid NGS showed 3 JAK2 variants: c.2047A>G p.(Arg683Gly) (variant allele frequency [VAF], 4%), c.2082C>G p.(Phe694Leu) (VAF, 3%), and c.1832T>C p.(Leu611Ser) (VAF, 11%). She had no phenotypic signs of DS. Immunophenotyping, gene expression, and in vitro drug sensitivity studies showed patterns similar to those of ML-DS and patterns different from those of non-DS myeloid leukemia (Figure 2).

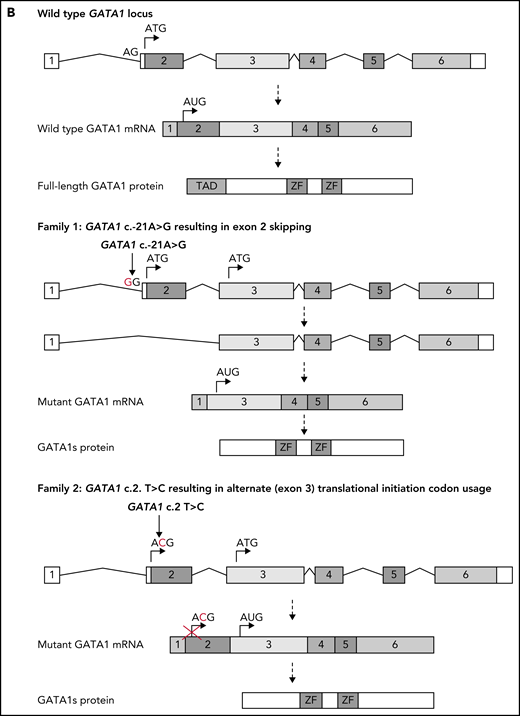
Identification of GATA1 mutations in affected members of 2 families. (A) Pedigree diagram of families. Arrows indicate index cases. (B) Schematic diagram of normal GATA1 gene splicing and translation and the impact of the c.-21 A>G and c.2 T>C variants. mRNA, messenger RNA; Wt, wild type.
Identification of GATA1 mutations in affected members of 2 families. (A) Pedigree diagram of families. Arrows indicate index cases. (B) Schematic diagram of normal GATA1 gene splicing and translation and the impact of the c.-21 A>G and c.2 T>C variants. mRNA, messenger RNA; Wt, wild type.
![Similarities between ML-DS and the leukemias that developed in the patients with germline GATA1 c.-21 A>G and c.2 T>C variants. (A) Immunochemical and histochemical characteristics of ML-DS and the patient leukemias. The overlap includes the unique characteristics of ML-DS compared with non-DS myeloid leukemia such as co-expression of myeloid markers (CD13, CD33, and CD38), early progenitor markers (CD34 and CD117), megakaryocytic/erythroid markers (CD41/61, CD42b, CD36, CD71, and CD235a), T-lymphocyte markers (CD4 and CD7), natural killer cell markers (CD56), but absence of monocytic and B-lymphocyte markers.16,17 CD7 expression (indicated in bold) is an important marker distinguishing ML-DS from non-DS AMKL.16,17 (B) Heat map showing gene expression (using Human Genome U133 Plus 2.0 Array) of samples from diagnostic blood and BM of AMKL in non-DS (3864-5036) and ML-DS (5809-7535) using unsupervised hierarchical clustering.18 Red, high gene expression; green, low gene expression. Patient IV-2 clusters with most of the ML-DS patients. (C) In vitro drug sensitivity (lethal concentration [LC50]) of the leukemia from family 1 patient IV-2 compared with ML-DS and non-DS myeloid leukemia patients.19 MPO, myeloperoxidase; p25-p75, 25th – 75th percentile; TdT, terminal deoxynucleotidyl transferase.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/21/10.1182_blood.2021011463/2/m_bloodbld2021011463f2.png?Expires=1769086305&Signature=E3qAkhN~17VfOQJCCNRPslKKufXODxNKR~4NOBuxPHrXbVa6TyAG-imSqTs5LE0VXAzydODE884-2gO3mO5tpt6LYzpSpjLCsj-qrA7ck6FeFgpCu6rUuagZUqFV7FUpONGr~HbM6zo5mBL4jKrRsBwZSkN2QDSbjGb3Sbgy9ynHWikL9kilejGhZh0VVuOKupALZKzWJ-n7vmThtogMpZS~Bmq3T5Uw2gr4xYJI1ybQxRbmJhJE4RMMcOK8hHxZTG-LKDHlHWH7dvBdYr5~busOtUGGrz4jBA1iM4vEuTxhrmeZZvrseQP6nLE~h1mKEke8SFqFvAeJjmCkoR8H~w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Similarities between ML-DS and the leukemias that developed in the patients with germline GATA1 c.-21 A>G and c.2 T>C variants. (A) Immunochemical and histochemical characteristics of ML-DS and the patient leukemias. The overlap includes the unique characteristics of ML-DS compared with non-DS myeloid leukemia such as co-expression of myeloid markers (CD13, CD33, and CD38), early progenitor markers (CD34 and CD117), megakaryocytic/erythroid markers (CD41/61, CD42b, CD36, CD71, and CD235a), T-lymphocyte markers (CD4 and CD7), natural killer cell markers (CD56), but absence of monocytic and B-lymphocyte markers.16,17 CD7 expression (indicated in bold) is an important marker distinguishing ML-DS from non-DS AMKL.16,17 (B) Heat map showing gene expression (using Human Genome U133 Plus 2.0 Array) of samples from diagnostic blood and BM of AMKL in non-DS (3864-5036) and ML-DS (5809-7535) using unsupervised hierarchical clustering.18 Red, high gene expression; green, low gene expression. Patient IV-2 clusters with most of the ML-DS patients. (C) In vitro drug sensitivity (lethal concentration [LC50]) of the leukemia from family 1 patient IV-2 compared with ML-DS and non-DS myeloid leukemia patients.19 MPO, myeloperoxidase; p25-p75, 25th – 75th percentile; TdT, terminal deoxynucleotidyl transferase.
Similarities between ML-DS and the leukemias that developed in the patients with germline GATA1 c.-21 A>G and c.2 T>C variants. (A) Immunochemical and histochemical characteristics of ML-DS and the patient leukemias. The overlap includes the unique characteristics of ML-DS compared with non-DS myeloid leukemia such as co-expression of myeloid markers (CD13, CD33, and CD38), early progenitor markers (CD34 and CD117), megakaryocytic/erythroid markers (CD41/61, CD42b, CD36, CD71, and CD235a), T-lymphocyte markers (CD4 and CD7), natural killer cell markers (CD56), but absence of monocytic and B-lymphocyte markers.16,17 CD7 expression (indicated in bold) is an important marker distinguishing ML-DS from non-DS AMKL.16,17 (B) Heat map showing gene expression (using Human Genome U133 Plus 2.0 Array) of samples from diagnostic blood and BM of AMKL in non-DS (3864-5036) and ML-DS (5809-7535) using unsupervised hierarchical clustering.18 Red, high gene expression; green, low gene expression. Patient IV-2 clusters with most of the ML-DS patients. (C) In vitro drug sensitivity (lethal concentration [LC50]) of the leukemia from family 1 patient IV-2 compared with ML-DS and non-DS myeloid leukemia patients.19 MPO, myeloperoxidase; p25-p75, 25th – 75th percentile; TdT, terminal deoxynucleotidyl transferase.
Acute myeloid leukemia (AML) therapy induced complete remission, including normal karyotype with disomy 21 in 1000 cells analyzed by fluorescence in situ hybridization. Macrocytosis and extreme lyonization (92%) were found during remission, although the dominant GATA1 allele was not identified. Myelodysplastic syndrome (MDS), which progressed to AML with 46,XX,-7,t(11;21)(q23;q21),+21[25] occurred 3.5 years from the end of therapy. Myeloid NGS identified a RUNX1 variant c.308_309dup, p.(Thr104Leufs*19) (VAF, 9%). She received a haploidentical hematopoietic stem cell transplantation (HSCT) with her mother as donor. She is in good health, including normal hematology 12.5 years from AML and 8.5 years from MDS diagnoses.
The father had had macrocytic anemia since childhood with normal neutrophil and platelet counts and moderate BM trilineage dysplasia. MDS with trilineage dysplasia was diagnosed at age 47 years. Karyotype was normal. Myeloid NGS identified an ASXL1 variant c.2632delA p.(Ser878fs) (VAF, 24%). Matched unrelated donor HSCT was performed. He is well with hematology within the normal range 14 months after HSCT.
Targeted GATA1 sequencing was performed on members of the extended family. The variant GATA1 c.-21A>G (NG_008846) located in the exon 2 splice acceptor site was identified in the index case, the father (III-2), paternal aunt (III-3), and paternal grandmother (II-3) (supplemental Figure 1). This causes exon 2 skipping and exclusive GATA1s production through an alternate exon 3 in-frame ATG initiation codon, as previously described9 (Figure 1B). The paternal aunt and grandmother with the GATA1 variant had normal complete blood counts apart from borderline thrombocytopenia in the grandmother (supplemental Table 1).
Family 2
The index case (III-2; Figure 1A; supplemental Table 1) presented at 18 months of age with AMKL and karyotype 48,XX,+11,+21/48,idem,t(1;14)(q21;q32)/49,idem+8. She had no phenotypic signs of DS, but the leukemia immunophenotype overlapped ML-DS (Figure 2A). AML therapy induced complete remission with normal karyotype including disomy 21. She remained in complete remission at follow-up 5 years from completion of chemotherapy, although her remission BM tests showed hypoplasia with mild erythroid and megakaryocytic dysplasia.
The proband’s younger brother (III-3) was diagnosed with congenital dyserythropoietic anemia at 6 months of age. He remained transfusion-dependent until undergoing unrelated umbilical cord HSCT at 6 years of age. The proband’s younger half-brother (III-4), different father, had transfusion-dependent anemia from early childhood. At 21 months, he developed AMKL with unavailable cytogenetics and immunophenotype overlapping ML-DS (Figure 2A). He underwent matched unrelated HSCT because of persistent residual disease after AML induction and is in good health 10 years after receiving a transplant. The proband’s mother (II-2) has lifelong macrocytic anemia without a need for transfusions. The proband’s maternal grandmother (I-2) died in her 40s as a result of a myocardial infarction, and the maternal great aunt (I-3) died as a result of leukemia as a teenager.
WES of peripheral blood from patients II-2, III-2 (in remission), III-3, and III-4 revealed the variant GATA1 c.2 T>C (NG_008846) in all individuals (supplemental Figure 2). This changes exon 2 translation initiator ATG to ACG (Figure 1B) and leads to nearly exclusive production of GATA1s, as previously described10 (Figure 1B).
Discussion
Germline GATA1s-producing variants have been reported as a cause of anemia in males.3,4 Our report indicates that these patients are also at increased risk for hematologic malignancies associated with acquisition of trisomy 21 or tetrasomy 21. In addition, a 4-year-old boy with a c.2T>C GATA1 mutation developed MDS with complex cytogenetics that included trisomy 21.11
Abnormal hematopoiesis in carrier females could result from extreme lyonization as seen in X-chromosome diseases.12 We were unable to determine definitively whether extreme lyonization favoring the GATA1-mutant allele played a role in the females’ diseases. Regardless, our report indicates that female carriers of GATA1s-generating mutations are at risk for hematology malignancy.
Leukemogenesis is generally considered as a series of events with acquisition of many mutations before the leukemia phenotype develops in adults.13 Leukemogenesis in children may be simpler, especially in DS in which only 2 documented aberrations, trisomy 21 and GATA1 mutations, are sufficient to cause DS-TAM, although additional aberrations may be needed for ML-DS,14 and complex karyotype is common.15
The AMKLs in our patients shared many features of ML-DS, including unique immunophenotype, gene expression, drug sensitivity, secondary JAK2 mutations,12 complex karyotype,15 and onset before 4 years of age. The disease in these patients suggests that the order of AML-associated oncogenic events may not be important because constitutional trisomy 21 with acquired GATA1 mutations results in a disease similar to constitutional GATA1 mutation with acquired trisomy 21.
Previous reports identified children with germline mosaicism for trisomy 21 who developed ML-DS with acquired GATA1s-generating mutations in the trisomy 21 cells.13 These findings in concert with our current report underscore the unique functional synergy between trisomy 21 and GATA1s in producing age-specific myeloid leukemia.14 The mechanisms underlying this cooperativity remain incompletely understood. GATA1s expression in human fetal liver hematopoietic stem cells cooperates with trisomy 21 to promote blast and megakaryocyte expansion in xenotransplantation models.15 The ERG, ETS2, miR-99a, miR-125b, and miR-155 genes located on human chromosome 21 cooperate with GATA1s to perturb hematopoiesis and drive the development of AMKL in experimental systems.15,16 Our data suggest that either order of mutations preserves this cooperative effect.
The 2 families presented here challenge our concept of initiating and secondary events and suggest that the combination of GATA1s-generating mutation and trisomy 21 results in a unique myeloid malignancy regardless of the order of events. Our observations also indicate that GATA1s-generating mutations should be added to the list of constitutional abnormalities that predispose to MDS and leukemias in both males and females. Signs of worsening cytopenias should prompt appropriate investigation.
Acknowledgments
The authors are grateful to the families for their participation in this study, Dorte Melsvik for performing Sanger sequencing and analysis, and Anni Aggerholm for the NGS studies.
This work (WES) was partially supported by the Broad Institute of the Massachusetts Institute of Technology and Harvard Center for Mendelian Genomics, by grants from the National Institutes of Health, National Human Genome Research Institute (R01 HG009141 and UM1 HG008900), the National Heart, Lung and Blood Institute (R01 HL146500 [V.G.S.], P01 HL32262-30 [A.B.C.]), the National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK103794), and the National Eye Institute, by a grant from the New York Stem Cell Foundation (V.G.S.), and by a gift from the Lodish Family to Boston Children’s Hospital (V.G.S.).
V.G.S. is a New York Stem Cell Foundation-Robertson Investigator.
The views and opinions expressed in this document are those of the authors and do not necessarily represent the views or opinions of the United States Government or the United States Office of Personnel Management.
Authorship
Contribution: H.H. and A.B.C. conceived the study, coordinated the analysis, and wrote the manuscript; C.G.N. and E.K. performed the genetic analyses for family 1; J.P. performed drug sensitivity studies; C.M.Z. performed genetic expression studies; H.H. and K.R.-J. provided clinical data for family 1; R.M.K. identified family 2 and coordinated the clinical analysis; N.F.N.-A.-R. and D.B. provided the clinical description of family 2; S.P.D. and K.R.C. coordinated the WES; J.M.V., S.P.D., and K.R.C. analyzed the WES data; V.G.S. designed the experiments for family 2 and analyzed the genomic data; and all authors commented on and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Henrik Hasle, Department of Pediatrics, Aarhus University Hospital, Palle Juul-Jensens Blvd 99, 8200 Aarhus N, Denmark; e-mail: hasle@dadlnet.dk; and Alan B. Cantor, Division of Pediatric Hematology-Oncology, Boston Children’s Hospital, 300 Longwood Ave, Boston, MA 02115; e-mail: alan.cantor@childrens.harvard.edu.
The WES data have been deposited at accession No. phs001272 in the Genotypes and Phenotypes database at the National Human Genome Research Institute’s Analysis, Visualization, and Informatics Laboratory-space (https://anvilproject.org/data?query=dataTypes%3DExome%26search%3Dphs001272).
For additional primary data, please contact Henrik Hasle via e-mail at hasle@dadlnet.dk or Alan B. Cantor at alan.cantor@childrens.harvard.edu.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal