

Key Points
STAT signaling is dispensable for survival of IgH-CRLF2-r Ph-like ALL cells.
A precision medicine approach based on mutational status, namely of RAS, is key for treatment of IgH-CRLF2-r Ph-like ALL.

Abstract
Acute lymphoblastic leukemia (ALL) harboring the IgH-CRLF2 rearrangement (IgH-CRLF2-r) exhibits poor clinical outcomes and is the most common subtype of Philadelphia chromosome-like acute lymphoblastic leukemia (Ph-like ALL). While multiple chemotherapeutic regimens, including ruxolitinib monotherapy and/or its combination with chemotherapy, are being tested, their efficacy is reportedly limited. To identify molecules/pathways relevant for IgH-CRLF2-r ALL pathogenesis, we performed genome-wide CRISPR-Cas9 dropout screens in the presence or absence of ruxolitinib using 2 IgH-CRLF2-r ALL lines that differ in RAS mutational status. To do so, we employed a baboon envelope pseudotyped lentiviral vector system, which enabled, for the first time, highly efficient transduction of human B cells. While single-guide RNAs (sgRNAs) targeting CRLF2, IL7RA, or JAK1/2 significantly affected cell fitness in both lines, those targeting STAT5A, STAT5B, or STAT3 did not, suggesting that STAT signaling is largely dispensable for IgH-CRLF2-r ALL cell survival. We show that regulators of RAS signaling are critical for cell fitness and ruxolitinib sensitivity and that CRKL depletion enhances ruxolitinib sensitivity in RAS wild-type (WT) cells. Gilteritinib, a pan-tyrosine kinase inhibitor that blocks CRKL phosphorylation, effectively killed RAS WT IgH-CRLF2-r ALL cells in vitro and in vivo, either alone or combined with ruxolitinib. We further show that combining gilteritinib with trametinib, a MEK1/2 inhibitor, is an effective means to target IgH-CRLF2-r ALL cells regardless of RAS mutational status. Our study delineates molecules/pathways relevant for CRLF2-r ALL pathogenesis and could suggest rationally designed combination therapies appropriate for disease subtypes.
Introduction
Philadelphia chromosome-like acute lymphoblastic leukemia (Ph-like ALL, also known as BCR-ABL1-like ALL) is a disease entity of B-cell ALL (B-ALL) that exhibits a gene expression profile similar to that of Philadelphia chromosome-positive ALL (Ph+ ALL).1-4 Ph-like ALL comprises 15% to 30% of childhood and adult B-ALL and generally exhibits poor clinical outcomes.4-7 Ph-like ALL is categorized into 2 disease subtypes: ABL-class and CRLF2/JAK pathway types, both of which harbor gene alterations that constitutively activate cytokine/growth factor-related signals.4,8 The ABL-class type harbors alterations in genes encoding tyrosine kinases, such as ABL1, ABL2, CSF1R, PDGFRA, and PDGFRB, while the CRLF2/JAK pathway type harbors rearrangements of genes such as CRLF2, EPOR, and JAK2.4,8
Ph-like ALL with CRLF2 rearrangements (IgH-CRLF2 and P2RY8-CRLF2) comprises more than half of Ph-like ALL cases,5,6,8 and those cases exhibit worse prognosis than those without CRLF2 rearrangements.5,6,9 Notably, nearly 60% of CRLF2-rearranged (CRLF2-r) ALL cases harbor pathogenic mutation(s) in JAK2 and/or Ras-related genes, including NRAS, KRAS, PTPN11, and NF1.10,11 The frequency of IgH-CRLF2-r ALL is reportedly more than twice that of P2RY8-CRLF2 and increases with age.8
Tyrosine kinase inhibitor (TKI)-based treatment regimens are effective in treating ABL-class type Ph-like ALL12; however, no standard regimen has been established for the CRLF2/JAK pathway type. While ruxolitinib, a JAK1/2 inhibitor, has been tested in preclinical models, and ruxolitinib monotherapy and/or its combination with chemotherapy are being tested in clinical trials,8 their efficacy is reportedly limited.13 Thus, novel approaches are needed to treat CRLF2/JAK pathway type Ph-like ALL, in particular for CRLF2-r ALL.5,6,9
In this study, we performed genome-wide CRISPR/Cas9 functional screens with or without ruxolitinib using 2 IgH-CRLF2-r ALL lines (MUTZ5 and KOPN49), which differ in RAS mutational status. We employed a baboon envelope pseudotyped lentiviral vector (BaEV-LV) system,14 which, for the first time, enabled high transduction efficiency amenable for genome-wide CRSPR/Cas9 screens. Accordingly, we identified molecules/pathways necessary for the survival of IgH-CRLF2-r ALL cells, enabling us to propose novel combination therapies against the disease.
Materials and methods
Cell lines
B-ALL cell lines were cultured in a medium supplemented with 1% penicillin streptomycin (Life Technologies) at 37°C in 5% CO2. MUTZ5 and MHH-CALL4 were purchased from the DSMZ and cultured in RPMI medium (FUJIFILM Wako Pure Chemical Corporation) supplemented with 20% fetal bovine serum (FBS) (Ω Scientific). KOPN49 cells15 were cultured in RPMI medium with 10% FBS. TVA1 cells16 (a kind gift from David A. Fruman), which harbor the ETV6-ABL1 fusion, were cultured in Iscove's Modified Dulbecco's Media (IMDM) (Life Technologies) with 20% FBS.
BaEV lentiviral transduction
HEK293T cells were cultured in DMEM (FUJIFILM Wako Pure Chemical Corporation) supplemented with 10% FBS and 1% penicillin streptomycin in 10 cm dishes. HEK293T cells were transfected at 80% confluence in 10 mL media with 8.6 µg psPAX2 (Addgene), 7.0 µg BaEVTR,17 and 8.6 μg lentiviral vectors using 60 µg linear polyethylenimine (Polysciences). The medium was changed 8 hours after transfection. Lentiviral supernatants were harvested at 48 and 72 hours posttransfection and concentrated by ultracentrifugation (100 000g for 2 hours at 4°C with a Hitachi CS100FNX S50A-2254 rotor). ALL cells were plated into 12 well plates (1-3 × 106 cells per well) with the medium supplemented with 8 μg/mL polybrene (Santa Cruz Biotechnology) and spin-infected at 2000 rpm for 2 hours at 37°C.
Generation of Cas9-expressing ALL cells
MUTZ5, KOPN49, and TVA1 cells were transduced with Cas9 via lentivirus encoding both the S. pyogenes Cas9 protein and the blasticidin resistance gene (lentiCas9-Blast, Addgene plasmid 52962). Blasticidin selection was initiated 24 hours later.
CRISPR screen
The Brunello library was purchased from Addgene (plasmid #73179). It includes 77 441 single-guide RNAs (sgRNAs), an average of 4 sgRNAs per gene, and 1000 nontargeting control sgRNAs. Plasmid pool preparation and sgRNA library transduction were performed as described.18 Cells were treated with puromycin 24 hours after transduction (on day 1), and 4 × 107 cells were harvested 2 days later (on day 3) to obtain input DNA. Cells were maintained for 7 days after puromycin selection and then divided into vehicle (DMSO)- or 100 nM Ruxolitinib-treated groups and incubated 10 more days. Cells were then harvested, and genomic DNA extracted. Screens were performed in duplicate in each condition.
Library preparation for next-generation sequencing
Library preparation was performed as described.18 sgRNA inserts were PCR-amplified from 110 μg genomic DNA using Herculase 2 fusion DNA polymerase (Agilent). Resulting PCR products were purified and sequenced on a NextSeq500 sequencer (Illumina) to assess changes in sgRNA abundance between initial and final cell populations and between cell populations treated with or without ruxolitinib (supplemental Table 2 available on the Blood Web site).
Domain-saturating mutagenesis scan
All NGG-restricted sgRNA targeting coding exons of CRLF2, JAK1, JAK2, JAK3, STAT3, STAT5A, STAT5B, IL7R were designed using DNA-Striker (supplemental Table 3).19 Five hundred nontargeting sgRNAs were included as the negative control. The sgRNA oligo pool was purchased from Integrated DNA Technologies. Each sgRNA oligo was cloned using Gibson Assembly Master Mix (New England Biolabs) into the lentiGuide-Puro plasmid (Addgene plasmid 52963), PCR-purified, and dephosphorylated. Electrocompetent cells (E cloni; Lucigen) were transformed with Gibson Assembly products, and sufficient colonies were isolated to ensure ∼50× coverage of the library. The plasmid library was deep-sequenced using Miseq to confirm representation. Read counts from final and initial time points were normalized to control nontargeting guides via the MAGeCK count function,20 which was used to calculate log2 fold-changes in guide abundance (supplemental Table 4). Guides were then mapped to the protein by mapping the double-stranded break site to the corresponding codon. Lastly, scores for each amino acid were then mapped onto each structure publicly available in the Protein Data Bank.
CRISPR/Cas9-mediated gene knockout
Two individual sgRNA sequence oligos derived from the Brunello library for each of JAK2, CRLF2, CRKL, and FLT3 were cloned into lentiGuide-Puro (Addgene) and/or lentiGuide-Crimson (Addgene plasmid 70683). Lentivirus transduction was performed as described above.
Immunoblotting
Cells were lysed with RIPA buffer (Nakarai) supplemented with 1% protease inhibitor and phosphatase inhibitor. Lysates were resolved by SDS-PAGE and transferred to PVDF membranes. Immunoblotting was performed with the following antibodies: JAK2 (3230; Cell Signaling Technology [CST]), CRLF2 (12-5499-82; Invitrogen), FLT3 (3462; CST), pSTAT5Y694 (4322; CST), STAT5 (94205; CST), pERK1/2T202/Y204 (4370; CST), ERK1/2 (4695; CST), pAKTS473 (4060; CST), AKT (4691; CST), pS6S235/236 (4858; CST), pS6S240/244 (5364; CST), S6 (2217; CST), pCRKLY207 (3181; CST), CRKL (38710; CST), and β-actin (A1978; Sigma-Aldrich). For western analysis of multiple proteins blotted onto a single membrane, we cut membranes into horizontal strips based on target protein molecular weight and performed antibody staining of separate strips. To assess target protein phosphorylation, we first performed antibody staining with phosphospecific antibodies and then stripped the membrane (using WB Stripping Solution, Nacalai) and reprobed it using an antibody that detects total target protein. Densitometric analysis of western blot signal intensity was performed using ImageJ software.
Cell viability and apoptosis assays
Cells were treated with ruxolitinib (100 nM or 500 nM) (LC Laboratories), gilteritinib (500 nM) (Selleck), trametinib (500 nM) (Selleck), or their combinations. Cell viability was assessed using a Cell Counting Kit-8 (DOJINDO). Annexin V Binding Buffer (BD Biosciences) and FITC-Annexin V antibody (BioLegend) were used to detect apoptotic cells.
Patient samples
Four de novo IgH-CRLF2-r ALL samples were obtained at Kyushu University Hospital under approved institutional research protocols after obtaining written informed consent, in accordance with the Declaration of Helsinki.
Mutational analysis
Somatic mutations in cells were assessed via an in-house gene panel test that targets 391 genes relevant for hematological/malignancies. Variants are called using the Genomon pipeline (https://genomon-project.github.io/GenomonPagesR/).
Xenotransplantation
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice were purchased from Charles River Laboratories. Mice were bred and maintained in individual ventilated cages and fed autoclaved food and water at the Kyushu University Animal Facility. All mice were bred and maintained under specific pathogen-free conditions. All experiments were conducted following guidelines of the Institutional Animal Committee of Kyushu University. ALL cells were injected into irradiated (220 cGy) NSG mice via the tail vein. After transplantation, mice were given sterile water containing prophylactic enrofloxacin (Baytril; Bayer HealthCare). DMSO, ruxolitinib (60 mg/kg),21-23 gilteritinib (30 mg/kg),24,25 trametinib (1 mg/kg),26,27 or their combination was administered intraperitoneally once daily for 14 days.
Results
A CRISPR/Cas9 screen identifies signaling pathways relevant for survival of IgH-CRLF2-r ALL cells
MUTZ5, a IgH-CRLF2-r ALL line, was transduced with the Cas9 nuclease using a BaEV-LV.14 We then performed genome-wide CRISPR/Cas9 dropout screens using the Brunello library.28 Changes in sgRNAs abundance before and after the 17-day culture period were assessed using the MAGeCK MLE program20 (Figure 1A).
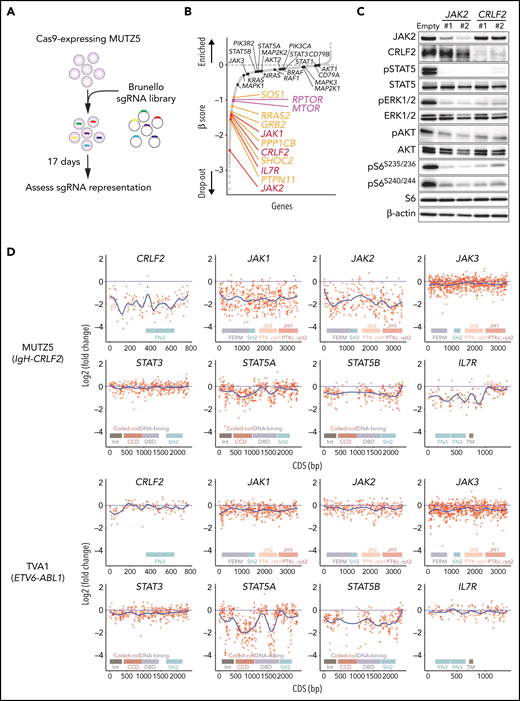
Genome-wide CRISPR-Cas9 screens identify genes/pathways relevant for IgH-CRLF2-r ALL cell survival. (A) Schematic representation of the CRISPR-Cas9 screen. MUTZ5 cells stably expressing Cas9 were transduced with the Brunello library, selected in puromycin, and cultured for 17 days. (B) Genes were significantly enriched after the 17-day incubation period. Genes essential for cell fitness are represented by negative β scores. Genes encoding positive regulators of the RAS (orange) or mTORC1 (purple) pathways were identified as essential genes. Genes encoding proteins involved in JAK/STAT signaling are indicated. (C) MUTZ5 cells stably expressing sgRNAs targeting JAK2 or CRLF2 were generated, and the activation status of signaling factors involved in JAK/STAT, RAS, and mTORC1 pathways was assessed via western blot. (D) Saturation mutagenesis scan was performed by targeting coding exons of indicated genes. TVA1 cells, which harbor the ETV6-ABL1 fusion, served as controls. Read counts from final and initial time points were normalized to nontargeting guides, and log2 fold-change in guide abundance was calculated.
Genome-wide CRISPR-Cas9 screens identify genes/pathways relevant for IgH-CRLF2-r ALL cell survival. (A) Schematic representation of the CRISPR-Cas9 screen. MUTZ5 cells stably expressing Cas9 were transduced with the Brunello library, selected in puromycin, and cultured for 17 days. (B) Genes were significantly enriched after the 17-day incubation period. Genes essential for cell fitness are represented by negative β scores. Genes encoding positive regulators of the RAS (orange) or mTORC1 (purple) pathways were identified as essential genes. Genes encoding proteins involved in JAK/STAT signaling are indicated. (C) MUTZ5 cells stably expressing sgRNAs targeting JAK2 or CRLF2 were generated, and the activation status of signaling factors involved in JAK/STAT, RAS, and mTORC1 pathways was assessed via western blot. (D) Saturation mutagenesis scan was performed by targeting coding exons of indicated genes. TVA1 cells, which harbor the ETV6-ABL1 fusion, served as controls. Read counts from final and initial time points were normalized to nontargeting guides, and log2 fold-change in guide abundance was calculated.
CRLF2 and IL7R heterodimerize upon TSLP binding and recruit JAK2 and JAK1 kinases, respectively.29,30 As expected, the abundance of sgRNAs targeting CRLF2, IL7R, JAK1, or JAK2 significantly decreased after the 17-day culture period (Figure 1B). Depletion of either JAK2 or CRLF2 significantly decreased downstream signaling through JAK-STAT, RAS-RAF-MEK-ERK, and PI3K-AKT-mTORC1 pathways, suggesting that these pathways primarily depend on CRLF2/JAK2-mediated signals (Figure 1C; supplemental Figure 1). Genes encoding positive regulators of RAS-RAF-MEK-ERK signaling (RAS pathway), including PTPN11, SHOC2, PPP1CB, GRB2, RRAS2, and SOS1, were significantly enriched among dropout genes. Similarly, genes encoding components of the mTORC1 complex (MTOR and RPTOR) were also identified as dropout genes (Figure 1B). In contrast, the abundance of sgRNAs targeting STAT5A, STAT5B, STAT3, or STAT1 barely changed, suggesting that the STAT signaling is largely dispensable for MUTZ5 cell survival (Figure 1B).
To confirm these findings, we next performed a negative selection CRISPR-Cas9 mutagenesis scan targeting coding exons of genes encoding JAK/STAT signaling components.31 TVA1, an ABL-class type Ph-like ALL line harboring the ETV6-ABL1 fusion,16 served as control. sgRNAs targeting CRLF2, JAK1, JAK2, or IL7R were depleted broadly across their coding exons in MUTZ5 but not TVA1 cells (Figure 1D). sgRNAs targeting STAT5A and STAT5B exons, specifically those encoding the DNA binding- and coiled-coiled-domains, were significantly depleted in TVA1 but not MUTZ5 cells (Figure 1D). Collectively, these observations confirm that STATs are largely dispensable for MUTZ5 cell survival, although STAT5 proteins are constitutively phosphorylated in that line (Figure 1C).
RAS activation leads to ruxolitinib resistance in MUTZ5 cells
To identify genes relevant for ruxolitinib resistance and those that exhibit synthetic lethal interaction with ruxolitinib treatment, we performed a genome-wide CRISPR/Cas9 screen in MUTZ5 cells in the presence of ruxolitinib or vehicle (DMSO) and assessed changes in abundance of each sgRNA using the MAGeCK MLE20 and DrugZ32 programs (Figure 2A). In this analysis, ruxolitinib was added to the medium at the IC50 concentration (supplemental Figure 2A-B). sgRNAs targeting LZTR1 (leucine zipper-like transcription regulator 1), which encodes an adaptor that functions in proteasomal degradation of RAS protein,33 were significantly enriched in the presence of ruxolitinib (Figure 2B-C). Similarly, NF1 and DUSP6, which encode negative regulators of the RAS pathway, were also among the top hits of ruxolitinib resistance genes (Figure 2B-C). These data suggest that the RAS pathway plays a major role in the survival and ruxolitinib sensitivity of MUTZ5 cells.
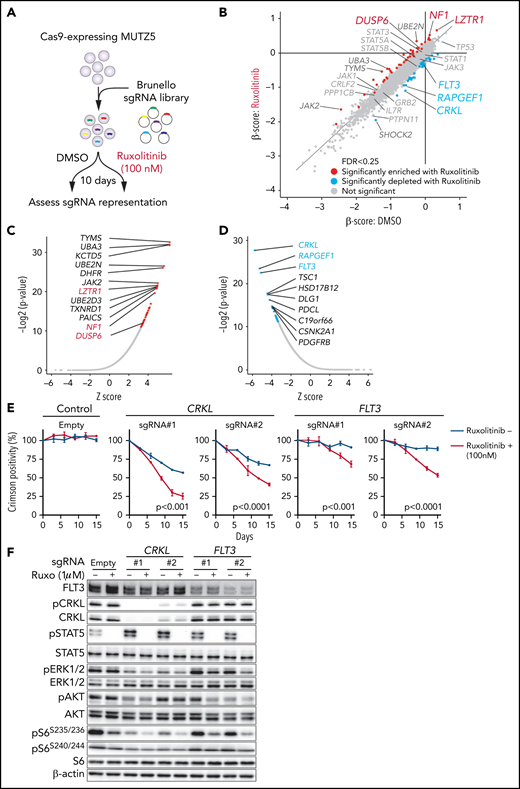
CRISPR screens identify genes relevant for ruxolitinib sensitivity. (A) Schematic representation of the CRISPR-Cas9 screen in the presence or absence of ruxolitinib. MUTZ5 cells were treated with ruxolitinib 7 days after library transduction and cultured for 10 more days. sgRNA abundance before and after culture was assessed as described.18 (B) Dot graph representing β scores of 19 114 genes in the presence or absence of ruxolitinib. Genes whose β scores exhibited statistically significant change upon ruxolitinib treatment (FDR <0.25) are depicted in red (sgRNA more enriched with ruxolitinib) or blue (sgRNA more depleted with ruxolitinib). (C) Genes whose sgRNA abundance was significantly altered by ruxolitinib treatment were assessed using the DrugZ program.32 Genes that possess positive (C) or negative (D) z scores are plotted according to statistical values. (E) MUTZ5 cells were transduced with a lentivirus vector encoding a sgRNA and the Crimson cassette and cultured with or without 100 nM ruxolitinib, which is the ruxolitinib concentration used for the CRISPR screen shown in (A). Crimson-positive cell fractions were measured at indicated times by FACS. At each time point, that proportion was normalized to the number of Crimson-positive cells present at day 0 (2 days after transduction). Empty vector served as control. Each condition was assessed in triplicate, and data are represented as means ± SD. P values were calculated using an unpaired t test. (F) MUTZ5 clones stably expressing sgRNA against CRKL or FLT3 were established, treated with or without ruxolitinib (Ruxo) (1 μM, 2 hours) as described43,50,58,65 and analyzed by western blotting.
CRISPR screens identify genes relevant for ruxolitinib sensitivity. (A) Schematic representation of the CRISPR-Cas9 screen in the presence or absence of ruxolitinib. MUTZ5 cells were treated with ruxolitinib 7 days after library transduction and cultured for 10 more days. sgRNA abundance before and after culture was assessed as described.18 (B) Dot graph representing β scores of 19 114 genes in the presence or absence of ruxolitinib. Genes whose β scores exhibited statistically significant change upon ruxolitinib treatment (FDR <0.25) are depicted in red (sgRNA more enriched with ruxolitinib) or blue (sgRNA more depleted with ruxolitinib). (C) Genes whose sgRNA abundance was significantly altered by ruxolitinib treatment were assessed using the DrugZ program.32 Genes that possess positive (C) or negative (D) z scores are plotted according to statistical values. (E) MUTZ5 cells were transduced with a lentivirus vector encoding a sgRNA and the Crimson cassette and cultured with or without 100 nM ruxolitinib, which is the ruxolitinib concentration used for the CRISPR screen shown in (A). Crimson-positive cell fractions were measured at indicated times by FACS. At each time point, that proportion was normalized to the number of Crimson-positive cells present at day 0 (2 days after transduction). Empty vector served as control. Each condition was assessed in triplicate, and data are represented as means ± SD. P values were calculated using an unpaired t test. (F) MUTZ5 clones stably expressing sgRNA against CRKL or FLT3 were established, treated with or without ruxolitinib (Ruxo) (1 μM, 2 hours) as described43,50,58,65 and analyzed by western blotting.
CRKL depletion sensitizes MUTZ5 cells to ruxolitinib
We next searched for genes whose loss sensitizes MUTZ5 cells to ruxolitinib. Depletion of CRKL, which encodes the adaptor protein CRKL (CRK-like),34 or RAPGEF1, which encodes Rap Guanine Nucleotide Exchange Factor 1, also known as C3G, exhibited synthetic lethal interaction with ruxolitinib treatment (Figure 2B-D). Of note, CRKL/RAPGEF forms a complex and activates RAP1 (RAS-proximate-1), a small GTPase that induces ERK activation,35-38 and RAP1-mediated signals are reportedly activated in IgH-CRLF2-r ALL cells.39 To confirm these results, we depleted CRKL or another synthetic lethal gene, FLT3 (Figure 2D), via lentivirus-based Crimson-tagged sgRNA and assessed effects on cell growth over 2 weeks with or without ruxolitinib. As expected, cells expressing sgRNA targeting CRKL or FLT3, but not those transduced with empty vector, demonstrated a proliferative disadvantage relative to nontransduced cells in the presence of ruxolitinib (Figure 2E). We next assessed the status of downstream signaling pathways in CRKL- or FLT3-depleted cells. To do so, we transduced MUTZ5 cells with sgRNAs targeting CRKL or FLT3 and determined phosphorylation status of STAT5, AKT, S6, and ERK1/2 proteins by western blot. That analysis revealed reduced levels of the ribosomal protein s6 phosphorylated at S235/236 sites (pS6S235/236), a modification that reportedly depends on both ERK and mTORC1 activity,40 and of phosphorylated extracellular signal-regulated kinase 1/2 (pERK1/2), a marker of RAS activation, in CRKL or FLT3 knockout (KO) relative to control cells. Both effects were more robust when CRKL or FLT3 KO cells were grown in the presence of ruxolitinib. By contrast, relative to controls, levels of S6 protein phosphorylated at S240/244 (pS6S240/244), a modification primarily dependent on mTORC1 activity,40 were barely changed among experimental conditions (Figure 2F; supplemental Figure 1). Collectively, these analyses suggest that inactivation of CRKL/RAPGEF1 or FLT3 signaling sensitizes MUTZ5 cells to ruxolitinib.
Gilteritinib treatment alone or combined with ruxolitinib suppresses IgH-CRLF2-r ALL cell proliferation
Our data suggest that ruxolitinib treatment in combination with CRKL/RAPGEF1 and/or FLT3 depletion suppresses MUTZ5 cell proliferation. Gilteritinib is an FDA-approved FLT3 inhibitor that also reportedly blocks multiple other kinases.24,41,42 Since there are no effective means to pharmacologically block CRKL function34 and FLT3 depletion enhances ruxolitinib sensitivity (Figure 2E), we assessed the effects of gilteritinib or a gilteritinib/ruxolitinib combination on activation of signaling factors in 3 IgH-CRLF2-r ALL lines. Of note, treatment with Quizartinib, a FLT3 inhibitor,41 did not decrease pCRKL levels relative to untreated samples (supplemental Figure 3). Gene panel testing indicated that all 3 lines harbored a JAK2 mutation (R683G or I682F) as well as mutations commonly observed in IGH-CRLF2-r ALL, including in IKZF1, TP53, and ETV6 (supplemental Table 1). As expected, ruxolitinib treatment reduced pSTAT5 levels in all 3 lines, while ERK1/2 activation was barely affected (Figure 3A; supplemental Figure 1). We also observed slightly reduced pS6 levels in all lines following ruxolitinib treatment, as previously reported.43 CRKL was constitutively phosphorylated at a steady state in both untreated and ruxolitinib-treated IgH-CRLF2-r ALL lines, while gilteritinib treatment alone reduced levels of phosphorylated CRKL (pCRKL) (Figure 3A; supplemental Figure 1). Gilteritinib treatment, either alone or combined with ruxolitinib, also significantly decreased pSTAT, pERK1/2, pAKT, and pS6S235/236 levels in all lines, although effects on pERK1/2 and pS6S235/236 levels were weaker in KOPN49 cells, a KRAS-mutated ALL line. pS6240/244 levels were slightly reduced following gilteritinib treatment in all lines tested (Figure 3A; supplemental Figure 1). We next assessed cell signaling after gilteritinib and/or ruxolitinib treatment in 4 primary IgH-CRLF2 ALL samples that harbor varying combinations of JAK2 and/or NRAS mutations (supplemental Table 1). As seen in cell lines, CRKL was phosphorylated in all samples, and gilteritinib treatment, either alone or combined with ruxolitinib, effectively downregulated pCRKL levels (Figure 3B; supplemental Figure 1). Only 1 sample (BALL142) was positive for pSTAT5 staining, and those levels decreased after either ruxolitinib or gilteritinib treatment. pAKT was not detected in any samples (not shown). As expected, ruxolitinib treatment alone had minimal effects on pERK1/2 and pS6 status in all 4 samples, while effects of gilteritinib or gilteritinib/ruxolitinib treatment on pERK1/2 and pS6 levels varied from sample to sample (Figure 3B; supplemental Figure 1). Of note, in RAS-mutated cases (such as KOPN49 in Figure 3A or BALL160 in Figure 3B), pERK1/2 levels were reduced only slightly following gilteritinib or gilteritinib/ruxolitinib treatment.
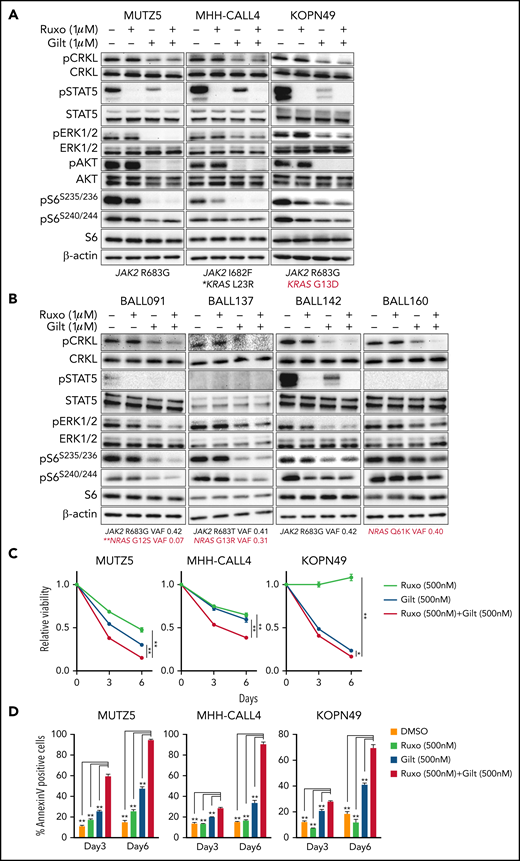
The gilteritinib/ruxolitinib combination effectively kills IgH-CRLF2-r ALL cells. Cells were treated with vehicle, gilteritinib (Gilt) (1 μM), ruxolitinib (Ruxo) (1 μM),43,50,58,65 or both, and protein samples were obtained 2 hours after treatment. Patient samples were cultured in RPMI medium supplemented with 20% FBS. Western blots were performed using antibodies against indicated proteins. Three cell lines (A) and 4 primary samples (B), all of which harbor the IgH-CRLF2 rearrangements, were used. Mutation status of JAK2 and RAS is indicated below. *Pathogenic significance of this mutation is unclear (COSMIC ID: COSM303853). **NRAS G12S mutation was detected only in a minor fraction (VAF 0.07). (C) Cells were treated with gilteritinib (500 nM), ruxolitinib (500 nM), or both and cultured for 6 days. Viability of drug-treated relative to vehicle (DMSO)-treated cells is plotted. Each condition was assessed in triplicate, and data are represented as means ± SD. Cells were treated with ruxolitinib at 500 nM, which represented the IC50 when MUTZ5 cell viability was assessed after 6 days of culture with drug. (D) Proportions of apoptotic cells were assessed by Annexin V stain via FACS on days 3 and 6 after DMSO or drug treatment. Each condition was assessed in triplicate, and data are represented as means + SD. P values were calculated using one-way ANOVA with Dunnett’s posttest for multiple comparisons. *P < .05, **P < .0001.
The gilteritinib/ruxolitinib combination effectively kills IgH-CRLF2-r ALL cells. Cells were treated with vehicle, gilteritinib (Gilt) (1 μM), ruxolitinib (Ruxo) (1 μM),43,50,58,65 or both, and protein samples were obtained 2 hours after treatment. Patient samples were cultured in RPMI medium supplemented with 20% FBS. Western blots were performed using antibodies against indicated proteins. Three cell lines (A) and 4 primary samples (B), all of which harbor the IgH-CRLF2 rearrangements, were used. Mutation status of JAK2 and RAS is indicated below. *Pathogenic significance of this mutation is unclear (COSMIC ID: COSM303853). **NRAS G12S mutation was detected only in a minor fraction (VAF 0.07). (C) Cells were treated with gilteritinib (500 nM), ruxolitinib (500 nM), or both and cultured for 6 days. Viability of drug-treated relative to vehicle (DMSO)-treated cells is plotted. Each condition was assessed in triplicate, and data are represented as means ± SD. Cells were treated with ruxolitinib at 500 nM, which represented the IC50 when MUTZ5 cell viability was assessed after 6 days of culture with drug. (D) Proportions of apoptotic cells were assessed by Annexin V stain via FACS on days 3 and 6 after DMSO or drug treatment. Each condition was assessed in triplicate, and data are represented as means + SD. P values were calculated using one-way ANOVA with Dunnett’s posttest for multiple comparisons. *P < .05, **P < .0001.
We next assessed the effects of ruxolitinib, gilteritinib, and their combination on cell viability using the 3 IgH-CRLF2 ALL lines. Ruxolitinib treatment alone had growth-suppressive effects in MUTZ5 and MHH-CALL4 lines, but not KOPN49 cells, which harbor KRAS G13D (Figure 3C). Gilteritinib treatment, either alone or combined with ruxolitinib, reduced viability of all lines tested (Figure 3C) and also induced apoptosis, based on Annexin V staining (Figure 3D).
Gilteritinib/ruxolitinib combination treatment has antileukemia effects in a human IgH-CRLF2-r ALL xenograft model
We next assessed potential antileukemia effects of the gilteritinib/ruxolitinib combination in vivo in a mouse xenograft model of human IgH-CRLF2-r ALL. As shown in the schematic in Figure 4A, after leukemia cell injection at day 1, we determined the proportion of ALL cells in white blood cells (WBCs) in peripheral blood (PB) over time by FACS, and at day 30, a point when ALL cells comprised 0.5% to 5.0% of WBCs in PB (supplemental Figure 4), we began treatment with vehicle (n = 6), gilteritinib (n = 6), ruxolitinib (n = 6), or their combination (n = 5) for 14 more days. We monitored the proportions of ALL cells in PB over the 14-day period and then killed mice to assess the proportions of ALL cells in the spleen and bone marrow (BM). In this analysis, gilteritinib/ruxolitinib combination treatment had antileukemic activity, as evidenced by the lower leukemia burden in PB relative to the vehicle- or single drug-treatment group (Figure 4B). Furthermore, spleen weight, an indicator of leukemia burden, as well as the number of ALL cells infiltrating the spleen, were significantly lower in mice treated with gilteritinib alone or gilteritinib combined with ruxolitinib compared with those treated with vehicle or ruxolitinib alone. Overall, gilteritinib/ruxolitinib combination treatment resulted in effects superior to those elicited by gilteritinib monotherapy (Figure 4C-D). Effects of drug treatments on BM leukemic burden were less pronounced than those observed in PB and spleen (Figure 4E).
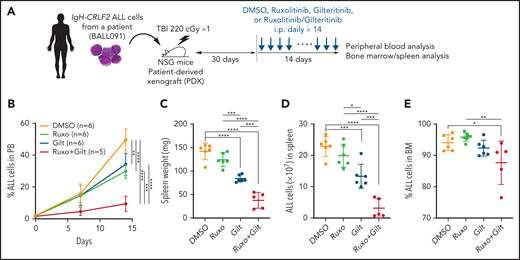
Gilteritinib combined with ruxolitinib has antileukemic activity in vivo. (A) Workflow of PDX experiments. Primary IgH-CRLF2 ALL cells (BALL091) were transferred to irradiated NSG mice, and drug treatment was initiated 30 days later. DMSO, gilteritinib (Gilt) (30 mg/kg), ruxolitinib (Ruxo) (60 mg/kg), or gilteritinib/ruxolitinib combination were administered intraperitoneally daily for 14 days. hCD19+ cells in PB were monitored over time by FACS, and leukemia burden in the spleen was assessed the day after the last drug injection. (B) Proportions of hCD19+ ALL cells in PB were examined by FACS over a 2-week period. Data are represented as means ± SD. Spleen weight (C), ALL cell numbers in spleen (D) and proportions of ALL cells in BM (E) were assessed the day after the last drug injection. Data are represented as individual values with mean ± SD bars. P values were calculated using one-way ANOVA with Tukey’s posttest for multiple comparisons. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Gilteritinib combined with ruxolitinib has antileukemic activity in vivo. (A) Workflow of PDX experiments. Primary IgH-CRLF2 ALL cells (BALL091) were transferred to irradiated NSG mice, and drug treatment was initiated 30 days later. DMSO, gilteritinib (Gilt) (30 mg/kg), ruxolitinib (Ruxo) (60 mg/kg), or gilteritinib/ruxolitinib combination were administered intraperitoneally daily for 14 days. hCD19+ cells in PB were monitored over time by FACS, and leukemia burden in the spleen was assessed the day after the last drug injection. (B) Proportions of hCD19+ ALL cells in PB were examined by FACS over a 2-week period. Data are represented as means ± SD. Spleen weight (C), ALL cell numbers in spleen (D) and proportions of ALL cells in BM (E) were assessed the day after the last drug injection. Data are represented as individual values with mean ± SD bars. P values were calculated using one-way ANOVA with Tukey’s posttest for multiple comparisons. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Gilteritinib/trametinib combination treatment has antileukemia effects against IgH-CRLF2-r ALL cells, regardless of RAS mutation status
In cell lines and primary ALL samples, including those that harbor RAS mutation, ruxolitinib had minimal effects on pERK1/2 levels (Figure 3A), and ruxolitinib monotherapy was ineffective against RAS-mutated ALL cells (Figure 3C). To examine differences in gene/pathway dependency between RAS-WT and RAS-mutated ALL, we performed a genome-wide CRISPR/Cas9 screen using KOPN49 cells, which harbor KRAS G13D and JAK2 R683G mutations (supplemental Table 1). Depletion of CRLF2, IL7R, JAK2, or JAK1 had negative effects on the growth of KOPN49 cells, while the degree of dependency on each gene was less than that seen in MUTZ5 cells (Figure 5A). While genes encoding positive regulators of RAS signaling, such as GRB2, SOS1, PTPN11, and SHOC2, were essential for MUTZ5 cell survival (Figure 1B), they were dispensable in KOPN49 cells presumably due to constitutive RAS signaling promoted by the G13D mutation (Figure 5A). The degree of dependency on MTOR was comparable between KOPN49 and MUTZ5 lines, and, as expected, KRAS depletion had negative effects on KOPN49 cell survival (Figure 5A).
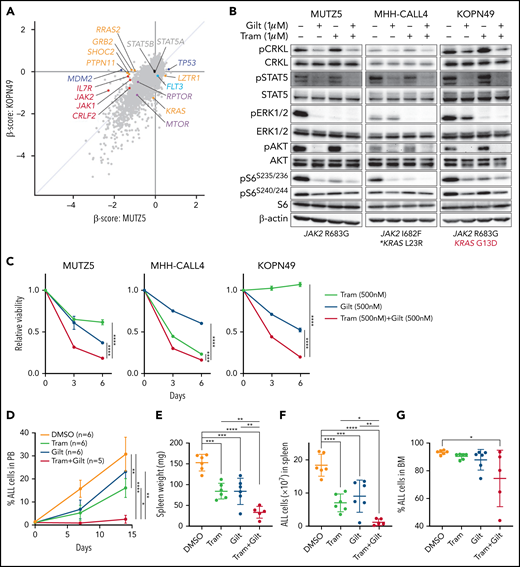
The gilteritinib/trametinib combination is effective against IgH-CRLF2-r ALL cells regardless of RAS mutation status. (A) Genome-wide CRISPR/Cas9 screen was performed using KOPN49 cells. Dot graph represents β scores obtained from KOPN49 and MUTZ5 screens. Positive regulators of the RAS pathway (depicted in orange), including RRAS2, GRAB2, SHOC2, and PTPN11, were relevant only in MUTZ5 cells. KRAS depletion had significant effects only in KOPN49 cells, which harbor the KRAS G13D mutation. (B) Cells were treated with vehicle, gilteritinib (Gilt) (1 μM), trametinib (Tram) (1 μM), or their combination, and protein samples were obtained 2 hours after treatment. Western blots were performed using 3 cell lines. (C) Growth curves were generated as described in Figure 3C. (D) The IgH-CRLF2-r ALL (BALL091) PDX model was treated with vehicle (DMSO), gilteritinib (30 mg/kg), trametinib (1 mg/kg), or the gilteritinib/trametinib combination as described in Figure 4A. Proportions of hCD19+ ALL cells in PB were examined by FACS over time. Data are represented as means ± SD. Spleen weight (E), ALL cell numbers in spleen (F), and proportions of ALL cells in BM (E) were assessed the day after the last drug injection. Data are represented as individual values with mean ± SD bars. P values were calculated using one-way ANOVA with Tukey’s posttest for multiple comparisons. *P < .05, **P < .01, ***P < .001, ****P < .0001.
The gilteritinib/trametinib combination is effective against IgH-CRLF2-r ALL cells regardless of RAS mutation status. (A) Genome-wide CRISPR/Cas9 screen was performed using KOPN49 cells. Dot graph represents β scores obtained from KOPN49 and MUTZ5 screens. Positive regulators of the RAS pathway (depicted in orange), including RRAS2, GRAB2, SHOC2, and PTPN11, were relevant only in MUTZ5 cells. KRAS depletion had significant effects only in KOPN49 cells, which harbor the KRAS G13D mutation. (B) Cells were treated with vehicle, gilteritinib (Gilt) (1 μM), trametinib (Tram) (1 μM), or their combination, and protein samples were obtained 2 hours after treatment. Western blots were performed using 3 cell lines. (C) Growth curves were generated as described in Figure 3C. (D) The IgH-CRLF2-r ALL (BALL091) PDX model was treated with vehicle (DMSO), gilteritinib (30 mg/kg), trametinib (1 mg/kg), or the gilteritinib/trametinib combination as described in Figure 4A. Proportions of hCD19+ ALL cells in PB were examined by FACS over time. Data are represented as means ± SD. Spleen weight (E), ALL cell numbers in spleen (F), and proportions of ALL cells in BM (E) were assessed the day after the last drug injection. Data are represented as individual values with mean ± SD bars. P values were calculated using one-way ANOVA with Tukey’s posttest for multiple comparisons. *P < .05, **P < .01, ***P < .001, ****P < .0001.
As noted, gilteritinib treatment significantly decreased levels of pERK1/2 and pS6S235/236 in IgH-CRLF2-r ALL cells, although effects were relatively weaker in RAS-mutated cells (Figure 3A-B). To effectively block signals downstream of mutated RAS in IgH-CRLF2-r ALL cells with RAS mutation, we evaluated the effects of gilteritinib combined with the MEK1/2 inhibitor trametinib. To do so, we treated the 3 IgH-CRLF2-r ALL lines assessed in Figure 3 with gilteritinib and/or trametinib. Trametinib, alone or combined with gilteritinib, reduced pERK1/2 levels more effectively than gilteritinib alone (Figure 5B; supplemental Figure 1). Trametinib alone had growth-suppressive effects in MUTZ5 and MHH-CALL4 cells without affecting pSTAT5 levels (Figure 5B), and those effects were more pronounced following gilteritinib/trametinib combination treatment in these 2 lines (Figure 5C). Gilteritinib treatment, either alone or combined with trametinib, reduced the viability of KOPN49 cells, while trametinib alone had no growth-suppressive effects in these cells (Figure 5C).
We next conducted a similar analysis in vivo using the xenograft model and protocol described in Figure 4A, except that mice were treated with vehicle (DMSO) (n = 6), trametinib (n = 6), gilteritinib (n = 6), or the trametinib/gilteritinib combination (n = 5). Trametinib, alone or combined with gilteritinib, significantly reduced leukemia burden in PB (Figure 5D). Furthermore, in contrast to ruxolitinib monotherapy (Figure 4C-D), trametinib or gilteritinib monotherapy had antileukemic activity, as evidenced by decreases in spleen weight and the number of ALL cells infiltrating the spleen, and as expected, the trametinib/gilteritinib combination had more pronounced antileukemic effects than either trametinib or gilteritinib monotherapy (Figure 5E-F). Effects of drug treatments on BM leukemic burden were less evident than those observed in PB and spleen (Figure 5G).
Discussion
Genome-wide CRISPR/Cas9 screening is a powerful tool to identify genes essential for cancer cell survival.18,44 The Dependency Map (DepMap: https://depmap.org/) is a data portal where researchers can interrogate cancer vulnerabilities using the CRISPR/Cas9 screen datasets of over 800 cancer cell lines, including 9 B-ALL lines (Public 21Q1). However, to our knowledge, such screens have never been performed using IgH-CRLF2-r ALL lines due, in part, to difficulties in lentivirus-mediated gene transduction of human B cells.45 Here, we employed a baboon envelope pseudotyped lentiviral vector system14 and performed, for the first time, genome-wide CRISPR/Cas9 screens in 2 IgH-CRLF2-r ALL lines, both of which harbor the JAK2 R683G but differ in RAS mutational status.
The IgH-CRLF2 rearrangement leads to “ectopic” expression of CRLF2 protein in immature B cells. CRLF2 protein is normally expressed in monocytes, basophils, and dendritic cells, but not in B cells,46,47 and in fact, B-cell development is grossly normal in Crlf2 KO mice.48 CRLF2 forms a heterodimer with IL7R (IL7 receptor α subunit) and functions as a receptor for TSLP (thymic stromal lymphopoietin). Upon TSLP binding, the CRLF2/IL7R heterodimer recruits JAK1 and JAK2, activating STAT5 in primary CD4+ T cells.29 Similarly, JAK/STAT signaling is reportedly activated in IgH-CRLF2-r ALL cells, as STAT5 is phosphorylated in these cells.49,50 Il7/Il7r-mediated signals are essential for early B-cell development in mice, and Stat5 plays a critical downstream role in this pathway. Misexpression of constitutively active STAT5A rescues differentiation defects in Il7r-null preproB cells.51 In contrast, the role of IL7/IL7R in human B-cell development is somewhat controversial,52-54 and it remains unclear whether STAT5-mediated signals are necessary for human B-cell development and/or survival of human B-ALL cells. The results of our unbiased CRISPR/Cas9 dropout screens (Figures 1B and 5A) and saturation mutagenesis scan (Figure 1D) suggest that STATs (STAT5A, STAT5B, STAT1, and STAT3) are largely dispensable for the survival of IgH-CRLF2-r ALL cells. Although functional redundancy of STAT5A and STAT5B may have affected our screening results, dependency on STAT5A or STAT5B was significantly weaker in MUTZ5 compared with TVA1 cells (Figure 1D). Our data also suggest that STAT5 phosphorylation status does not necessarily reflect its requirement in cell survival: ruxolitinib treatment failed to suppress KOPN49 cell growth but reduced pSTAT5 to undetectable levels with no effect on total protein (Figure 3A,C). Similarly, AZD1480, an ATP-competitive small-molecule inhibitor of JAK1, JAK2, and JAK3, reportedly exhibits modest antileukemia effects in vitro and in vivo, while it effectively inhibits JAK/STAT signaling in human B-ALL xenografts.55
Ph-like ALL is categorized as “ABL-class” or “CRLF2/JAK pathway” types.4,8 The ABL-class type harbors fusions involving genes encoding tyrosine kinases and frequently harbors additional mutations, such as in IKZF1, PAX5, and EBF1.10,11 In contrast, CRLF2/JAK pathway-type Ph-like ALL are more genetically heterogenous, consisting of CRLF2-r (IgH-CRLF2 and P2RY8–CRLF2), EPOR-r (IgH-EPOR etc), JAK2-r (PAX5-JAK2, BCR-JAK2, etc) and forms with mutations in genes such as IL7R, FLT3, SH2B3, JAK1, JAK3, or CRLF2.10,11 Furthermore, JAK2 mutations are also present in 40% to 60% of IgH-CRLF2 ALL cases, and mutations in the RAS-related genes KRAS, NRAS, PTPN11, and NF1 are observed in nearly half of the cases, regardless of JAK2 mutation status.11 Although various combination therapies have been tested in preclinical models,39,43,49,50,55-57 and ruxolitinib, either as monotherapy or combined with chemotherapy, is being tested in clinical trials, genomics-based precision medicine approaches are necessary to improve clinical outcomes given the genomic heterogeneity of CRLF2/JAK pathway-type ALL. Our analysis showed that RAS mutation significantly altered dependency to signal modulators downstream of the CRLF2/IL7R complex as well as ruxolitinib sensitivity. While positive regulators of the RAS pathway, such as GRB2, SOS1, PTPN11, and SHOC2, were essential for the survival of RAS WT MUTZ5 cells (Figure 1B), they were dispensable in KRAS-mutated KOPN49 cells (Figure 5A). Similarly, levels of dependency on CRLF2, IL7R, JAK2, or JAK1, all of which encode proteins upstream of RasGTPases, were significantly low in KOPN49 cells (Figure 5A). Ruxolitinib treatment had minimal effects on RAS- and/or mTORC1-related signals in RAS-mutated ALL cells (Figure 3A-B), and ruxolitinib monotherapy failed to eliminate RAS-mutated ALL cells (Figure 3C). Of note, depletion of either MTOR or RPTOR, which encode components of the mTORC1 complex, significantly altered cell fitness regardless of RAS mutation status, suggesting that the mTORC1 pathway plays a key role in the survival of IgH-CRLF2 ALL cells, as reported.43,56-58 Collectively, our data suggest that ruxolitinib may not be the appropriate treatment choice in the case of RAS-mutated IgH-CRLF2-r ALL, but rather that treatment should be stratified based on cooccurring mutations, namely those of the RAS-related genes.
Our data, as well as that of others,16,43,49,55-57 suggest that inhibiting both RAS and mTORC1 pathways is critical to treat IgH-CRLF2 ALL. In RAS WT IgH-CRLF2 ALL cells, both RAS and mTORC1 pathways are activated via IL7R/CRLF2/JAK1/2- and receptor-type tyrosine kinase (RTK)-mediated signals (Figure 6), and we show that combining ruxolitinib with gilteritinib treatment effectively blocks both pathways and has antileukemic activity (Figures 3 and 4). Although we used gilteritinib, which is a FLT3 inhibitor with relatively broad TKI activity, a different FLT3 inhibitor, such as midostaurin, would likely promote comparable effects as it also inhibits multiple kinases.41,42 In RAS-mutated IgH-CRLF2 ALL cells, ruxolitinib efficacy was limited (Figure 3). RAS reportedly activates both RAF-MEK-ERK and PI3K-AKT-mTORC1 pathways, and inhibition of one could lead to compensatory activation of the other.59 For example, resistance to MEK inhibitors is associated with PI3K-AKT-mTORC1 activation in KRAS-mutated cell lines.60 Moreover, MEK inhibition leads to RTK activation via compensatory mechanisms in breast cancer cells.61 Thus, simultaneous targeting of 2 pathways, using combined MEK and mTOR inhibitors, is a reasonable approach to treat RAS-mutated cancers, although toxicity has limited such approaches in clinical settings.59 One way to overcome this problem is to use a TKI combined with a RAS pathway inhibitor.60-62 In fact, we show that gilteritinib combined with the MEK1/2 inhibitor trametinib effectively blocked both RAS and mTORC1 pathways and had significant antileukemic activity (Figure 5). We propose that the use of the trametinib/gilteritinib combination is a reasonable approach to treat CRLF2-r ALL, regardless of RAS mutational status. In RAS-WT CRLF2-r ALL cells, trametinib blocks downstream RAS signaling by inhibiting MEK activity, while gilteritinib inhibits RTK-mediated signals upstream of RAS that positively modulate its activity (Figure 6). In RAS-mutated CRLF2-r ALL cells, the efficacy of trametinib monotherapy is limited by compensatory activation of the mTORC1 pathway (Figure 5B)59; however, gilteritinib inhibits RTK-mediated mTORC1 activation and prevents feedback RTK activation caused by MEK inhibition.61 Thus, simultaneous inhibition of MEK and RTKs via the trametinib/gilteritinib combination could block both RAS and mTORC1 pathways and promote significant antileukemic activity in RAS-mutated CRLF2-r ALL cells (Figure 6).
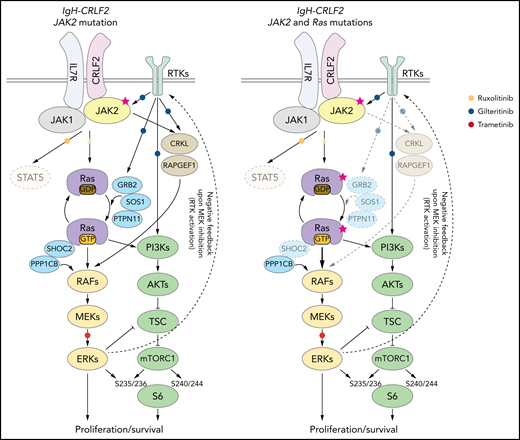
Proposed model of signaling pathways relevant to RAS-WT or RAS -mutated IgH-CRLF2-r ALL cells. STAT signaling is largely dispensable regardless of RAS mutation status. (Left) In RAS-WT/JAK2-mutated-IgH-CRLF2-r ALL cells, the IL7R/CRLF2/JAK2/JAK1 complex plays a major role in relaying signals toward downstream RAS/mTORC1 pathways. Adaptor molecules (CRKL and RAPGEF), which relay RTK-mediated signals (eg, FLT3), and modulators of the RAS/mTORC1 pathway (GRB2, SOS1, PTPN11, and SHOC2) are necessary for cell growth. (Right) By contrast, they are largely dispensable in RAS-mutated IgH-CRLF2-r ALL cells. Furthermore, RAS-mutated ALL cells depend less on the IL7R/CRLF2/JAK2/JAK1 complex than do RAS-WT cells. Signaling pathways targeted by ruxolitinib, gilteritinib, and trametinib are also depicted. Phosphorylation of S6 protein at S235/236 is mediated by both ERK and mTORC1 activity,40 while S6 phosphorylation at S240/244 is primarily mTORC1-dependent.40 The upstream RTK pathway is reportedly activated by MEK inhibition.61
Proposed model of signaling pathways relevant to RAS-WT or RAS -mutated IgH-CRLF2-r ALL cells. STAT signaling is largely dispensable regardless of RAS mutation status. (Left) In RAS-WT/JAK2-mutated-IgH-CRLF2-r ALL cells, the IL7R/CRLF2/JAK2/JAK1 complex plays a major role in relaying signals toward downstream RAS/mTORC1 pathways. Adaptor molecules (CRKL and RAPGEF), which relay RTK-mediated signals (eg, FLT3), and modulators of the RAS/mTORC1 pathway (GRB2, SOS1, PTPN11, and SHOC2) are necessary for cell growth. (Right) By contrast, they are largely dispensable in RAS-mutated IgH-CRLF2-r ALL cells. Furthermore, RAS-mutated ALL cells depend less on the IL7R/CRLF2/JAK2/JAK1 complex than do RAS-WT cells. Signaling pathways targeted by ruxolitinib, gilteritinib, and trametinib are also depicted. Phosphorylation of S6 protein at S235/236 is mediated by both ERK and mTORC1 activity,40 while S6 phosphorylation at S240/244 is primarily mTORC1-dependent.40 The upstream RTK pathway is reportedly activated by MEK inhibition.61
In Ph-like B-ALL xenograft models, efficacies of drug treatment are generally assessed by examining PB and/or spleen, not BM.10,43,57,63 We observed less robust antileukemic effects in BM relative to that seen in PB or spleen after drug treatment (Figures 4 and 5). Although we cannot yet account for these findings, several explanations come to mind. For example, the leukemia cell niche in BM could differ between humans and mice: the mouse BM microenvironment could activate signaling pathways in leukemia cells in a manner that increases drug resistance. It is also possible that drug bioavailability in mouse BM differs from that in humans.64
In summary, our genome-wide functional screens delineate genes/pathways relevant for IgH-CRLF2-r ALL, leading to rational design of combination therapies. Our study sheds light on future precision medicine approaches to treat Ph-like ALL, which currently exhibits poor clinical outcomes with available therapeutic modalities.
Acknowledgments
The authors thank the members of the Department of Medicine and Biosystemic Science and the Division of Precision Medicine at Kyushu University for assistance and helpful discussion, Shinobu Tsuzuki for advice, and Elise Lamar for critical reading of the manuscript. This work is supported, in part, by a Grant-in-Aid for Young Scientists (18K16089) (Y.S.), a Grant-in-Aid for Young Scientists (19K17859), Research Grant of KANAE Foundation, MSD Life Science Foundation, The Yasuda Medical Foundation, Mochida Memorial Foundation for Medical and Pharmaceutical Research, The Shinnihon Foundation of Advanced Medical Treatment Research, the Takeda Science Foundation (T.Y.), a Grant-in-Aid for Scientific Research (S)(16H06391) (K.A.), a Grant-in-Aid for Scientific Research (A) (17H01567 and 20H00540), AMED under grant number 18063889, and a Grant-in-Aid for Scientific Research (S)(20H05699) (T.M.).
Authorship
Contribution: K.S., Y.S., T.Y., and T.M. designed CRISPR-Cas9 screen experiments; K.S., Y.S., T.Y., J.N., K.A., and T.M. reviewed CRISPR screen data; K.S., Y.S., T.Y., H.I., T.T., and F.N. executed CRISPR-Cas9 experiments, cell biology experiments, and in vivo mouse studies; Y.S. and J.N. analyzed CRISPR saturation mutagenesis data; E.V. supervised lentiviral transduction; K.A. and T.I. provided KOPN49 cells; and K.S. and T.M. wrote the manuscript with help from all authors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Takahiro Maeda, Division of Precision Medicine, Kyushu University Graduate School of Medical Sciences, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan; e-mail: maeda.takahiro.294@m.kyushu-u.ac.jp.
There is a Blood Commentary on this article in this issue.
The online version of this article contains a data supplement
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal