

Key Points
Individuals with clonal hematopoiesis are more likely to have COPD and more severe disease.
Mice lacking Tet2 (often mutated in CHIP) in blood cells have accelerated development of emphysema in models of pulmonary inflammation.

Abstract
Chronic obstructive pulmonary disease (COPD) is associated with age and smoking, but other determinants of the disease are incompletely understood. Clonal hematopoiesis of indeterminate potential (CHIP) is a common, age-related state in which somatic mutations in clonal blood populations induce aberrant inflammatory responses. Patients with CHIP have an elevated risk for cardiovascular disease, but the association of CHIP with COPD remains unclear. We analyzed whole-genome sequencing and whole-exome sequencing data to detect CHIP in 48 835 patients, of whom 8444 had moderate to very severe COPD, from four separate cohorts with COPD phenotyping and smoking history. We measured emphysema in murine models in which Tet2 was deleted in hematopoietic cells. In the COPDGene cohort, individuals with CHIP had risks of moderate-to-severe, severe, or very severe COPD that were 1.6 (adjusted 95% confidence interval [CI], 1.1-2.2) and 2.2 (adjusted 95% CI, 1.5-3.2) times greater than those for noncarriers. These findings were consistently observed in three additional cohorts and meta-analyses of all patients. CHIP was also associated with decreased FEV1% predicted in the COPDGene cohort (mean between-group differences, −5.7%; adjusted 95% CI, −8.8% to −2.6%), a finding replicated in additional cohorts. Smoke exposure was associated with a small but significant increased risk of having CHIP (odds ratio, 1.03 per 10 pack-years; 95% CI, 1.01-1.05 per 10 pack-years) in the meta-analysis of all patients. Inactivation of Tet2 in mouse hematopoietic cells exacerbated the development of emphysema and inflammation in models of cigarette smoke exposure. Somatic mutations in blood cells are associated with the development and severity of COPD, independent of age and cumulative smoke exposure.
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic lung disease that affects approximately 15 million adults and is the fourth leading cause of death in the United States.1,2 The pathophysiologic drivers of COPD are complex, including both innate and adaptive immune cells and increased levels of inflammatory cytokines.3-5 Cigarette smoking and age contribute to this aberrant inflammatory pathophysiology and are two of the strongest risk factors for the development and progression of COPD.1,6,7
Clonal hematopoiesis of indeterminate potential (CHIP) is a common, age-related phenomenon in which somatic mutations in hematopoietic stem cells result in clonal outgrowth of a mutant population of blood cells in individuals without a hematologic malignancy.8 CHIP, which is associated with an increased risk of leukemia and ischemic cardiovascular diseases, is characterized by an aberrant inflammatory state.9-12 We therefore hypothesized that CHIP may also be a causal risk factor for the development and/or progression of COPD.
Weak associations between CHIP and COPD have been observed in previous studies, but definitive conclusions were precluded by small sample size, quality of COPD phenotyping and smoking data, and a potential confounding association between CHIP and smoking.13-16 In this study, we sought to definitively assess the association between CHIP and COPD through analyses of whole-genome sequencing and whole-exome sequencing data from several large, extensively phenotyped cohorts of COPD patients and controls. We then used mouse models to determine whether there is a causal relationship between CHIP and emphysema, one of the most common manifestations of COPD.
Methods
Study samples
Spirometry data were available for all patients included in this study. The primary study cohort was COPDGene, the largest COPD study in the Trans-Omics for Precision Medicine (TOPMed) program.17 We also included the following three additional cohorts: (1) TOPMed studies that comprised population- and family-based cohorts and an additional COPD-enriched study18: Atherosclerosis Risk in Communities (ARIC), Cleveland Family Study (CFS), Framingham Heart Study (FHS), Jackson Heart Study (JHS), the Multi-Ethnic Study of Atherosclerosis (MESA) Lung Study, and the Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE); (2) two family-based cohorts: the International COPD Genetics Network (ICGN) study and the Boston Early-Onset COPD study (EOCOPD)19 (ICGN-EOCOPD); and (3) the subset of UK Biobank participants with whole-exome sequencing and spirometry data.20
COPD and COPD severity were defined on the basis of spirometry grades from the Global Initiative for Chronic Obstructive Lung Disease (GOLD), in which GOLD 2 is classified as moderate, GOLD 3 as severe, and GOLD 4 as very severe.21 Age and cigarette smoking history (pack-years) were available for all analyzed patients (Table 1; supplemental Tables 1 and 2; supplemental Figure 1 [available on the Blood Web site]). Further information about these cohorts is included in the supplemental Data.
Cohort characteristics
| Characteristic | Cohort | Total (N = 48 835) | |||
|---|---|---|---|---|---|
| COPDGene (n = 8395) | Additional TOPMed studies (n = 11 269) | ICGN and EOCOPD (n = 1554) | UK Biobank (n = 27 617) | ||
| Median age, y (IQR) | 58.7 (51.8-66.3) | 60.0 (53.0-67.0) | 56.0 (49.7-63.1) | 58 (51.0-63.0) | 58.6 (51.3-64.0) |
| Patients with CHIP | 478 (5.7) | 566 (5.0) | 92 (5.9) | 1,588 (5.8) | 2,724 (5.6) |
| COPD GOLD status | |||||
| 2-4 | 3421 | 2282 | 1282 | 1459 | 8444 |
| 3-4 | 1698 | 945 | 853 | 149 | 3645 |
| Mean predicted FEV1% (SD) | 73 (25.6) | 87.7 (23) | 47.7 (26.8) | 100.8 (16.6) | 91.3 (24.2) |
| Smoking status | |||||
| Smokers | 8395 | 6515 | 1462 | 12 321 | 28 693 |
| Mean pack-years (SD) | 44.3 (25.0) | 16.1 (24.7) | 42.0 (26.7) | 6.2 (13.12) | 16.1 (24.1) |
| Characteristic | Cohort | Total (N = 48 835) | |||
|---|---|---|---|---|---|
| COPDGene (n = 8395) | Additional TOPMed studies (n = 11 269) | ICGN and EOCOPD (n = 1554) | UK Biobank (n = 27 617) | ||
| Median age, y (IQR) | 58.7 (51.8-66.3) | 60.0 (53.0-67.0) | 56.0 (49.7-63.1) | 58 (51.0-63.0) | 58.6 (51.3-64.0) |
| Patients with CHIP | 478 (5.7) | 566 (5.0) | 92 (5.9) | 1,588 (5.8) | 2,724 (5.6) |
| COPD GOLD status | |||||
| 2-4 | 3421 | 2282 | 1282 | 1459 | 8444 |
| 3-4 | 1698 | 945 | 853 | 149 | 3645 |
| Mean predicted FEV1% (SD) | 73 (25.6) | 87.7 (23) | 47.7 (26.8) | 100.8 (16.6) | 91.3 (24.2) |
| Smoking status | |||||
| Smokers | 8395 | 6515 | 1462 | 12 321 | 28 693 |
| Mean pack-years (SD) | 44.3 (25.0) | 16.1 (24.7) | 42.0 (26.7) | 6.2 (13.12) | 16.1 (24.1) |
Data represent no. (%) unless otherwise specified.
IQR, interquartile range; SD, standard deviation.
Whole-genome sequencing and whole-exome sequencing
We obtained whole-genome sequencing or whole-exome sequencing data from DNA in peripheral blood samples. The raw sequencing data were processed, and somatic mutation calling was performed by using Mutect2 as previously described.11,22 We identified cases of CHIP by using a prespecified list of variants predicted or reported to be pathogenic and drivers of myeloid malignancies. Details regarding the sequencing procedures are provided in the supplemental Data.
Mouse models
We transplanted lethally irradiated mice with bone marrow from Tet2 wild-type (WT) or Tet2 knockout (KO) donors, exposed the recipient animals to air, cigarette smoke (CS), or CS and polyinosinic:polycytidylic acid (poly(I:C)), and quantified the development of emphysema by using morphometric methods.23 Single-cell RNA-sequencing was performed on the 10x platform (10x Genomics). Details on the experimental approach, including quantification of occurrences of emphysema and single-cell analyses are provided in the supplemental Data.
Statistical analysis
We examined the associations of five COPD-related phenotypes with CHIP, including moderate-to-severe COPD (GOLD 2-4 vs GOLD 0), severe or very severe COPD (GOLD 3-4 vs GOLD 0), severe-to-very-severe COPD within the group of patients with moderate-to-severe COPD (GOLD 3-4 vs GOLD 2), and percent predicted forced expiratory volume in 1 second (FEV1%p), and FEV1%p within patients who had moderate-to-severe COPD (supplemental Tables 4, 7-9, and 11). We also examined the effect of smoking on CHIP.
For the COPDGene and UK Biobank data, we used multivariable logistic regression to examine the associations between COPD (GOLD 2-4 vs GOLD 0) and CHIP, adjusting for age at blood draw, age2, sex, sequencing center, number of pack-years, smoking status, and the top 10 principal components of genetic ancestry. Linear regression tested the association between CHIP and FEV1%p, a quantitative measure of airflow obstruction in patients with COPD, adjusted for the same set of covariates using all samples (including patients with GOLD 1). To examine the association between the presence of CHIP (as a binary outcome) and smoking, we used number of pack-years as the independent variable in the logistic regression, adjusting for age, sex, current smoking status, sequencing center, and top 10 principal components of genetic ancestry.
For the additional TOPMed, ICGN, and EOCOPD cohorts, we used a generalized linear mixed-effects model and linear mixed-effects models to account for family relatedness between patients in the GENESIS R package with the same covariates as described above.24 Random effects meta-analyses were conducted for the additional TOPMed cohorts for all the phenotypes by using the metafor package.25
The 95% confidence intervals (CIs) reported for the primary cohort (COPDGene) were adjusted for the 6 phenotypes we examined (supplemental Table 4), and the significance level was defined as 0.05/6 = 0.0083 for the primary cohort. Replication was defined as reaching a nominal significance level of 0.05 using a one-sided test with consistent direction of effect in at least one of the three replication cohorts. The reported 95% CIs for the additional cohorts (additional TOPMed, ICGN, EOCOPD, and UK Biobank cohorts) were not adjusted for multiple testing. Details regarding the statistical analyses are provided in the Extended Methods of the supplemental Data. Informed consent was obtained, per the Declaration of Helsinki.
Results
Association between CHIP and COPD in COPDGene
In our primary cohort (COPDGene), we identified CHIP mutations in 478 (5.7%) of the 8395 individuals (Table 1). The most common mutations were in DNMT3A, TET2, and ASXL1, genes that encode for epigenetic regulators and are the most commonly mutated genes in CHIP (Figure 1; supplemental Figure 2; supplemental Table 3).22 In a multivariable model that included age, smoking, and sex, CHIP was associated with odds ratios (ORs) of 1.6 (adjusted 95% CI, 1.1-2.2; P = .0003) for GOLD 2-4 and 2.2 (adjusted 95% CI, 1.5-3.2; P < .0001) for GOLD 3-4 COPD (Figure 2A-B; supplemental Table 4). Among the 3421 patients with GOLD 2-4 disease, CHIP was more common in those with GOLD 3-4 (OR, 1.6; adjusted 95% CI, 1.1-2.4; P = .0009) (Figure 2C; supplemental Table 4).
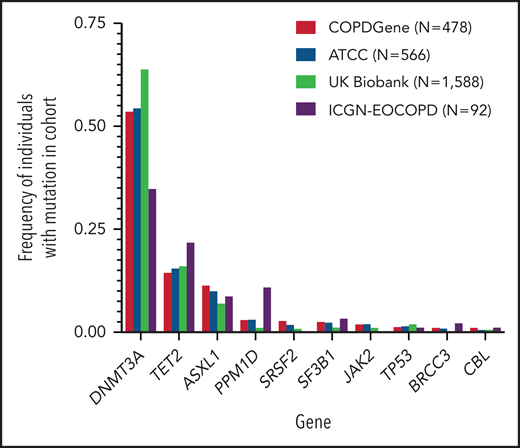
Distribution of clonal hematopoietic mutations in studied cohorts. Clonal hematopoietic mutations were identified from sequencing of whole genomes (COPDGene, additional TOPMed cohorts) or exomes (ICGN-EOCOPD, UK Biobank) found in samples of peripheral blood. Shown are the 10 most frequently mutated genes in COPDGene. N represents the number of CHIP carriers identified in each cohort. In patients with multiple mutations, the mutated gene with the largest variant allele frequency is shown.
Distribution of clonal hematopoietic mutations in studied cohorts. Clonal hematopoietic mutations were identified from sequencing of whole genomes (COPDGene, additional TOPMed cohorts) or exomes (ICGN-EOCOPD, UK Biobank) found in samples of peripheral blood. Shown are the 10 most frequently mutated genes in COPDGene. N represents the number of CHIP carriers identified in each cohort. In patients with multiple mutations, the mutated gene with the largest variant allele frequency is shown.
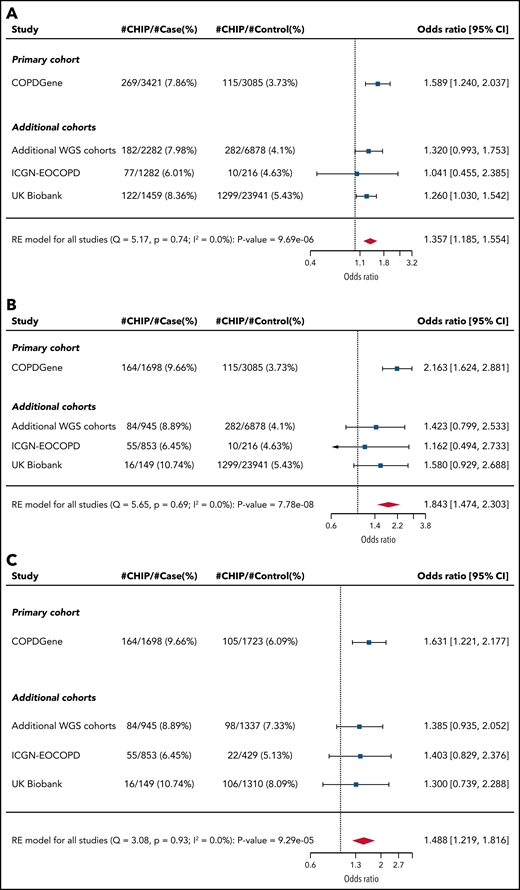
Association between CHIP status and COPD. Forest plots showing logistic regression results for association between CHIP status and (A) GOLD 2-4 and (B) GOLD 3-4 COPD among all patients. (C) Forest plot showing logistic regression results for association between CHIP status and severity (GOLD 3-4) among patients with GOLD 2-4 COPD. The arrowhead indicates that the limit of the CI is beyond the range annotated on the OR axis at the bottom. Note that the CIs listed for all cohorts are unadjusted. RE, random effects; WGS, whole-genome sequencing.
Association between CHIP status and COPD. Forest plots showing logistic regression results for association between CHIP status and (A) GOLD 2-4 and (B) GOLD 3-4 COPD among all patients. (C) Forest plot showing logistic regression results for association between CHIP status and severity (GOLD 3-4) among patients with GOLD 2-4 COPD. The arrowhead indicates that the limit of the CI is beyond the range annotated on the OR axis at the bottom. Note that the CIs listed for all cohorts are unadjusted. RE, random effects; WGS, whole-genome sequencing.
These associations remained significant in COPDGene after controlling for the COPD polygenic risk score, a metric that integrates many germline risk alleles (OR for GOLD 2-4 COPD, 1.57; P = .0006; OR for GOLD 3-4 COPD, 2.15; P < .0001), and when adjusting for incident lung cancer, lung cancer mortality, other self-reported cancers, cardiovascular disease, and the highest degree of school completed as a measure of socioeconomic status (OR for GOLD 2-4 COPD, 1.53; P = .001) (supplemental Table 5).26 Examining the individual genes, we found that patients in COPDGene with DNMT3A or TET2 mutations had significantly higher odds of having GOLD 3-4 disease (OR, 1.8; adjusted 95% CI, 1.03-3.06; P = .004 and OR, 3.7; adjusted 95% CI, 1.01-13.4; P = .005, respectively) (supplemental Table 6).
Association between CHIP and COPD in the ICGN, EOCOPD, and UK Biobank additional TOPMed cohorts
We next analyzed three additional cohorts, all of which included COPD phenotyping, smoking, and sequencing data. The additional TOPMed cohorts included six studies totaling 11 269 patients (2282 with COPD). ICGN-EOCOPD are family-based studies of younger patients and included 1554 patients (1282 with COPD). The UK Biobank has a population-based cohort and included 27 617 individuals (1459 with COPD). In the additional TOPMed cohorts, ICGN-EOCOPD, and the UK Biobank, CHIP was identified in 5.0%, 5.9%, and 5.8% of individuals, respectively.
Across all cohorts, CHIP was more common in the patients with COPD (supplemental Tables 7-9). In both UK Biobank and the additional TOPMed cohorts, CHIP was associated with an OR of 1.3 (P = .025 and one-sided P = .028, respectively) for GOLD 2-4 COPD, but was not significantly associated in ICGN-EOCOPD (Figure 2; supplemental Figure 3). The association in the UK Biobank remained after adjusting for lung cancer, lung cancer mortality, presence of other cancer, cardiovascular disease, and highest degree of school completed (OR, 1.3; P = .03) (supplemental Table 10). We also observed the association between TET2 and GOLD 3-4 COPD in the UK Biobank cohort (OR, 2.67; 1-sided P = .029).
In an exploratory analysis of the UK Biobank using 14 International Classification of Diseases 10th Revision (ICD-10) codes (see the supplemental Data), we found that the presence of CHIP was significantly associated with an increased incidence of COPD with variant allele frequencies >0.1, which is equivalent to >10% of DNA reads carrying a mutation (hazard ratio [HR], 1.69; 95% CI, 1.03-2.76; P = .036) but not with variant allele frequencies <0.1 (HR, 0.66; 95% CI, 0.33-1.24, P = .18) (supplemental Figure 4).
Association between CHIP and COPD in all patients
We combined all of the cohorts and performed random effects meta-analyses (Figure 2; supplemental Table 11). Across all 48 835 individuals, CHIP was associated with ORs of 1.4 (95% CI, 1.2-1.6; P < .0001) for GOLD 2-4 COPD and 1.8 (95% CI, 1.5-2.3; P < .0001) for GOLD 3-4 COPD (Figure 2A-B). Among the 8444 patients with GOLD 2-4 disease, CHIP was more common in those with GOLD 3-4 (OR, 1.5; 95% CI, 1.2-1.8; P < .0001) (Figure 2C). To quantify the effect of CHIP on COPD in terms of CS exposure, we compared the estimated OR for COPD with the presence of CHIP from the meta-analysis with the OR for COPD per 10 pack-years of smoking from the population-based BOLD study (estimated OR, between 1.16 and 1.28).27,28 The effect of CHIP on the odds of GOLD 2-4 disease was equivalent to 12 to 21 pack-years of smoking, and the effect on the odds of GOLD 3-4 disease was equivalent to 25 to 41 pack-years of smoking.
To further interrogate the relationship between COPD and the intensity of cigarette smoking, we generated six strata: never-smokers, ever-smokers, current-smokers, former-smokers, heavy smokers (pack-years >30), and light smokers (pack-years <15) (supplemental Table 12; supplemental Figure 7). We observed a significant association between CHIP and GOLD 2-4 COPD in ever-smokers, former-smokers, and heavy-smokers and a nominal association in light-smokers. The OR in never-smokers and current-smokers was directionally consistent with the other strata but did not reach statistical significance, potentially because the former was underpowered with few cases of COPD in never-smokers and the latter was potentially confounded by the “healthy smoker phenomenon,” in which patients with the worst COPD are more likely to quit smoking, causing a paradoxical finding that current-smoking status is protective for COPD and COPD exacerbations in COPDGene.
In the meta-analyses, GOLD 2-4 COPD was significantly associated with mutations in DNMT3A (OR, 1.30; adjusted 95% CI, 1.02-1.67, P = .003). GOLD 3-4 COPD was significantly associated with mutations in DNMT3A (OR, 1.7; adjusted 95% CI, 1.1-2.6, P = .001), TET2 (OR, 2.4; adjusted 95% CI, 1.0-5.6, P = .004), and TP53 (OR, 9.2; adjusted 95% CI, 1.5-57.7; P = .001) (supplemental Table 13).
Association between CHIP and quantitative impairment of lung function
We next analyzed the associations between CHIP and FEV1%p (Figure 3). In COPDGene, CHIP was significantly associated with decreased FEV1%p among all patients (mean between-group difference, −5.8%; adjusted 95% CI, −8.7% to −2.8%; P < .0001) and when restricted to those with GOLD 2-4 COPD (mean between-group difference, −2.6%; adjusted 95% CI, −5.2% to −0.07%; P = .008).
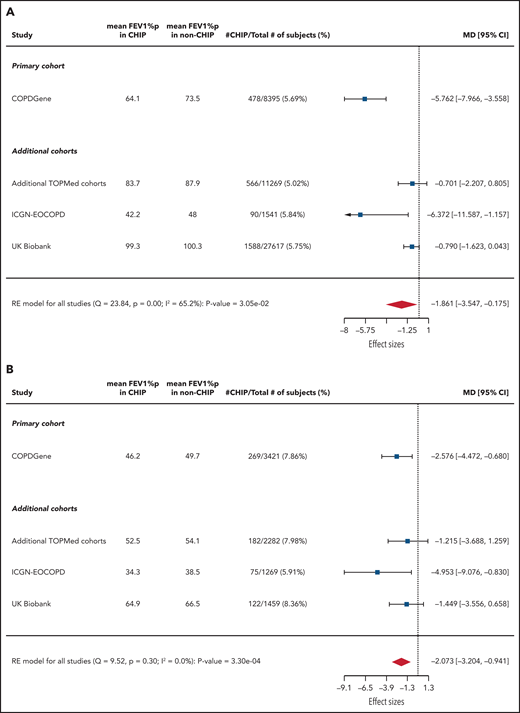
Association between CHIP status and FEV1%p. Forest plots showing linear regression results of association between CHIP status and FEV1%p (A) across all patients and (B) restricted to those with GOLD 2-4 COPD. Note that the CIs listed for all cohorts are unadjusted. MD, mean difference (%).
Association between CHIP status and FEV1%p. Forest plots showing linear regression results of association between CHIP status and FEV1%p (A) across all patients and (B) restricted to those with GOLD 2-4 COPD. Note that the CIs listed for all cohorts are unadjusted. MD, mean difference (%).
CHIP was significantly associated with decreased FEV1%p in ICGN-EOCOPD (mean between-group difference, −6.4%; 95% CI, −11.6% to −1.2%; P = .02) and UK Biobank (mean between-group difference, −0.8%; one-sided P = .03) but not in the additional TOPMed cohorts. When restricted to those with GOLD 2-4 COPD, CHIP was associated with decreased FEV1%p in ICGN-EOCOPD (mean between-group difference, −5.0%; 95% CI, −9.1% to −0.8%; P = .02) but not in the additional TOPMed or UK Biobank cohorts.
Using a random effects meta-analysis, CHIP was associated with decreased FEV1%p among all 48 835 patients (mean between-group difference, −1.9%; 95% CI, −3.5% to −0.2%; P = .03) and the 8444 patients with GOLD 2-4 COPD (mean between-group difference, −2.1%; 95% CI, −3.2% to −0.9%; P = .0003).
Association between CHIP and smoking
Smoking has been reported as a risk factor for CHIP, but these reports generally used discrete metrics of smoking (yes/no or former/current/never) and often have missing data.13,14 To address these issues, we examined the association between smoking status and CHIP by using the comprehensive smoking history available for all patients (supplemental Figure 5). After controlling for age, there was no significant association between CHIP status and number of pack-years of smoking in COPDGene, ICGN-EOCOPD, or UK Biobank cohorts. In contrast, CHIP was associated with smoking in the additional TOPMed cohorts (OR, 1.007; 95% CI, 1.003-1.01; P = .0005). A random effects model of all patients revealed a small but significant association (OR, 1.003; 95% CI, 1.001-1.005; P = .0037) corresponding to an OR of 1.03 for every 10 pack-years of smoking (supplemental Figure 5; supplemental Table 14). Taken together, these data suggest that there is a small but significant relationship between cumulative CS exposure and CHIP.
Hematopoietic Tet2 loss accelerates the development of emphysema in a mouse model via alteration in inflammation and cytokine signaling
On the basis of observed associations in humans, we hypothesized that mutated hematopoietic cells could promote an inflammatory phenotype to accelerate the pathogenesis of COPD. To test this hypothesis directly, we used Tet2 KO mice to model CHIP because TET2 was commonly mutated in and independently associated with COPD in the human cohorts (supplemental Table 13), and loss of Tet2 in the blood cells of mice faithfully recapitulates inflammatory phenotypes observed in humans with CHIP.9,10,29,30 Mice carrying Tet2 WT or Tet2 KO hematopoietic cells were generated by bone marrow transplantation from 8-week-old donors that were aged for an additional 6 months and then exposed to CS for 6 weeks with poly(I:C) treatment during weeks 5 and 6 (Figure 4A).4 This model of smoking-induced COPD incorporates poly(I:C), a synthetic oligonucleotide that stimulates toll-like receptor 3 (TLR3) and is critical for the immune response to numerous viral infections, which are more common in patients with COPD.31 The mice were then euthanized and the lungs were analyzed.
![Effect of hematopoietic Tet2 KO on emphysema development. (A) Schematic of experimental approach for mice treated with CS and poly(I:C). (B) Representative images of airspace destruction (Gill’s stain imaged at ×10 magnification) in Tet2 WT and Tet2 KO mice (left) and quantification of emphysema in Tet2 WT (n = 10) and Tet2 KO (n = 10) mice exposed to CS and poly(I:C). Error bars indicate standard error of the mean. (C) UMAP visualization of single-cell RNA sequencing data shows clustering of 25 cell types that were identified in the lungs of Tet2 WT and Tet2 KO mice. (D) Assessment of signature scores in the single-cell transcriptional data of mice exposed to CS and poly(I:C) across all cells for IFN and tumor growth factor β (TGFB) gene sets obtained from Hallmark (H), Reactome (R), Biocarta (B), KEGG (K), or Wikipathways (W). A positive value (blue) represents higher scores in Tet2 WT (WT), and a negative value (orange) reflects higher scores in Tet2 KO mice. The P values were calculated using a Wilcoxon test and adjusted (adj) using a Bonferroni correction. (E-F) For each cluster of cells, the mean of cell signature scores for (E) IFN-α and (F) IFN-γ was determined and plotted for Tet2 WT (y-axis) and Tet2 KO (x-axis) animals. Clusters above the line of identity are enriched for the signature in Tet2 WT mice; clusters that fall below are enriched for the signature in Tet2 KO mice. Clusters in red have significantly higher scores in Tet2 KO mice with the size of each cluster representing the significance of difference between the Tet2 KO and Tet2 WT groups. AM, alveolar macrophage; AT1, alveolar type 1; AT2, alveolar type 2; CEC, circulating endothelial cell; DC, dendritic cell; DN, double negative; EC, endothelial cell; IM, interstitial macrophage; LEC, lymphatic endothelial cell; Mig DC, migratory dendritic cell; NK, natural killer [cell]; P, Pvalue; pDC, plasmacytoid dendritic cell; SMC, smooth muscle cell; Treg, regulatory T cell. pIpC, poly(I:C).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/3/10.1182_blood.2021013531/3/m_bloodbld2021013531f4.png?Expires=1767705145&Signature=4i2mzf37ZgZlYwdV6f7mfiqk51XvdFL~ZLv8u7V1~ieTNKZrjOBsT17kKzY0lhcZsjlR3cHLCs1I6xVtaslZJBo4wkdXFqbefeXr6gEYRnMW0gd7xvURZiIm1OleQnBse5wbWhgOOIftA25PwrqdX3ei0VViw15nT5THNCK1ZBFMf4gGebso8TTIeCbG2yGNlr5Qf3OkeTWdy5U9mTEqW4UihAXv5An8nUi04WKBTdkeD-hiQYmXD~pwl2zS6O1w94-y7aqttpqtSig9Tqawq818JRgJyrijalXaZQCh7bxLjkqyvZpA6MJ1wIg7xNR8cE4P0MWU-za9J4gDo6FlPA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of hematopoietic Tet2 KO on emphysema development. (A) Schematic of experimental approach for mice treated with CS and poly(I:C). (B) Representative images of airspace destruction (Gill’s stain imaged at ×10 magnification) in Tet2 WT and Tet2 KO mice (left) and quantification of emphysema in Tet2 WT (n = 10) and Tet2 KO (n = 10) mice exposed to CS and poly(I:C). Error bars indicate standard error of the mean. (C) UMAP visualization of single-cell RNA sequencing data shows clustering of 25 cell types that were identified in the lungs of Tet2 WT and Tet2 KO mice. (D) Assessment of signature scores in the single-cell transcriptional data of mice exposed to CS and poly(I:C) across all cells for IFN and tumor growth factor β (TGFB) gene sets obtained from Hallmark (H), Reactome (R), Biocarta (B), KEGG (K), or Wikipathways (W). A positive value (blue) represents higher scores in Tet2 WT (WT), and a negative value (orange) reflects higher scores in Tet2 KO mice. The P values were calculated using a Wilcoxon test and adjusted (adj) using a Bonferroni correction. (E-F) For each cluster of cells, the mean of cell signature scores for (E) IFN-α and (F) IFN-γ was determined and plotted for Tet2 WT (y-axis) and Tet2 KO (x-axis) animals. Clusters above the line of identity are enriched for the signature in Tet2 WT mice; clusters that fall below are enriched for the signature in Tet2 KO mice. Clusters in red have significantly higher scores in Tet2 KO mice with the size of each cluster representing the significance of difference between the Tet2 KO and Tet2 WT groups. AM, alveolar macrophage; AT1, alveolar type 1; AT2, alveolar type 2; CEC, circulating endothelial cell; DC, dendritic cell; DN, double negative; EC, endothelial cell; IM, interstitial macrophage; LEC, lymphatic endothelial cell; Mig DC, migratory dendritic cell; NK, natural killer [cell]; P, Pvalue; pDC, plasmacytoid dendritic cell; SMC, smooth muscle cell; Treg, regulatory T cell. pIpC, poly(I:C).
Effect of hematopoietic Tet2 KO on emphysema development. (A) Schematic of experimental approach for mice treated with CS and poly(I:C). (B) Representative images of airspace destruction (Gill’s stain imaged at ×10 magnification) in Tet2 WT and Tet2 KO mice (left) and quantification of emphysema in Tet2 WT (n = 10) and Tet2 KO (n = 10) mice exposed to CS and poly(I:C). Error bars indicate standard error of the mean. (C) UMAP visualization of single-cell RNA sequencing data shows clustering of 25 cell types that were identified in the lungs of Tet2 WT and Tet2 KO mice. (D) Assessment of signature scores in the single-cell transcriptional data of mice exposed to CS and poly(I:C) across all cells for IFN and tumor growth factor β (TGFB) gene sets obtained from Hallmark (H), Reactome (R), Biocarta (B), KEGG (K), or Wikipathways (W). A positive value (blue) represents higher scores in Tet2 WT (WT), and a negative value (orange) reflects higher scores in Tet2 KO mice. The P values were calculated using a Wilcoxon test and adjusted (adj) using a Bonferroni correction. (E-F) For each cluster of cells, the mean of cell signature scores for (E) IFN-α and (F) IFN-γ was determined and plotted for Tet2 WT (y-axis) and Tet2 KO (x-axis) animals. Clusters above the line of identity are enriched for the signature in Tet2 WT mice; clusters that fall below are enriched for the signature in Tet2 KO mice. Clusters in red have significantly higher scores in Tet2 KO mice with the size of each cluster representing the significance of difference between the Tet2 KO and Tet2 WT groups. AM, alveolar macrophage; AT1, alveolar type 1; AT2, alveolar type 2; CEC, circulating endothelial cell; DC, dendritic cell; DN, double negative; EC, endothelial cell; IM, interstitial macrophage; LEC, lymphatic endothelial cell; Mig DC, migratory dendritic cell; NK, natural killer [cell]; P, Pvalue; pDC, plasmacytoid dendritic cell; SMC, smooth muscle cell; Treg, regulatory T cell. pIpC, poly(I:C).
Compared with the WT controls, mice carrying Tet2 KO hematopoietic cells exhibited significantly increased development of emphysema (mean alveolar cord length [a measure of alveolar diameter], 16.0 vs 17.4 µM; P = .008) (Figure 4B; supplemental Table 15). In a separate model that more closely resembles the clonal size of mutant hematopoietic cells found in human CHIP, mice with ∼15% of hematopoietic cells lacking Tet2 were exposed to 6 months of CS and also showed enhanced emphysema (18.0 vs 20.0 µM; P = .02) (supplemental Figure 8A-B; supplemental Table 16).9,23 CS exposure did not lead to significant changes in the frequency of Tet2 KO cells in the blood over 6 months, neither model showed a significant difference in alveolar cord length in the air controls, and there were no significant differences between genotypes upon exposure to CS for 6 weeks without poly(I:C) or to poly(I:C) without CS (supplemental Figure 8C-G).
We processed lungs from Tet2 WT and Tet2 KO mice exposed to either air or CS/poly(I:C) and performed single-cell RNA-sequencing, collecting 41 870 high-quality transcriptomes (supplemental Table 17). Clustering and annotation by canonical marker genes revealed 25 cell types. We did not find any significant differences in cell type frequencies between the groups (Figure 4C; supplemental Figure 9A-B; supplemental Table 18). We then scored all single-cell transcriptomes for the 50 well-established transcriptional signatures of different cellular programs to investigate gene expression changes between the groups.32 The most significant changes were upregulation of interferon type I (IFN-I) and IFN-II signaling in Tet2 KO compared with Tet2 WT mice, which we confirmed by using gene set enrichment analyses. We also identified natural killer and T cells as the predominant source of IFN-γ (Figure 4D-F; supplemental Figures 9C-D, and 10A).32,33 In contrast, transforming growth factor-β (TGF-β) signaling was significantly lower in the Tet2 KO animals, with differing cellular sources of TGF-β depending on the isoform (supplemental Figure 10B).
To examine mediators of inflammation, we measured the levels of 44 secreted inflammatory cytokines and chemokines from Tet2 WT or Tet2 KO pulmonary macrophages cultured ot after exposure to poly(I:C) for 24 hours. Seven proteins (Cxcl1, Cxcl2, Cxcl9, Ccl5, Ccl11, Ccl20, and Tnfa) were secreted at significantly higher levels from the Tet2 KO macrophages (supplemental Figure 10C). Moreover, in the single-cell transcriptional data, RNA expression of these 7 proteins was enriched in Tet2 KO alveolar macrophages and monocytes relative to Tet2 WT controls (supplemental Figure 10D). Taken together, these data show that Tet2 loss in hematopoietic cells enhances pulmonary inflammation, increases IFN signaling, decreases TGF-β signaling, and accelerates the development of emphysema in a mouse model.
Discussion
By using data from nearly 50 000 individuals in four large cohorts, we found that CHIP is independently associated with COPD, COPD severity, and quantitative impairment in pulmonary function. Moreover, we observed accelerated development of emphysema in mice with loss of Tet2 in hematopoietic cells after two different stimuli. Taken together, our data suggest that the presence of somatic mutations in blood cells contributes to the development and severity of COPD. These findings also highlight that the inflammatory sequelae of CHIP extend beyond those established in cardiovascular disease and can influence pathophysiologic changes in other tissues.9,10
Risk factors for COPD include age, smoking, rare α1-antitrypsin mutations, and inherited polygenic risk score.26,34,35 Our data indicate that CHIP increases the risk of COPD and disease severity independent of age, exposure to CS, or inherited polygenic risk score. CHIP confers a risk equivalent to ∼20 to 40 pack-years of exposure to CS for the development of severe or very severe COPD. Given the high prevalence of and health implications of both COPD and CHIP, these findings could inform risk mitigation strategies, including smoking cessation, more intensive pulmonary observation, and therapeutic interventions targeting inflammatory pathways.
Although previous studies have reported smoking as a risk factor for developing CHIP, sample size and variable smoking data have complicated their interpretation.13 For example, a recent analysis of the UK Biobank cohort reported a significant association between CHIP and current smoking status but did not quantify the association per pack-year and did not find a significant association between past smoking status and CHIP.14 In a meta-analysis, we confirmed a significant association between pack-years of smoking and the risk of having CHIP. The per-pack-year risk is small, but it remains important, particularly in heavy smokers, because an individual with a 50 pack-year history has 16% higher odds of having CHIP. Given these findings, we included pack-year data as a covariate in all analyses of CHIP and COPD, and we found an effect of CHIP on COPD independent of smoking history. Furthermore, in contrast to other reported associations between COPD and CHIP, our study incorporates pack-year smoking data, includes thousands of patients and controls from four distinct cohorts, and uses detailed phenotyping, including severity of disease by both GOLD and quantitative spirometric measurements.
In mouse models of CHIP and COPD, Tet2 KO in blood cells exacerbated the development of emphysema, enhanced inflammatory signaling via IFN-I and IFN-II, and attenuated TGF-β signaling. The changes we observed in these pathways are consistent with previous studies from mouse models of emphysema and from human genetics studies of COPD.36-40 Moreover, genome-wide association studies have identified variants near TET2 associated with COPD and FEV1.6,41 The causal gene and variant have not been identified, but this association raises the possibility that TET2 is an effector gene at this locus. Additional murine experiments in which the ages of the animals are varied or other genes mutated in clonal hematopoiesis such as Dnmt3a are altered, may provide further insights into the timing and mechanism by which mutant hematopoietic cells influence pulmonary inflammation and emphysema development.
The strength of associations between CHIP and COPD was most significant in the COPDGene cohort compared with other cohorts. COPDGene patients undergo comprehensive phenotyping including chest computed tomography scans and exclusions for other lung diseases, and the study includes more than twice the number of moderate and severe or very severe cases of COPD than any other cohort. The remaining COPD cohorts are smaller, are unbalanced (ECLIPSE, few controls), or represent specific subsets (ICGN-EOCOPD, younger individuals with more severe disease). The remaining TOPMed cohorts and the population-based UK Biobank are not as well phenotyped, and each contain less than half the number of GOLD 2-4 and less than one-tenth the number of GOLD 3-4 COPD patients compared with COPDGene. These factors may explain the stronger signal in COPDGene. Despite these limitations, we found consistent associations in the other cohorts.
One limitation of this study is the use of cross-sectional instead of incident COPD data, precluding calculations of HRs and the inference of causality from the observed human associations between CHIP and COPD. However, COPD is typically a disease that develops over decades, few cohorts have sufficient samples and follow-up using lung function, and incident case determination using self-reported or ICD-coded diagnoses are highly unreliable. Indeed, our ability to make COPD determinations using the standard definition requiring spirometry data and without using self-reported or ICD-coded diagnoses differentiates our study from many previous reports and further strengthens our confidence in the observed associations.
In summary, our data indicate that age-associated somatic mutations in blood cells are independently associated with the presence and severity of COPD. Identification of these mutations may provide important prognostic information for healthy individuals, those at high-risk for development of COPD, and those with COPD. Our results also highlight the potential therapeutic value of targeting CHIP in the treatment or prevention of COPD, including efforts to reduce the development or progression of clonal hematopoiesis, inhibition of the inflammatory response in mutant hematopoietic cells, and targeting elevated inflammatory cytokine levels present in patients with CHIP.
Acknowledgments
The authors thank the staffs and participants of the Jackson Heart Study (JHS) and those of the Atherosclerosis Risk in Communities (ARIC) study for their important contributions; they also acknowledge the dedication of the Framingham Heart Study (FHS) participants without whom this research would not be possible, and the studies and participants who provided biological samples and data for TOPMed.
This work was supported by grants from the National Institutes of Health (NIH), National Heart, Lung, and Blood Institute (NHLBI) (K01-HL129039 [D.Q.]; R01-HL068111 [Y.T.]; U01-HL089856 [E.K.S.]; R01-HL113264, R01-HL135142, and T32-HL094301-07 [M.H.C.]; R01-HL082945 [B.L.E.]; T32-HL007427 [M.M.]), from the National Cancer Institute (NCI) (K12-CA087723 [A.S.S.], P01-CA108631 [B.L.E.]), and from the Office of the Director (DP5-OD02958 [A.G.B.]), by the Evans Foundation (P.G.M. and B.L.E.), the American Society of Hematology (P.G.M.), the Burroughs Wellcome Foundation (A.G.B. and S.J.), the Knut and Alice Wallenberg Foundation (A.N.), and the Howard Hughes Medical Institute (B.L.E.). S.J.L. is supported by a grant from the NIH Intramural Research Program, National Institute of Environmental Health Sciences (Z01ES043012-10). The COPDGene trial (NCT00608764) was supported by grants from the NHLBI (U01-HL089897, U01-HL089856) and by the COPD Foundation through contributions made to an Industry Advisory Board comprising AstraZeneca, Boehringer-Ingelheim, Genentech, GlaxoSmithKline, Novartis, Pfizer, Siemens, and Sunovion. The Atherosclerosis Risk in Communities (ARIC) study was supported in whole or in part by grants from the NHLBI, NIH, Department of Health and Human Services (DHHS) (HHS-N268201700001I, HHS-N268201700002I, HHS-N268201700003I, HHS-N268201700004I, HHS-N268201700005I, HHS-N268200625226C), from NHLBI (R01-HL087641, R01-HL086694), and from the National Human Genome Research Institute (NHGRI) (U01-HG004402). The Framingham Heart Study (FHS) was supported by grants from (N01-HC25195), from DHHS (HHS-N268201500001I), from (75N92019D00031), and a grant supplement from NHLBI (R01-HL092577-06S1). The Jackson Heart Study (JHS) was supported by grants from the DHHS (HHS-N268201800013I [Jackson State University]; HHS-N268201800014I [Tougaloo College]; HHS-N268201800015I [Mississippi State Department of Health]; HHS-N268201800010I, HHS-N268201800011I, and HHS-N268201800012I [University of Mississippi Medical Center]), the NHLBI, and the National Institute on Minority Health and Health Disparities (NIMHD). JHS was conducted in collaboration with Jackson State University and the DHHS. The Multi-Ethnic Study of Atherosclerosis (MESA) Lung Study was supported by grants from (75N92020D00001, 75N92020D00002, 75N92020D00003, 75N92020D00004, 75N92020D00005, 75N92020D00006, 75N92020D00007), from (N01-HC95159, N01-HC95160, N01-HC95161, N01-HC95162, N01-HC95163, N01-HC95164, N01-HC95165, N01-HC95166, N01-HC95167, N01-HC95168, N01-HC95169), from DHHS (HHS-N2682015000031) from the National Center for Advancing Translational Sciences (NCATS), Clinical and Translational Science Institute (CTSI) (UL1-TR001881, UL1-TR000040, UL1-TR001079, UL1-TR001420), from DHHS (HHS-N268201500003I), from NHLBI (R01-HL077612, R01-HL098031), and from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Diabetes Research Center (DRC) (DK063491 [Southern California Diabetes Endocrinology Research Center]). The MESA Family study was supported by grants from the NHLBI (R01-HL071051, R01-HL071205, R01-HL071250, R01-HL071251, R01-HL071258, R01-HL071259), and by the National Center for Research Resources (NCRR) (UL1-RR033176). MESA, the MESA Lung study, the MESA SHARe project, and the MESA Family study were conducted by the NHLBI in collaboration with MESA investigators. The Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) trial (NCT00292552) and the International COPD Genetics Network (ICGN) study were supported by GlaxoSmithKline. The Boston Early-Onset COPD (EOCOPD) study was supported by grants from the NHGRI (1U54-HG006493) and the NHLBI. VSR is supported in part by the Evans Medical Foundation and the Jay and Louis Coffman Endowment from the Department of Medicine, Boston University School of Medicine. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NHLBI or the NIH.
Genome sequencing for the NHLBI TOPMed studies (and their phs accession numbers) was performed at various centers under grants from various NIH institutes as follows: Northwest Genome Center (NWGC): “Whole Genome Sequencing and Related Phenotypes in the Genetic Epidemiology of COPD Study” (phs000951) (NHLBI: 3R01-HL089856-08S1) and “Whole Genome Sequencing and Related Phenotypes in the Jackson Heart Study” (phs000964) (DHHS: HHS-N268201100037C). Broad Genomics: “Whole Genome Sequencing and Related Phenotypes in the Genetic Epidemiology of COPD Study” (phs000951) (DHHS: HHS-N268201500014C), “Whole Genome Sequencing and Related Phenotypes in the Framingham Heart Study” (phs000974) (NHLBI: 3R01-HL092577-06S1; NHGRI: 3U54-HG003067-12S2), “Whole Genome Sequencing and Related Phenotypes in the Atherosclerosis Risk in Communities Study VTE Cohort” (phs001211) (NHLBI: 3R01-HL092577-06S1), “Whole Genome Sequencing and Related Phenotypes in the Multi-Ethnic Study of Atherosclerosis” (phs001416) (DHHS: HHS-N268201500014C; NHGRI: 3U54-HG003067-13S1). Baylor: “Whole Genome Sequencing and Related Phenotypes in the Atherosclerosis Risk in Communities Study VTE Cohort” (phs001211) (NHGRI: 3U54-HG003273-12S2; DHHS: HHS-N268201500015C). McDonnell Genome Institute (MGI): “Whole Genome Sequencing and Related Phenotypes in the Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-Points (ECLIPSE)” (phs001472) (DHHS: HHS-N268201600037I).
Molecular data for the TOPMed program was supported by the NHLBI. Core support for centralized genomic read mapping, genotype calling, variant quality metrics, and filtering was provided by the TOPMed Informatics Research Center (NHLBI: 3R01-HL117626-02S1; DHHS: HHS-N268201800002I); core support for phenotype harmonization, data management, sample-identity quality control, and general program coordination was provided by the TOPMed Data Coordinating Center (NHLBI: R01-HL120393, U01-HL120393; DHHS: HHS-N268201800001I). Sequencing for the EOCOPD was provided by the University of Washington Center for Mendelian Genomics. Provision of genotyping data was supported in part by the NCATS, CTSI (UL1-TR001881), and the NIDDK DRC (DK063491 [Southern California Diabetes Endocrinology Research Center]). Some images in this manuscript were created using Biorender (Biorender.com).
Authorship
Contribution: P.G.M., D.Q., E.K.S., P.N., Y.T., M.H.C., and B.L.E. designed and conceived the study; D.Q., M.C.H., P.G.M., L.W., D.N., A.S.S., and M.H.C. performed the statistical analyses and analyzed and interpreted the data; A.G.B., A.N., C.J.G., M.L., and S.J. generated the somatic mutation calls; P.G.M., J.R.-Q., A.S.S., M.E.M., B.S., K.V., B.D.L., L.S., W.S., C.A.O., B.C.M., P.v.G., and Y.T. performed the mouse experiments and analyzed the data; M.M. generated the polygenic risk score information; B.E.C., R.G.B., A.C., L.A.C., S.A.G., L.A.L., S.J.L., A.M., G.T.O., E.C.O., D.J., S.M.G., S.R., S.S.R., J.I.R., V.R., and B.Y. provided the TOPMed cohort data; and D.Q., P.G.M., M.C.H., D.N., E.K.S., M.H.C., and B.L.E. drafted the manuscript.
Conflict-of-interest disclosure: In the past 3 years, B.C.M. received consulting fees from Foundation Medicine. C.A.O. is currently employed by Vertex Pharmaceuticals. E.K.S. received grant support from GlaxoSmithKline and Bayer. Y.T. received grant support from GlaxoSmithKline, Sunovion (formerly Sepracor), and AstraZeneca and speaking fees from Bayer and AstraZeneca. M.H.C. received grant support from GlaxoSmithKline and Bayer and consulting and speaking fees from Illumina and AstraZeneca. B.L.E. received research funding from Celgene and Deerfield Ventures, consulting fees from GRAIL, and is on the scientific advisory boards for Exo Therapeutics and Skyhawk Therapeutics. The remaining authors declare no competing financial interests.
A complete list of the members of the COPDGene Study Investigators, National Heart, Lung, and Blood Institute Trans-Omics for Precision Medicine Consortium appears in “Appendix.”
Correspondence: Benjamin L. Ebert, Dana-Farber Cancer Institute, D1610A, 450 Brookline Ave, Boston, MA 02215; e-mail: benjamin_ebert@dfci.harvard.edu; and Michael H. Cho, Channing Division of Network Medicine, Brigham and Women’s Hospital, 181 Longwood Ave, Boston, MA 02115; e-mail: remhc@channing.harvard.edu.
Data can be found at accession numbers phs000951, phs001416, phs000974, phs001211, phs000964, phs001472, phs000954, phs000946, and phs000917.v1.p1.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Appendix
The members of the COPDGene Study Investigators, National Heart, Lung, and Blood Institute Trans-Omics for Precision Medicine Consortium are: Namiko Abe, Gonçalo Abecasis, Francois Aguet, Christine Albert, Laura Almasy, Alvaro Alonso, Seth Ament, Peter Anderson, Pramod Anugu, Deborah Applebaum-Bowden, Kristin Ardlie, Dan Arking, Donna K. Arnett, Allison Ashley-Koch, Stella Aslibekyan, Tim Assimes, Paul Auer, Dimitrios Avramopoulos, Najib Ayas, Adithya Balasubramanian, John Barnard, Kathleen Barnes, R. Graham Barr, Emily Barron-Casella, Lucas Barwick, Terri Beaty, Gerald Beck, Diane Becker, Lewis Becker, Rebecca Beer, Amber Beitelshees, Emelia Benjamin, Takis Benos, Marcos Bezerra, Larry Bielak, Joshua Bis, Thomas Blackwell, John Blangero, Eric Boerwinkle, Donald W. Bowden, Russell Bowler, Jennifer Brody, Ulrich Broeckel, Jai Broome, Deborah Brown, Karen Bunting, Esteban Burchard, Carlos Bustamante, Erin Buth, Brian Cade, Jonathan Cardwell, Vincent Carey, Julie Carrier, Cara Carty, Richard Casaburi, Juan P. Casas Romero, James Casella, Peter Castaldi, Mark Chaffin, Christy Chang, Yi-Cheng Chang, Daniel Chasman, Sameer Chavan, Bo-Juen Chen, Wei-Min Chen, Seung Hoan Choi, Lee-Ming Chuang, Mina Chung, Ren-Hua Chung, Clary Clish, Suzy Comhair, Matthew Conomos, Elaine Cornell, Carolyn Crandall, James Crapo, L. Adrienne Cupples, Joanne Curran, Jeffrey Curtis, Brian Custer, Coleen Damcott, Dawood Darbar, Sean David, Colleen Davis, Michelle Daya, Mariza de Andrade, Lisa de Las Fuentes, Paul de Vries, Michael DeBaun, Ranjan Deka, Dawn DeMeo, Scott Devine, Huyen Dinh, Harsha Doddapaneni, Qing Duan, Shannon Dugan-Perez, Ravi Duggirala, Jon Peter Durda, Susan K. Dutcher, Charles Eaton, Lynette Ekunwe, Adel El Boueiz, Patrick Ellinor, Leslie Emery, Serpil Erzurum, Charles Farber, Jesse Farek, Tasha Fingerlin, Matthew Flickinger, Myriam Fornage, Nora Franceschini, Chris Frazar, Mao Fu, Stephanie M. Fullerton, Lucinda Fulton, Stacey Gabriel, Weiniu Gan, Shanshan Gao, Yan Gao, Margery Gass, Heather Geiger, Bruce Gelb, Mark Geraci, Soren Germer, Robert Gerszten, Auyon Ghosh, Richard Gibbs, Chris Gignoux, Mark Gladwin, David Glahn, Stephanie Gogarten, Da-Wei Gong, Harald Goring, Sharon Graw, Kathryn J. Gray, Daniel Grine, Colin Gross, C. Charles Gu, Yue Guan, Namrata Gupta, David M. Haas, Jeff Haessler, Michael Hall, Yi Han, Patrick Hanly, Daniel Harris, Nicola L. Hawley, Jiang He, Ben Heavner, Susan Heckbert, Ryan Hernandez, David Herrington, Craig Hersh, Bertha Hidalgo, James Hixson, Brian Hobbs, John Hokanson, Elliott Hong, Karin Hoth, Chao Agnes Hsiung, Jianhong Hu, Yi-Jen Hung, Haley Huston, Chii Min Hwu, Marguerite Ryan Irvin, Rebecca Jackson, Deepti Jain, Cashell Jaquish, Jill Johnsen, Andrew Johnson, Craig Johnson, Rich Johnston, Kimberly Jones, Hyun Min Kang, Robert Kaplan, Sharon Kardia, Shannon Kelly, Eimear Kenny, Michael Kessler, Alyna Khan, Ziad Khan, Wonji Kim, John Kimoff, Greg Kinney, Barbara Konkle, Charles Kooperberg, Holly Kramer, Christoph Lange, Ethan Lange, Leslie Lange, Cathy Laurie, Cecelia Laurie, Meryl LeBoff, Jiwon Lee, Sandra Lee, Wen-Jane Lee, Jonathon LeFaive, David Levine, Dan Levy, Joshua Lewis, Xiaohui Li, Yun Li, Henry Lin, Honghuang Lin, Xihong Lin, Simin Liu, Yongmei Liu, Yu Liu, Ruth J. F. Loos, Steven Lubitz, Kathryn Lunetta, James Luo, Ulysses Magalang, Michael Mahaney, Barry Make, Ani Manichaikul, Alisa Manning, JoAnn Manson, Lisa Martin, Melissa Marton, Susan Mathai, Rasika Mathias, Susanne May, Patrick McArdle, Merry-Lynn McDonald, Sean McFarland, Stephen McGarvey, Daniel McGoldrick, Caitlin McHugh, Becky McNeil, Hao Mei, James Meigs, Vipin Menon, Luisa Mestroni, Ginger Metcalf, Deborah A. Meyers, Emmanuel Mignot, Julie Mikulla, Nancy Min, Mollie Minear, Ryan L. Minster, Braxton D. Mitchell, Matt Moll, Zeineen Momin, May E. Montasser, Courtney Montgomery, Donna Muzny, Josyf C. Mychaleckyj, Girish Nadkarni, Rakhi Naik, Take Naseri, Pradeep Natarajan, Sergei Nekhai, Sarah C. Nelson, Bonnie Neltner, Caitlin Nessner, Deborah Nickerson, Osuji Nkechinyere, Kari North, Jeff O'Connell, Tim O'Connor, Heather Ochs-Balcom, Geoffrey Okwuonu, Allan Pack, David T. Paik, James Pankow, George Papanicolaou, Cora Parker, Gina Peloso, Juan Manuel Peralta, Marco Perez, James Perry, Ulrike Peters, Patricia Peyser, Lawrence S. Phillips, Jacob Pleiness, Toni Pollin, Wendy Post, Julia Powers Becker, Meher Preethi Boorgula, Michael Preuss, Bruce Psaty, Pankaj Qasba, Dandi Qiao, Zhaohui Qin, Nicholas Rafaels, Laura Raffield, Mahitha Rajendran, Vasan S. Ramachandran, D. C. Rao, Laura Rasmussen-Torvik, Aakrosh Ratan, Susan Redline, Robert Reed, Catherine Reeves, Elizabeth Regan, Alex Reiner, Muagututi'a Sefuiva Reupena, Ken Rice, Rebecca Robillard, Nicolas Robine, Dan Roden, Carolina Roselli, Ingo Ruczinski, Alexi Runnels, Pamela Russell, Sarah Ruuska, Kathleen Ryan, Ester Cerdeira Sabino, Danish Saleheen, Shabnam Salimi, Sejal Salvi, Steven Salzberg, Kevin Sandow, Vijay G. Sankaran, Jireh Santibanez, Karen Schwander, David Schwartz, Frank Sciurba, Christine Seidman, Jonathan Seidman, Frédéric Sériès, Vivien Sheehan, Stephanie L. Sherman, Amol Shetty, Aniket Shetty, Wayne Hui-Heng Sheu, M. Benjamin Shoemaker, Brian Silver, Edwin Silverman, Robert Skomro, Jennifer Smith, Josh Smith, Nicholas Smith, Tanja Smith, Sylvia Smoller, Beverly Snively, Michael Snyder, Tamar Sofer, Nona Sotoodehnia, Adrienne M. Stilp, Garrett Storm, Elizabeth Streeten, Jessica Lasky Su, Yun Ju Sung, Jody Sylvia, Adam Szpiro, Daniel Taliun, Hua Tang, Margaret Taub, Matthew Taylor, Simeon Taylor, Marilyn Telen, Timothy A. Thornton, Machiko Threlkeld, Lesley Tinker, David Tirschwell, Sarah Tishkoff, Hemant Tiwari, Catherine Tong, Russell Tracy, Michael Tsai, Dhananjay Vaidya, David Van Den Berg, Peter VandeHaar, Scott Vrieze, Tarik Walker, Robert Wallace, Avram Walts, Fei Fei Wang, Heming Wang, Jiongming Wang, Karol Watson, Jennifer Watt, Daniel E. Weeks, Joshua Weinstock, Bruce Weir, Scott T. Weiss, Lu-Chen Weng, Jennifer Wessel, Cristen Willer, Kayleen Williams, L. Keoki Williams, Carla Wilson, James Wilson, Lara Winterkorn, Quenna Wong, Joseph Wu, Huichun Xu, Lisa Yanek, Ivana Yang, Ketian Yu, Seyedeh Maryam Zekavat, Yingze Zhang, Snow Xueyan Zhao, Wei Zhao, Xiaofeng Zhu, Michael Zody, and Sebastian Zoellner.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal