

Key Points
Heparin and danaparoid inhibit VITT Ab-mediated thrombus formation and procoagulant platelet generation.
Heparin and danaparoid interfere with the binding of VITT Abs to PF4, leading to the dissociation of preformed VITT–Ab/PF4 complexes.

Abstract
Life-threatening thrombotic events at unusual sites have been reported after vector-based vaccinations against severe acute respiratory syndrome coronavirus 2. This phenomenon is now termed vaccine-induced immune thrombotic thrombocytopenia (VITT). The pathophysiology of VITT is similar to that of heparin-induced thrombocytopenia (HIT) and is associated with platelet-activating antibodies (Abs) against platelet factor 4 (PF4). Therefore, current guidelines suggest nonheparin anticoagulants to treat VITT patients. In this study, we investigated the interactions of heparin, danaparoid, fondaparinux, and argatroban with VITT–Ab/PF4 complexes using an ex vivo model for thrombus formation as well as in vitro assays to analyze Ab binding and platelet activation. We found that immunoglobulin Gs (IgGs) from VITT patients induce increased adherent platelets/thrombus formation in comparison with IgGs from healthy controls. In this ex vivo flow-based model, the procoagulant activity of VITT IgGs was effectively inhibited with danaparoid and argatroban but also by heparin. Interestingly, heparin and danaparoid not only inhibited IgG binding to PF4 but were also able to effectively dissociate the preformed PF4/IgG complexes. Fondaparinux reduced the in vitro generation of procoagulant platelets and thrombus formation; however, it did not affect platelet aggregation. In contrast, argatroban showed no effect on procoagulant platelets and aggregation but significantly inhibited VITT-mediated thrombus formation. Taken together, our data indicate that negatively charged anticoagulants can disrupt VITT–Ab/PF4 interactions, which might serve as an approach to reduce Ab-mediated complications in VITT. Our results should be confirmed, however, in a clinical setting before a recommendation regarding the selection of anticoagulants in VITT patients could be made.
Introduction
The ongoing COVID-19 pandemic has led to a significant amount of loss to human lives during the last 24 months.1,2 However, a major breakthrough has been achieved in tackling the pandemic by rapid development, rollout, and administration of many vaccines against severe acute respiratory syndrome coronavirus 2.1,2 Although the vaccines are largely effective and considered safe from adverse events, with an increasing number of vaccinations across the world, there have been reports of serious illnesses after vaccine administration. Some of the most significant adverse events are reported in people vaccinated with the ChAdOx1 nCoV-19 vaccine (Vaxzevria: University of Oxford/AstraZeneca) and collectively termed vaccine-induced immune thrombotic thrombocytopenia (VITT).3-6 Patients with VITT develop severe thrombocytopenia and thrombosis 1 to 4 weeks after vaccination, such as cerebral venous sinus thrombosis or splanchnic vein thrombosis (SVT), pulmonary embolism (PE), and deep vein thrombosis (DVT), with a mortality rate of 30%.7 Serological findings in VITT patients resemble clinical manifestation of spontaneous heparin-induced thrombocytopenia (HIT), which is an immune-mediated thrombotic and thrombocytopenic condition developing even without any previous heparin exposure. Similar to HIT, antibodies (Abs) directed against platelet factor 4 (PF4) are detected in the sera of patients with VITT.6-8 These Abs bind to PF4 and form immune complexes, which in turn activate platelets through Fc γ receptor IIA (FcγRIIA).6,9
A number of guidelines have been issued regarding the diagnosis and treatment of VITT.10,11 The recommendations are mainly based on the experience with HIT due to the similarities in the pathophysiology of HIT and VITT. To prevent any potential thrombotic incidences because of binding with immunoglobulin G (IgG)–PF4 immune complexes, VITT guidelines recommend avoidance of anticoagulation with heparin and rather to use nonheparin anticoagulants such as dabigatran, fondaparinux, and argatroban.10,11 Although growing evidence suggests that the use of nonheparin anticoagulants is beneficial for VITT patients, their use in thrombocytopenic patients, especially those with increased risk for occlusive cerebral bleeding, remains a clinical challenge in the absence of an efficient antidote.
To our best knowledge, the effect of heparin and nonheparin anticoagulants on the interaction between VITT Abs and PF4, platelet activation, and thrombus formation has not been addressed. Awaiting in vivo models for VITT, ex vivo models could not only improve our understanding of the pathomechanistic events of the disease but also evaluate therapeutic approaches to prevent thrombosis progression in affected patients. In this study, we systemically analyzed the interactions of heparin, danaparoid, fondaparinux, and argatroban with VITT–IgG/PF4 complexes using a microfluidic system as well as several functional and immunoassays to investigate the ability of these anticoagulants to interfere with Ab binding as well as thrombus formation. Since IV immune globulin (IVIG) is used to treat VITT patients, it was also included in some experimental settings to analyze any synergistic effects.
Methods
Patient cohort
Patients with suspected VITT were referred to our hospital after vaccination with ChAdOx1 nCoV-19 (AstraZeneca, London, United Kingdom). Blood samples obtained from patients with confirmed VITT were included in this study. Laboratory and clinical data were reviewed by 3 physicians (G.U., K.A., and T.B.). The serological investigation included blood count, D-Dimer, PF4/Hep enzyme immunoassay (EIA), and heparin-induced platelet activation assay (HIPA). The diagnosis of VITT was considered confirmed if IgG Abs against PF4 were detected in EIA, and the modified HIPA revealed a positive reaction within 30 minutes in the presence of PF4 with ≥2 out of 4 platelet donors.
Investigations of VITT Ab-mediated thrombus formation
To assess the impact of VITT IgGs, a flow-based ex vivo model for platelet adhesion/thrombus formation in whole blood was established utilizing citrated blood which was recalcified and hirudin. A microfluidic system (BioFlux 200; Fluxion Biosciences, Alameda, CA) was used according to the recommendations of the International Society on Thrombosis and Haemostasis standardization committee for biorheology.12 Briefly, microfluidic channels were coated with collagen (100 µg/mL, Collagen Horn; Takeda, Linz, Austria) overnight at 4°C and blocked with 2.5% of human serum albumin (Kedrion, Barga, Italy).
Whole blood samples of healthy individuals of blood group O were collected and allowed to rest for 30 minutes at room temperature (RT). After splitting the whole blood into aliquots of 200 µL, platelet-rich plasma (PRP) was prepared via centrifugation (20 minutes, 120 g, at RT, no break). IgG fractions were isolated from sera of VITT patients as described in the supplemental Methods. Afterward, 45 µL of the supernatant PRP was gently separated and incubated with 5 µL of IgG fraction from VITT or healthy control sera and incubated for 90 minutes at RT under rotating conditions. Platelets were fluorescently labeled by adding Calcein-FITC (4 µM; Thermo Fisher Scientific, Waltham, MA) 15 minutes before the end of the incubation period. Subsequently, PRP was gently added back to reconstitute whole blood samples. Where indicated, platelets were incubated with anticoagulants: heparin (unfractionated heparin, 0.1 and 0.5 U/mL), danaparoid (0.8 U/mL), fondaparinux (1 and 8 µg/mL), and argatroban (0.8 and 8 µg/mL).
To assess the impact of IVIG on thrombus formation, microfluidic channels were prepared as described above, and where indicated, IgG-treated PRP was coincubated with IVIG in the presence or absence of anticoagulants. Finally, reconstituted whole blood samples were perfused at a shear rate of 250 s−1 (10 dyne) for a maximum of 15 minutes. Immunofluorescence images were acquired from randomly selected microscopic fields (Olympus IX73, Olympus GmbH, or Zeiss Axio Observer, Karl Zeiss, Germany). Thrombus formation was determined by measuring surface area coverage (SAC) by adherent platelets on collagen. Adherent platelets were quantified as the percent (%SAC) of 5 images via ImageJ (National Institutes of Health, Bethesda, MD) as previously described.13
Assessment of Ab-mediated procoagulant platelet generation and platelet aggregation
The impact of anticoagulants on the ability of VITT sera to induce procoagulant phenotype and platelet aggregation was assessed using flow cytometry and the HIPA assay, respectively, as described previously.6,13 For more details, see supplemental Methods.
Interaction between VITT Abs and anticoagulants in immunoassays
Patient sera were tested for anti-PF4 Abs using an in-house PF4 enzyme-linked immunosorbent assay (PF4–EIA). Detailed information is available in the supplemental Materials. In brief, microtitre plates were coated overnight with PF4 (25 μg/mL) before blocking with 3% bovine serum albumin. After subsequent washing, plates were incubated with patient sera (1:50 diluted, 1 hour, RT), followed by further washing and incubation with 1:1000 diluted peroxidase-conjugated anti-human IgG (RT, 1 hour). Substrate tetramethylbenzidine was added, and the reaction was stopped with H2SO4. Absorbance was measured at 450 nm with a microplate reader.
Inhibition assay
The ability of anticoagulants to inhibit VITT–IgG binding to PF4 was analyzed by adding heparin, danaparoid, fondaparinux, and argatroban to the patient serum (illustration in Figure 3A) before the incubation with PF4-coated plates as previously described.14 Anticoagulants were used at a concentration of 0.07, 0.13, 0.26, 0.52, 1.04, 2.08, 4.17, 8.33, 16.67, and 33.33 U/mL or μg/mL if not indicated otherwise. Vehicle controls were used in the same dilution series as a control. IgG Ab binding was determined as described above. Test results were normalized to buffer.
Dissociation assay
IgG/PF4 complex dissociation was determined with a slight modification in the PF4–EIA assay (illustration in Figure 3B). VITT–IgG-containing sera were first incubated on the PF4 plates for 1 hour to facilitate the complex formation. This step was followed by incubation of serially diluted anticoagulants (1 hour, RT), and remaining bound IgG Abs were detected as described above. Test results were normalized to buffer.
Assessment of IgG binding to platelet surface
IgG binding to platelets after incubation with sera from VITT patients was assessed using flow cytometry (illustration in Figure 3C-D) in the presence or absence of exogenous PF4. In brief, cell suspensions were preincubated with PF4 (25 µg/mL) before incubation of anticoagulants and patient sera for 1.5 hours at RT with gentle rotation. FITC-conjugated mouse anti-human IgG (BD Biosciences) was added and directly analyzed by flow cytometry. IgG binding was expressed as a fold increase normalized to healthy controls.
Binding kinetics of Abs using biolayer interferometry (BLI)
Purified PF4 (ChromaTec, Greifswald, Germany) was biotinylated with Sulfo-NHS-LC-LC-Biotin (Thermo Fisher Scientific, Waltham, MA) in 5 molar excess at ambient temperature for 30 minutes. Excess biotin was removed by size exclusion chromatography using Zeba Spin Desalting Columns 7K MWCO 0.5 mL (Thermo Fisher Scientific) according to the manufacturer’s protocol. Analysis of binding kinetics of PF4-specific Abs in heat-inactivated serum samples or purified IgG fractions was performed using the Octet RED96e system (Sartorius, Goettingen, Germany) as per the manufacturer’s recommendations. Data were analyzed using the Octet Data Analysis HT 12.0 software applying the 2:1 heterogeneous ligand-binding model. For quantification, the averages of binding between 475 s and 480 s of the association step and 460 s and 462 s of the dissociation step was used to calculate the percent Residual Binding T560s. The binding profile response of each sample is illustrated as the mean wavelength shift in nm. For details, see supplemental Material.
Ethics statement
The study was conducted in accordance with the declaration of Helsinki. The study protocol was approved by the Institutional Review Board of the University of Tuebingen (236/2021BO2, 224/2021BO2).
Statistical analyses
The statistical analysis was performed using GraphPad Prism, Version 8.0 (GraphPad, La Jolla, CA), and violin plots were generated. Comparison between groups was performed by paired or samples t test. P values <.05 were considered statistically significant. Data in the text are presented as mean ± SEM or n (%).
Results
Patient characteristics
In this study, we used leftover serum samples from 8 patients (4 female, 4 male; median age, 38) with suspected thrombotic complications after the first vaccination with ChAdOx1 nCoV-19. The mean duration between vaccination and first symptoms was 9 days (range, 5-16 days). All patients had severe thrombocytopenia (mean platelet count: 41 × 109/L; range, 8-60) and increased d-dimer (mean, 29 µg/mL; range, 9-54). At admission, thrombotic events were observed in all patients, including 6 SVTs and 2 DVT/PE (for more details, seeTable 1).
Patient characteristics
| Patient # | Sex | First symptoms after vaccination (d) | Thrombosis | Platelet count | D-Dimer | PF4–heparin–EIA (OD) | Platelet activation with PF4 (modified HIPA) |
|---|---|---|---|---|---|---|---|
| 1 | M | 16 | SVT/DVT/PE | 60 | 12 | 3.4 | + |
| 2 | M | 8 | SVT | 22 | n.a | 3.1 | + |
| 3 | F | 6 | SVT | 40 | n.a | 2.2 | + |
| 4 | F | 9 | SVT | 27 | 54 | 3.2 | + |
| 5 | F | 7 | SVT | 53 | 35 | 3.1 | + |
| 6 | F | 5 | SVT | 56 | 9 | 2.1 | + |
| 7 | M | 9 | DVT/PE | 8 | >35 | 3.2 | + |
| 8 | M | 12 | DVT/PE | 60 | n.a. | 3.0 | + |
| Patient # | Sex | First symptoms after vaccination (d) | Thrombosis | Platelet count | D-Dimer | PF4–heparin–EIA (OD) | Platelet activation with PF4 (modified HIPA) |
|---|---|---|---|---|---|---|---|
| 1 | M | 16 | SVT/DVT/PE | 60 | 12 | 3.4 | + |
| 2 | M | 8 | SVT | 22 | n.a | 3.1 | + |
| 3 | F | 6 | SVT | 40 | n.a | 2.2 | + |
| 4 | F | 9 | SVT | 27 | 54 | 3.2 | + |
| 5 | F | 7 | SVT | 53 | 35 | 3.1 | + |
| 6 | F | 5 | SVT | 56 | 9 | 2.1 | + |
| 7 | M | 9 | DVT/PE | 8 | >35 | 3.2 | + |
| 8 | M | 12 | DVT/PE | 60 | n.a. | 3.0 | + |
F, female; M, male; OD, optical density.
IgG from VITT patients induce ex vivo thrombus formation
We investigated the ability of VITT–IgG fractions to induce adherent platelets/thrombi using a flow-based microfluidic system. To mimic venous flow conditions, we selected a shear stress of 250 s−1 (10 dyne). Our analyses were focused on the role of IgG/platelet interplay. Therefore, platelets from healthy individuals were preincubated with IgGs from VITT patients or healthy controls, added back to autologous whole blood samples, and finally perfused through collagen-covered microfluidic channels. As shown in Figure 1, IgGs from VITT patients caused increased platelet adhesion. Overall, significantly higher SAC by platelets was observed in the presence of VITT IgGs in comparison with IgGs from healthy controls (mean percent, SAC ± SEM: 11.59 ± 0.57 vs 1.99 ± 0.34, respectively; P < .001) (Figure 1A). Similar findings were also observed when IgG was added directly to whole blood samples (supplemental Figure 1).
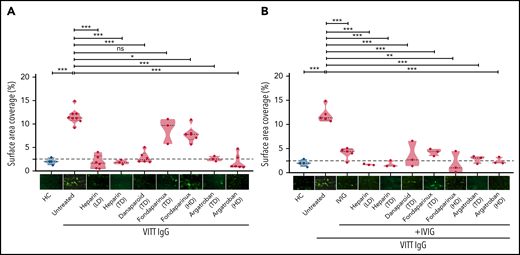
Impact of anticoagulants on VITT–IgG-mediated platelet adhesion/thrombus formation. (A) Platelets from healthy individuals were incubated with IgGs from healthy controls (HCs) or VITT patients before calcein staining and reconstitution with autologous whole blood and perfusion through BioFlux microfluidic channels. IgG concentrations were measured using nanodrop (mean IgG concentration, 15.10 ± 2.14 mg/mL). Where indicated, samples were treated with different concentrations of anticoagulants: low-dose heparin (0.1 U/mL), therapeutic-dose heparin (0.5 U/mL), TD danaparoid (0.8 U/mL), TD and HD fondaparinux (1 and 8 µg/mL), and TD and HD of argatroban (0.8 and 8 µg/mL). (B) IVIG was added alone or in combination with anticoagulants. Each dot represents 1 VITT patient IgG. Images were acquired at 20× magnification using an inverted Olympus IX73 or Zeiss Axio Observer 7 fluorescence microscope. Thrombus formation was determined as the %SAC by adherent platelets using ImageJ. Images were processed identically using automated or manually adjusted threshold settings and exclusion of image artifacts. Violin plots showing the distribution of the values were generated using Graphpad Prism 8. This figure represents values from the same experimental setup. The figure is split into 2 different panels (1A and 1B) for easier interpretation of the results. Therefore values and representative images in the HCs group and some in untreated VITT IgGs are presented in both panels. HD, high dose; LD, low dose; ns, not significant; TD, therapeutic dose. *P < .05, **P < .01, and ***P < .001.
Impact of anticoagulants on VITT–IgG-mediated platelet adhesion/thrombus formation. (A) Platelets from healthy individuals were incubated with IgGs from healthy controls (HCs) or VITT patients before calcein staining and reconstitution with autologous whole blood and perfusion through BioFlux microfluidic channels. IgG concentrations were measured using nanodrop (mean IgG concentration, 15.10 ± 2.14 mg/mL). Where indicated, samples were treated with different concentrations of anticoagulants: low-dose heparin (0.1 U/mL), therapeutic-dose heparin (0.5 U/mL), TD danaparoid (0.8 U/mL), TD and HD fondaparinux (1 and 8 µg/mL), and TD and HD of argatroban (0.8 and 8 µg/mL). (B) IVIG was added alone or in combination with anticoagulants. Each dot represents 1 VITT patient IgG. Images were acquired at 20× magnification using an inverted Olympus IX73 or Zeiss Axio Observer 7 fluorescence microscope. Thrombus formation was determined as the %SAC by adherent platelets using ImageJ. Images were processed identically using automated or manually adjusted threshold settings and exclusion of image artifacts. Violin plots showing the distribution of the values were generated using Graphpad Prism 8. This figure represents values from the same experimental setup. The figure is split into 2 different panels (1A and 1B) for easier interpretation of the results. Therefore values and representative images in the HCs group and some in untreated VITT IgGs are presented in both panels. HD, high dose; LD, low dose; ns, not significant; TD, therapeutic dose. *P < .05, **P < .01, and ***P < .001.
Inhibition of ex vivo adhesion/thrombus formation by anticoagulants
We further investigated the impact of anticoagulants on SAC by VITT IgGs in our ex vivo model. In comparison with untreated controls, SAC was significantly reduced when platelets were preincubated with 0.1 and 0.5 U/mL heparin (mean percent, SAC ± SEM: 11.59 ± 0.57 vs 1.85 ± 0.56 and 1.86 ± 0.28, respectively; both P < .001).
Furthermore, a strong reduction in SAC was observed with danaparoid, as well as argatroban at 0.8 and 8 µg/mL (mean percent, SAC ± SEM: 2.82 ± 0.50; P < .001; 2.56 ± 0.31 and 1.84 ± 0.66; P < .001, respectively) (Figure 1A). Fondaparinux slightly but significantly reduced SAC at 8 but not 1 µg/mL (mean percent, SAC ± SEM: 7.84 ± 0.701; P = .01; and 8.82 ± 1.6) (Figure 1A). Furthermore, the administration of IVIG resulted in inhibition of SAC by VITT IgGs in our microfluidic system in the presence of all anticoagulants including fondaparinux (mean percent, SAC ± SEM: untreated 12.04 ± 0.75; vs IVIG alone: 4.07 ± 0.51; P < .001; and IVIG in combination with: heparin 0.1 and 0.5 U/mL, 1.71 ± 0.08, and 1.77 ± 0.41; P < .001; danaparoid 3.55 ± 1.57; P < .001; fondaparinux 1 and 8 µg/mL, 4.25 ± 0.43 and 1.89 ± 1.32; P < .001; and argatroban 0.8 and 8 µg/mL, 2.79 ± 0.48 and 2.44 ± 0.41; P < .001) (Figure 1B).
When IgGs from HIT patients were tested, heparin at low concentration had no inhibitory effect on HIT–IgG-induced thrombus formation, and rather, increased SAC was observed (supplemental Figure 2).
The effect of anticoagulants on VITT sera-mediated platelet activation
To explore the mechanisms underlying the inhibition of thrombus formation, we thought to investigate the impact of anticoagulants on the generation of procoagulant phenotype and platelet aggregation.
VITT sera were incubated with washed platelets in the presence of buffer, varying concentrations of heparin, danaparoid, fondaparinux, and argatroban. Flow cytometry analyses revealed that sera from VITT patients induced significantly higher amount of CD62p/phosphatidylserine (PS)-positive procoagulant platelets in comparison with healthy controls (mean percent, CD62p/PS-positive platelets 40.82 ± 7.02 vs 0.50 ± 0.16, respectively; P ≤ .001) (Figure 2A). The generation of procoagulant platelets was reduced by 0.1 U/mL heparin (mean percent, CD62p/PS-positive platelets: 8.04 ± 3.16; P = .03) (Figure 2A). Procoagulant platelet formation was completely inhibited with further increasing concentrations of heparin to 0.5, 1, 5, and 10 U/mL (mean percent CD62p/PS-positive platelets: 12.59 ± 4.77; 4.41 ± 0.86; 4.71 ± 1.07; and 3.96 ± 1.51, respectively; all P < .01) (Figure 2A). VITT sera-mediated procoagulant platelet formation was also significantly inhibited after incubation with low- and high-dose danaparoid (mean percent CD62p/PS-positive platelets: 3.03 ± 0.69; P = .01; and 2.14 ± 0.98; P = .03, respectively) (Figure 2A). Addition of 8 µg/mL fondaparinux resulted in notable reduction but not complete inhibition of procoagulant platelets (24.36 ± 5.18) but not 0.8 µg/mL (59.85 ± 11.02). On the other hand, treatment of platelets with argatroban had no effect on procoagulant platelets for both the doses tested (mean percent CD62p/PS-positive platelets: 60.67 ± 12.42 and 38.65 ± 7.73; P > .05).
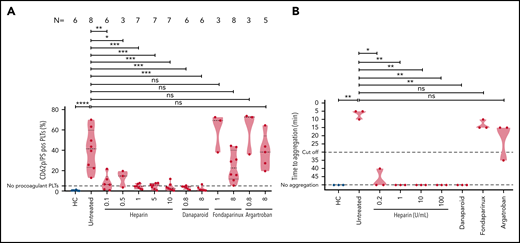
VITT–IgG-mediated procoagulant platelets and platelet activation in the presence of anticoagulants. (A) Procoagulant platelets (CD62P/PS-positive cells) were analyzed with flow cytometry after incubation of healthy platelets with VITT sera or sera from HCs in different settings and staining with annexin V–FITC and CD62p–APC Ab. Where indicated, platelets were coincubated with different concentrations of anticoagulants: heparin (unfractionated heparin, 0.1, 0.5, 1, 5, and 10 U/mL), danaparoid (0.8 and 8 U/mL), fondaparinux (1 and 8 µg/mL), and argatroban (0.8 and 8 µg/mL). The percent of CD62p/PS-double–positive platelets is shown as violin plots. Each dot represents sera from an individual VITT patient, and the number of sera tested is reported in each graphic. Violin plots showing the distribution of the values were generated using Graphpad Prism 8. ns, not significant. *P < .05, **P < .01, ***P < .001, and ***P < .0001. (B) Results of the platelet activation assay (HIPA). Each dot represents the median value of 2 different healthy platelet donors. VITT patients showed strong platelet activation with buffer alone in comparison with HCs, which was inhibited with varying concentrations of heparin. Where indicated, platelets were treated with different concentrations of anticoagulants: heparin (unfractionated heparin, 0.2, 1, 10, and 100 U/mL), danaparoid (0.8 U/mL), fondaparinux (8 µg/mL), and argatroban (8 µg/mL). Data are presented as time to aggregate. Violin plots showing the distribution of the values were generated using Graphpad Prism 8. *P < .05 and **P < .01.
VITT–IgG-mediated procoagulant platelets and platelet activation in the presence of anticoagulants. (A) Procoagulant platelets (CD62P/PS-positive cells) were analyzed with flow cytometry after incubation of healthy platelets with VITT sera or sera from HCs in different settings and staining with annexin V–FITC and CD62p–APC Ab. Where indicated, platelets were coincubated with different concentrations of anticoagulants: heparin (unfractionated heparin, 0.1, 0.5, 1, 5, and 10 U/mL), danaparoid (0.8 and 8 U/mL), fondaparinux (1 and 8 µg/mL), and argatroban (0.8 and 8 µg/mL). The percent of CD62p/PS-double–positive platelets is shown as violin plots. Each dot represents sera from an individual VITT patient, and the number of sera tested is reported in each graphic. Violin plots showing the distribution of the values were generated using Graphpad Prism 8. ns, not significant. *P < .05, **P < .01, ***P < .001, and ***P < .0001. (B) Results of the platelet activation assay (HIPA). Each dot represents the median value of 2 different healthy platelet donors. VITT patients showed strong platelet activation with buffer alone in comparison with HCs, which was inhibited with varying concentrations of heparin. Where indicated, platelets were treated with different concentrations of anticoagulants: heparin (unfractionated heparin, 0.2, 1, 10, and 100 U/mL), danaparoid (0.8 U/mL), fondaparinux (8 µg/mL), and argatroban (8 µg/mL). Data are presented as time to aggregate. Violin plots showing the distribution of the values were generated using Graphpad Prism 8. *P < .05 and **P < .01.
To investigate the impact of anticoagulants on platelet aggregation, the HIPA assay was used. Sera were incubated with washed platelets in the presence of buffer (untreated), heparin in different concentrations (0.2, 1, 10, and 100 U/mL), danaparoid, fondaparinux, and argatroban. We observed strong platelet activation in the presence of buffer in all VITT patients (median time to platelet aggregation: 5 minutes, range 5-10 minutes) (Figure 2B). However, the reaction was weaker in the presence of 0.2 U/mL heparin (median time to aggregate: 40-50 minutes ⟶ no aggregation). All reactions were further inhibited by higher doses of heparin (1, 10, and 100 U/mL; P value = .001). Danaparoid also inhibited platelet aggregation but not fondaparinux and argatroban.
Heparin and danaparoid interfere with VITT–IgG binding to PF4
Next, we intended to understand how anticoagulants interfere with (Ab) function in VITT. To decipher this, PF4 binding to VITT IgGs without any anticoagulants was arbitrarily set at 100% (baseline). Heparin showed a strong inhibition of PF4 binding to VITT IgGs, and almost 50% inhibition was observed even at a lower concentration of 0.07 U/mL (Figure 3A). Danaparoid also showed significant inhibition of VITT–IgG binding to PF4, and 50% inhibition was observed at 8.33 U/mL (Figure 3A). However, no differences were observed when fondaparinux or argatroban were added with patient sera in EIA.
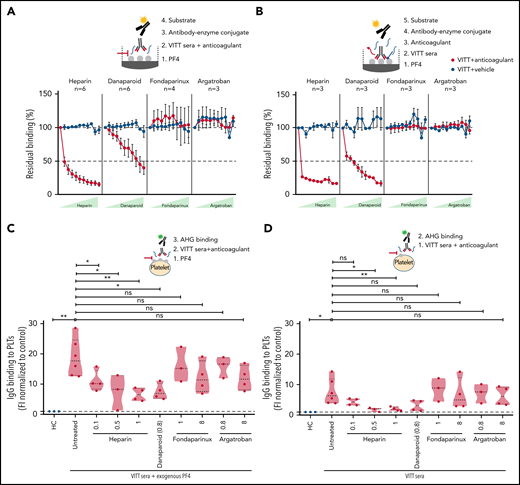
Heparin and danaparoid, but not fondaparinux or argatroban, interfere with anti-PF4 Ab binding to PF4. (A) Sera from VITT patients with anti-PF4–IgG were tested in an in-house PF4 enzyme-linked immunoassay (ELISA). Sera were incubated on PF4 (25 µg/mL)-coated microplates. Where indicated, increasing concentrations (0.07, 0.13, 0.26, 0.52, 1.04, 2.08, 4.17, 8.33, 16.67, and 33.33) of heparin and danaparoid (both U/mL) or fondaparinux and argatroban (both µg/mL) were added as described previously.14 Heparin and danaparoid equally reduced the Ab binding to PF4. However, fondaparinux and argatroban had no effect. Each dot represents the mean value of VITT patient sera tested, and the numbers are reported in each graphic for each anticoagulant (n = 3-6). The signal of anti-PF4 Ab binding without anticoagulants was set to 100%. Data are shown as mean ± SEM. (B) Sera from VITT patients with anti-PF4–IgG were tested in an in-house PF4 ELISA to analyze the VITT–IgG/PF4 complex dissociation by anticoagulants. Patient sera were first incubated in PF4-coated microplates for 1 hour to facilitate VITT–IgG/PF4 binding and complex formation. Increasing amounts (0.07-33.33 U or µg/mL) of anticoagulants were added to VITT–IgG/PF4 complex containing microplates. While heparin and danaparoid potently disrupted the IgGs bound to PF4, no disruption was observed with fondaparinux and argatroban. Each dot represents the mean value of VITT patient sera tested, and the numbers are reported in each graphic for each anticoagulant (n = 3). The signal of anti-PF4 Ab binding without anticoagulants was set to 100%. Data are shown as mean ± SEM. (C) IgG binding to healthy washed platelets after incubation with sera from VITT patients in the presence of PF4 (25 µg/mL) or without PF4 (D) was assessed by flow cytometry and expressed as fold increase normalized to HCs. VITT patients showed significantly higher binding at the baseline in comparison with HCs, which was significantly inhibited by heparin and danaparoid. Fondaparinux and argatroban also showed slight displacement of IgG binding. Violin plots showing the distribution of the values were generated using Graphpad Prism 8. ns, not significant. *P < .05 and **P < .01.
Heparin and danaparoid, but not fondaparinux or argatroban, interfere with anti-PF4 Ab binding to PF4. (A) Sera from VITT patients with anti-PF4–IgG were tested in an in-house PF4 enzyme-linked immunoassay (ELISA). Sera were incubated on PF4 (25 µg/mL)-coated microplates. Where indicated, increasing concentrations (0.07, 0.13, 0.26, 0.52, 1.04, 2.08, 4.17, 8.33, 16.67, and 33.33) of heparin and danaparoid (both U/mL) or fondaparinux and argatroban (both µg/mL) were added as described previously.14 Heparin and danaparoid equally reduced the Ab binding to PF4. However, fondaparinux and argatroban had no effect. Each dot represents the mean value of VITT patient sera tested, and the numbers are reported in each graphic for each anticoagulant (n = 3-6). The signal of anti-PF4 Ab binding without anticoagulants was set to 100%. Data are shown as mean ± SEM. (B) Sera from VITT patients with anti-PF4–IgG were tested in an in-house PF4 ELISA to analyze the VITT–IgG/PF4 complex dissociation by anticoagulants. Patient sera were first incubated in PF4-coated microplates for 1 hour to facilitate VITT–IgG/PF4 binding and complex formation. Increasing amounts (0.07-33.33 U or µg/mL) of anticoagulants were added to VITT–IgG/PF4 complex containing microplates. While heparin and danaparoid potently disrupted the IgGs bound to PF4, no disruption was observed with fondaparinux and argatroban. Each dot represents the mean value of VITT patient sera tested, and the numbers are reported in each graphic for each anticoagulant (n = 3). The signal of anti-PF4 Ab binding without anticoagulants was set to 100%. Data are shown as mean ± SEM. (C) IgG binding to healthy washed platelets after incubation with sera from VITT patients in the presence of PF4 (25 µg/mL) or without PF4 (D) was assessed by flow cytometry and expressed as fold increase normalized to HCs. VITT patients showed significantly higher binding at the baseline in comparison with HCs, which was significantly inhibited by heparin and danaparoid. Fondaparinux and argatroban also showed slight displacement of IgG binding. Violin plots showing the distribution of the values were generated using Graphpad Prism 8. ns, not significant. *P < .05 and **P < .01.
Heparin and danaparoid dissociate VITT IgGs bound to PF4
Finally, we analyzed the ability of anticoagulants to dissociate VITT IgGs, which are already bound to PF4. VITT Abs were allowed to bind on PF4 coated microtiter plates before anticoagulants were added. Heparin effectively dissociated the complex, and a 75% reduction in IgG binding was observed at a low concentration (0.07 U/mL). Danaparoid also significantly displaced the VITT–Ab/PF4 complex, and 50% dissociation was observed starting from 0.26 U/mL. In contrast, fondaparinux and argatroban had no effect on bound VITT IgGs at all concentrations tested (Figure 3B). Similarly, when VITT sera were incubated with washed platelets from healthy donors in the presence or absence of exogenous PF4, heparin and danaparoid resulted in higher inhibition of IgG binding compared with fondaparinux and argatroban (Figure 3C-D).
Binding kinetics of VITT Abs
To further investigate the influence of the anticoagulants, particularly on the dissociation of VITT-specific IgGs, we performed BLI analysis. Therefore, we immobilized biotinylated PF4 on streptavidin biosensors and measured the association of IgGs from serum samples or purified IgG fractions, followed by determining their dissociation in the presence of the various anticoagulants (Figure 4). While binding of PF4-specific Abs to the biosensor was observed for all VITT samples (n = 5) tested (Figure 4; supplemental Figure 7A-F), we found remarkable changes in their dissociation after the addition of the various anticoagulants. To highlight differences between anticoagulants, quantification of residual binding at a defined time point (560 s) of dissociation was calculated. In particular, in the presence of heparin (0.1 U/mL, 1 U/mL), we observed increased dissociation of PF4-binding Abs, resulting in a significant difference in residual binding compared with no anticoagulant (Figure 4C; supplemental Table 1). A slight but nonsignificant increase in dissociation was measured for danaparoid, whereas the addition of fondaparinux or argatroban did not appear to affect the dissociation rate. Notably, serum samples and purified IgG fractions showed similar binding kinetics (supplemental Figure 7A-B), confirming that PF4-specific Ab responses were visualized. In comparison, the investigated HIT samples (n = 5) applied in the same experimental setting showed high variabilities regarding their dissociation in the presence of the different anticoagulants (supplemental Figure 7G).
![Binding kinetics of PF4-specific Abs from VITT samples in the presence of different anticoagulants. Biotinylated PF4 (5 µg/mL) was immobilized on streptavidin biosensor tips, and binding kinetics of serum samples from patients with VITT (n = 5) were analyzed using BLI. For dissociation, different anticoagulants (heparin, 0.1 U/mL [pink]; heparin, 1 U/mL [dark red]; danaparoid, 0.8 U/mL; fondaparinux 8 µg/mL; argatroban 8 µg/mL) or Octet buffer were applied. Curves and data points represent the mean of technical duplicates (n = 2). (A) Schematic illustration of the experimental setup. (B) Representative binding responses of PF4-specific Abs from a VITT patient serum (VITT1). (C) Quantification of residual binding in percent at time point 560 secondsof the dissociation (percent Residual Binding T560s) of VITT serum samples.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/23/10.1182_blood.2021013839/3/m_bloodbld2021013839f4.png?Expires=1769134647&Signature=HDcjokt8YDNZ0xOT3ybV5P9allPaTR7GfWSVZHl6LloToLPlNkdZJ60meK7~kRnCxwIVYZkty2YTHxSYANxI2PFaaPptfY6q89XzOvMcW0vH8c-on28OvY-nw3XuTQYEz7Ju6556paQLKUa0qJ7Q3zsBSYmdjviMf7t4UuzayN2KXqWTDBhxm3IicxbPlxG8X2AaEX5FGT3xzBSEMtfOEOAHdmBaOvRind-0YHJbuiULnJ7GAfkY59sXbO80lLoXYwO7d7QJTdtzaRwrjKzV1D86U7IzZaYznaKHdAEMEdutr0bZM8AQDb3FygP7-hMwKQYt3kTuHEqLA0dnqNNtyg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Binding kinetics of PF4-specific Abs from VITT samples in the presence of different anticoagulants. Biotinylated PF4 (5 µg/mL) was immobilized on streptavidin biosensor tips, and binding kinetics of serum samples from patients with VITT (n = 5) were analyzed using BLI. For dissociation, different anticoagulants (heparin, 0.1 U/mL [pink]; heparin, 1 U/mL [dark red]; danaparoid, 0.8 U/mL; fondaparinux 8 µg/mL; argatroban 8 µg/mL) or Octet buffer were applied. Curves and data points represent the mean of technical duplicates (n = 2). (A) Schematic illustration of the experimental setup. (B) Representative binding responses of PF4-specific Abs from a VITT patient serum (VITT1). (C) Quantification of residual binding in percent at time point 560 secondsof the dissociation (percent Residual Binding T560s) of VITT serum samples.
Binding kinetics of PF4-specific Abs from VITT samples in the presence of different anticoagulants. Biotinylated PF4 (5 µg/mL) was immobilized on streptavidin biosensor tips, and binding kinetics of serum samples from patients with VITT (n = 5) were analyzed using BLI. For dissociation, different anticoagulants (heparin, 0.1 U/mL [pink]; heparin, 1 U/mL [dark red]; danaparoid, 0.8 U/mL; fondaparinux 8 µg/mL; argatroban 8 µg/mL) or Octet buffer were applied. Curves and data points represent the mean of technical duplicates (n = 2). (A) Schematic illustration of the experimental setup. (B) Representative binding responses of PF4-specific Abs from a VITT patient serum (VITT1). (C) Quantification of residual binding in percent at time point 560 secondsof the dissociation (percent Residual Binding T560s) of VITT serum samples.
Discussion
In this study, we analyzed the interactions of heparin, danaparoid, fondaparinux, and argatroban with VITT–IgG/PF4 complexes. We observed that IgGs from VITT patients are able to induce strong thrombus formation in the absence of anticoagulants. The negatively charged anticoagulants, danaparoid and heparin, were found to be able to interfere with the binding of VITT IgGs to PF4, leading to their dissociation, which in turn inhibits the generation of procoagulant platelets and the subsequent thrombus formation. Moreover, the inhibitory effect of anticoagulants on thrombus formation was more pronounced when IVIG was coadministered. IVIG has been previously shown to inhibit FcγRIIA-mediated activation of platelets by VITT Abs and is also recommended to use in therapy.8,15
Heparin is usually preferred as one of the most widely used anticoagulants for the prevention and treatment of thrombotic events due to its rapid action, low cost, and availability of antidote.16,17 The use of heparin is, however, discouraged in VITT patients because of clinical similarities observed between VITT and HIT. In contrast to HIT, where low concentrations of heparin enhance platelet responses,18 Ab-mediated platelet activation was inhibited in the case of VITT in our study. Recently, successful treatment with heparin was reported in 1 patient with VITT19 and in a case series involving 3 female VITT patients who developed intracranial venous sinus thrombosis after their first vaccination with ChAdOx1 nCov-19.20 In a recent study, Smith and colleagues also observed that heparin blocks VITT serum-mediated platelet aggregation.9 Recently, VITT Abs were shown to recognize epitopes within the heparin-binding site on PF4.21 In fact, VITT Abs were shown to mimic the effect of heparin by causing PF4 tetramers clustering.20 In our studies, we found that increasing heparin can disrupt the binding of VITT Abs with PF4 (Figure 3A-B). Using BLI, a rapid dissociation of the IgG–PF4 complexes was observed when heparin was added. Other anticoagulants were less effective in disrupting the prebound complexes. Due to the polyclonal nature of the Ab pool within serum samples, a calculation of a defined affinity was not possible. However, as BLI allows measurement of the association and dissociation of target-specific Abs, we believe that a clear conclusion about the influence of heparin on the dissociation rate of PF4-specific Abs could be drawn.
It is interesting, but outside the scope of our current work, to investigate whether heparin blocked the VITT epitopes on PF4 or induced conformational changes in PF4 that might have reduced the Ab binding avidity. However, in light of our findings along with other similar recent observations, more studies are warranted to explore the suitability of heparin in VITT patients. Danaparoid is a chemically distinct low-molecular–weight heparinoid devoid of heparin. Danaparoid has different protein-binding properties, making it an excellent candidate for the treatment of HIT and the prevention of postoperative DVT.22,23 In our experimental settings, danaparoid could effectively inhibit VITT sera-mediated platelet responses and thrombus formation, similar to heparin. However, in comparison with heparin, a higher amount of danaparoid was required to inhibit VITT Ab binding to immobilized PF4 in the enzyme-linked immunoassay(Figure 3A-B). Of note, danaparoid inhibited the binding of HIT Abs to PF4 and also disrupted PF4/VITT–IgG-complexes (supplemental Figure 4A-B). Our results are in accordance with the findings of Krauel and colleagues, which showed that danaparoid inhibits the binding of anti-PF4/heparin Abs to PF4/heparin complexes up to 30% at prophylactic doses and 50% at therapeutic doses.24 Our data indicate that, in addition to its anticoagulatory effects, danaparoid can also interfere with Ab-mediated platelet activation and might protect against new thrombotic events in patients with VITT. The prompt onset of action might be another advantage of this anticoagulant during acute VITT due to the ability to disassociate PF4/VITT–IgG complexes.
Another interesting finding in our study was that argatroban showed no direct effect on the formation of procoagulant platelets, platelet activation, and disruption of VITT Ab binding to PF4 but significantly inhibited the ex vivo thrombus formation. Fondaparinux, a selective factor Xa inhibitor, is another nonheparin anticoagulant used in patients with HIT25 and is also recommended in the treatment of VITT.26 Among nonheparin anticoagulants, fondaparinux has been shown to have the lowest thrombotic risk and all-cause-mortality rate in HIT.25 Fondaparinux showed slight inhibition of ex vivo thrombus formation, which was significantly increased when IVIG was coadminstrated. While fondaparinux reduced the formation of procoagulant platelets and thrombus formation, it failed to interfere with the formation of immune complexes from PF4 and VITT or HIT Abs (Figure 3A; supplemental Figure 4A). Similar to our observations, Krauel and colleagues found that fondaparinux at therapeutic concentrations does not inhibit Ab binding to PF4/heparin complexes and platelet activation in serotonin release assay in HIT.23 The differences between these polyanions might be due to different degrees of sulfation. The latter was shown to be responsible for structural changes in PF4, resulting in antiparallel β-sheet content >30% that makes PF4/polyanion complexes, for example, antigenic for the HIT Abs.27-29 While our data provide new insight into the interaction between anticoagulants and VITT–PF4 immune complexes, clinical studies are needed to investigate whether anticoagulants that showed higher inhibition of thrombus formation ex vivo might be more efficient to treat patients with VITT.
Nevertheless, our studies show that heparin and danaparoid interfere with in vitro Ab binding to PF4 and platelets leading to sufficient inhibition of platelet activation and generation of procoagulant phenotype. In the absence of the procoagulant platelet phenotype, no thrombus was observed in our ex vivo model. This finding indicates that Ab-mediated generation of procoagulant platelets is essential for the development of thrombosis in VITT. On the other hand, thrombin inhibition prevented the thrombus formation despite the lack of impact on Ab binding and subsequent platelet activation. These data suggest that thrombus formation in our ex vivo model is dependent on thrombin. In contrast to argatroban, heparin and danaparoid might inhibit VITT-induced thrombosis by interfering with 3 steps: Ab binding, platelet activation/generation of procoagulant phenotype, and factor X activation, while fondaparinux seems to exist mainly on anti-Xa activity.
While our ex vivo microfluidic model might provide preclinical information on the performance of therapeutic approaches to thrombosis, further investigations on the role of complement activation in VITT are needed to understand the disease pathophysiology and better management. In vivo animal models, when available, will be able to show the capacity of the VITT Abs to induce thrombosis and the ability of anticoagulants to block it. In addition, the influence of other cell types (eg, endothelial cells) and other in vivo factors (eg, distribution of the drug within the animal's body and binding of the drug to tissues) can only be addressed in vivo.
Conclusions
Our study suggests that heparin and danaparoid can inhibit thrombus formation in VITT patients in part through the inhibition of the interaction of VITT Abs with PF4 and subsequent platelet activation. The effect of argatroban is independent of the interaction of VITT Abs with PF4. To our best knowledge, this is the first study to investigate systematically the interactions of heparin and nonheparin anticoagulants with VITT–Ab/PF4 complexes. Our results should be confirmed, however, in a clinical setting before a recommendation regarding the selection of anticoagulation in VITT patients could be made.
Acknowledgments
This work was supported by grants from the German Research Foundation and from the Herzstiftung to T.B. (BA5158/4 and TSG-Study), by special funds from the state of Baden-Württemberg for coagulation research, and by the Blood Donation Service of the German Red Cross to T.B.
Authorship
Contribution: A.S. and T.B. designed the study; A.S., F.T., T.R.W., L.P., J.Z., V.F., and K.W. performed the experiments; G.U., K.A., S.N.-H., and T.B. provided clinical data; A.S., F.T., G.U., T.R.W., U.R., K.A., and T.B. analyzed the data, interpreted the results, and wrote the manuscript; and all authors read and approved the manuscript before submission.
Conflict-of-interest disclosure: T.B. has received research funding from CoaChrom Diagnostica GmbH, DFG, Robert Bosch GmbH, Stiftung Transfusionsmedizin und Immunhämatologie e.V.: Ergomed, Surrey, DRK Blutspendedienst, Deutsche Herzstiftung, Ministerium für Wissenschaft, Forschung und Kunst Baden-Wuerttembergm; has received lecture honoraria from Aspen Germany GmbH, Bayer Vital GmbH, Bristol-Myers Squibb GmbH & Co., Doctrina Med AG, Meet The Experts Academy UG, Schoechl medical education GmbH, Mattsee, Stago GmbH, Mitsubishi Tanabe Pharma GmbH, Novo Nordisk Pharma GmbH; has provided consulting services to Terumo; has provided expert witness testimony relating to heparin-induced thrombocytopenia (HIT) and non‐HIT thrombocytopenic and coagulopathic disorders. All of these are outside the current work. The remaining authors declare no competing financial interests.
Correspondence: Tamam Bakchoul, Institute for Clinical and Experimental Transfusion Medicine, Medical Faculty of Tuebingen, Otfried-Müller Str. 4/1, 72076 Tuebingen, Germany; e-mail: tamam.bakchoul@med.uni-tuebingen.de.
Data may be requested for academic collaborations from the corresponding author.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal