

Key Points
Excess systemic heme in mice with SCD upregulates Hmox1 and promotes cardiac damage.
Elevated Hmox1 in mice with SCD exacerbates iron overload and induces cardiac ferroptosis.

Abstract
Sickle cell disease (SCD) is characterized by increased hemolysis, which results in plasma heme overload and ultimately cardiovascular complications. Here, we hypothesized that increased heme in SCD causes upregulation of heme oxygenase 1 (Hmox1), which consequently drives cardiomyopathy through ferroptosis, an iron-dependent non-apoptotic form of cell death. First, we demonstrated that the Townes SCD mice had higher levels of hemopexin-free heme in the serum and increased cardiomyopathy, which was corrected by hemopexin supplementation. Cardiomyopathy in SCD mice was associated with upregulation of cardiac Hmox1, and inhibition or induction of Hmox1 improved or worsened cardiac damage, respectively. Because free iron, a product of heme degradation through Hmox1, has been implicated in toxicities including ferroptosis, we evaluated the downstream effects of elevated heme in SCD. Consistent with Hmox1 upregulation and iron overload, levels of lipid peroxidation and ferroptotic markers increased in SCD mice, which were corrected by hemopexin administration. Moreover, ferroptosis inhibitors decreased cardiomyopathy, whereas a ferroptosis inducer erastin exacerbated cardiac damage in SCD and induced cardiac ferroptosis in nonsickling mice. Finally, inhibition or induction of Hmox1 decreased or increased cardiac ferroptosis in SCD mice, respectively. Together, our results identify ferroptosis as a key mechanism of cardiomyopathy in SCD.
Introduction
Erythroid disorders like sickle cell disease (SCD) are characterized by excess heme because of increased hemolysis.1 Increased heme in hemolytic disorders has been linked to inflammation and organ damage including cardiovascular disorders.2,3 Heme is catabolized by heme oxygenase 1 (Hmox1) to release antioxidant metabolites: carbon monoxide and biliverdin.4 However, heme catabolism by Hmox1 also releases free iron, which has been associated with oxidative stress and cardiac damage. Although Hmox1 was thought to be a protective antioxidant enzyme, recent studies have demonstrated its role in promoting iron overload in β-thalassemia5 and driving anthracycline cardiotoxicity.6 Ferroptosis is a regulated form of cell death that is characterized by iron-dependent lipid peroxidation and is distinct from other cell death processes like apoptosis.7 Although ferroptosis was first studied in cancer cells, subsequent studies have demonstrated its implications in several disorders including ischemic stroke and cardiomyopathy.6-8 Recognizing the impact of ferroptosis on metabolism and disease, we sought to examine the effects of excess heme on cardiac ferroptosis in SCD.
Methods
Mice
All animal protocols were approved by the Institutional Animal Care and Use Committees of Northeastern University, University of Massachusetts Lowell, and Beth Israel Deaconess Medical Center. We used the Townes mouse model of SCD, including HbSS sickling mice (16-24 weeks old) and their age-matched nonsickling HbAS littermates. Townes SCD transgenic mice that express humanized sickle cell genes9 were purchased from Jackson Laboratories (Bar Harbor, ME; stock #013071). Genotypes of homozygous sickling (HbSS) and heterozygous nonsickling (HbAS) littermate controls were confirmed using polymerase chain reaction (PCR). Animals were maintained on a 12:12-hour light:dark cycle and had ad libitum access to water and facility chow. Mice were euthanized at 16 to 24 weeks of age by isoflurane overdose, followed by exsanguination and tissue collection.
In vivo drug administration
Deferoxamine (DFO, 100 mg/kg), ferrostatin-1 (Fer-1, 1 mg/kg), erastin (20 mg/kg), hemin (25 mg/kg), or tin protoporphyrin-IX (SnPP, 12 mg/kg) or corresponding vehicle (saline or 5% dimethyl sulfoxide) was administered intraperitoneally to 12-week-old HbSS and HbAS mice 3 times per week for 4 weeks. Human hemopexin (Hx, 4 mg/kg in saline) was administered intraperitoneally once a week for 4 weeks to 12-week-old HbSS and HbAS mice.
Serum heme measurement
Blood was collected from the submandibular vein and centrifuged at 1200 g for 12 minutes to obtain serum. Serum heme levels were measured using a commercially available assay kit (KA1617; Abnova) as per manufacturer’s instructions.10
Serum hemopexin measurement
Serum Hx was measured using a commercially available enzyme-linked immunosorbent assay (ELISA; Aviva Systems Biology, OKIA00097) as per manufacturer’s instructions.11
Real-time qPCR
RNA was extracted from cardiac tissue using TRI reagent (Sigma-Aldrich). Single strand complementary DNA was synthesized using Maxima First Strand cDNA Synthesis Kit. Quantitative PCR (qPCR) was performed in QuantStudio 3 (Applied Biosystems) using the iTaq Universal SYBR Green Supermix (Bio-Rad). Relative messenger RNA (mRNA) levels were normalized to β-actin using the 2-ΔΔCTmethod and expressed as relative fold change as compared to HbAS mice or saline-treated HbAS mice. The primers used for real-time qPCR were: ANP, forward GAACCTGCTAGACCACCT, reverse CCTAGTCCACTCTGGGCT12; BNP, forward AAGCTGCTGGAGCTGATAAGA, reverse GTTACAGCCCAAACGACTGAC13; Ptgs2, forward CTGCGCCTTTTCAAGGATGG, reverse GGGGATACACCTCTCCACCA6; and Gpx4, forward TCTGTGTAAATGGGGACGATGC, reverse TCTCTATCACCTGGGGCTCCTC.14
Serum CK-MB measurement
Serum creatinine kinase MB (CK-MB) was measured by ELISA using a commercial assay kit (MBS705293; MyBioSource) as per manufacturer’s instructions.15
Determination of lipid peroxidation
Lipid peroxidation in heart tissues was measured using OxiSelect MDA Adduct competitive ELISA (Cell Biolabs) according to the manufacturer’s protocol.16
Western blot analysis
Protein was extracted from cardiac tissue using RIPA and separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis. The proteins were transferred to a PVDF membrane. Membranes were incubated overnight at 4°C with primary antibodies (Hmox1, Abcam ab13248, 1:1000; ALAS1, Abcam ab84962, 1:500; caspase-3, Abcam ab13847, 1:2000; Bax, Cell Signaling Technologies #2772, 1:1000; Bcl-2, Cell Signaling Technologies #3498, 1:1000; and tubulin, Abcam ab4074, 1:2000).
Statistical analyses
Data were presented as mean ± standard error of the mean. Groups were compared using the Student t test for 2-group comparison or 1-way analysis of variance (ANOVA) with Tukey’s honestly significant difference (HSD) post hoc test for comparison among 3 to 6 groups. Differences with P < .05 were considered significant.
Results and Discussion
Similar to patients with SCD, HbSS mice displayed increased serum heme levels (Figure 1A) and depleted Hx (Figure 1B), the heme scavenger protein,17,18 which resulted in increased non-Hx bound (free) heme levels (Figure 1C). Considering the association between serum heme and cardiac inflammation and hypertrophy,19 we determined if excess heme promotes cardiomyopathy in SCD. HbSS mice displayed increased heart weight (supplemental Figure 1A available on the Blood Web site) and cardiac expression of the hypertrophic markers atrial and brain natriuretic peptide (ANP and BNP; Figure 1D) and serum CK-MB (Figure 1E). In patients with SCD, heart disease manifests as hypertrophic cardiomyopathy with preserved systolic function.20 Similarly, echocardiography revealed that HbSS mice developed left ventricular hypertrophy (supplemental Figure 1B). Interestingly, exogenous Hx administration restored serum heme levels and corrected cardiac damage in HbSS mice, while demonstrating no effects in HbAS mice (Figure 1F,G; supplemental Figures 1D and 2G). These results indicate that cardiomyopathy in SCD is driven by excess heme, which is reversed by Hx administration.
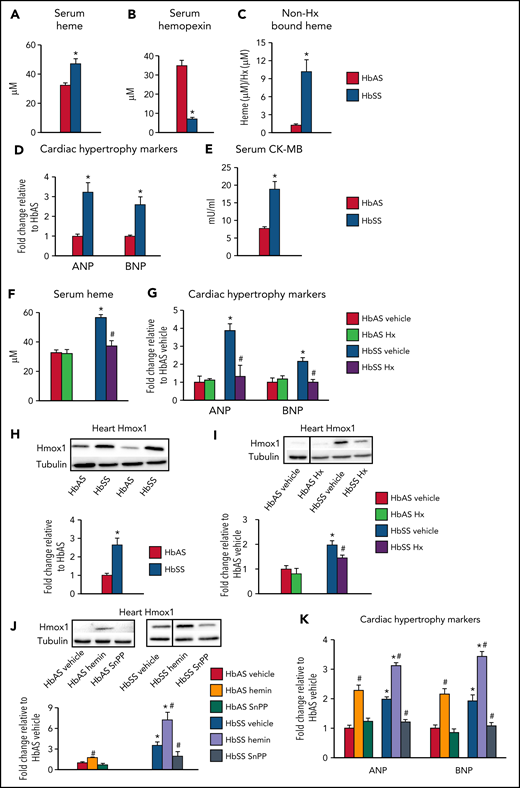
Excess heme leads to cardiomyopathy in mouse model of sickle cell disease through Hmox1 upregulation. (A-E,H) Sickling (HbSS) mice and nonsickling (HbAS) littermates (males and females, 16-24 weeks old) fed facility chow were euthanized to collect tissues. (A) Serum heme and (B) hemopexin (Hx) levels were determined by colorimetric analysis and ELISA, respectively. (C) Serum heme was normalized to hemopexin to determine relative amounts of Hx-free heme. (D) Relative mRNA expression was determined using qPCR in cardiac tissue and normalized to β-actin. (E) Serum creatine kinase-MB (CK-MB) levels were measured by ELISA. (F-G, I) HbAS and HbSS mice were injected with human Hx (4 mg/kg) once a week for 4 weeks. (F) Serum heme levels were determined by colorimetric analysis. (G) Relative mRNA expression was determined using qPCR in cardiac tissue and normalized to β-actin. (H-I) Relative protein expression of Hmox1 was determined using cardiac tissue and normalized to α-tubulin. Discontinuities between nonadjacent lanes of the same membrane were indicated by a solid line. (J-K) HbAS and HbSS mice were injected with hemin (25 mg/kg) or SnPP (12 mg/kg) intraperitoneally 3 times per week for 4 weeks, and cardiac Hmox1 expression was determined using western blot analysis. Discontinuities between nonadjacent lanes of the same membrane are indicated by a solid line. (K) Relative mRNA expression in cardiac tissue was determined by qPCR and normalized to β-actin. Results are representative of n = 7-11 per group. Data are expressed as mean ± standard error of the mean. Statistical significance was assessed using the (A-E, H) Student t test for 2-group comparison or (F-G, I-K) 1-way ANOVA followed by Tukey HSD post hoc test for comparison among 3 to 6 groups. *P < .05 vs HbAS mice of the same treatment. #P < .05 vs vehicle-treated mice of the same genotype.
Excess heme leads to cardiomyopathy in mouse model of sickle cell disease through Hmox1 upregulation. (A-E,H) Sickling (HbSS) mice and nonsickling (HbAS) littermates (males and females, 16-24 weeks old) fed facility chow were euthanized to collect tissues. (A) Serum heme and (B) hemopexin (Hx) levels were determined by colorimetric analysis and ELISA, respectively. (C) Serum heme was normalized to hemopexin to determine relative amounts of Hx-free heme. (D) Relative mRNA expression was determined using qPCR in cardiac tissue and normalized to β-actin. (E) Serum creatine kinase-MB (CK-MB) levels were measured by ELISA. (F-G, I) HbAS and HbSS mice were injected with human Hx (4 mg/kg) once a week for 4 weeks. (F) Serum heme levels were determined by colorimetric analysis. (G) Relative mRNA expression was determined using qPCR in cardiac tissue and normalized to β-actin. (H-I) Relative protein expression of Hmox1 was determined using cardiac tissue and normalized to α-tubulin. Discontinuities between nonadjacent lanes of the same membrane were indicated by a solid line. (J-K) HbAS and HbSS mice were injected with hemin (25 mg/kg) or SnPP (12 mg/kg) intraperitoneally 3 times per week for 4 weeks, and cardiac Hmox1 expression was determined using western blot analysis. Discontinuities between nonadjacent lanes of the same membrane are indicated by a solid line. (K) Relative mRNA expression in cardiac tissue was determined by qPCR and normalized to β-actin. Results are representative of n = 7-11 per group. Data are expressed as mean ± standard error of the mean. Statistical significance was assessed using the (A-E, H) Student t test for 2-group comparison or (F-G, I-K) 1-way ANOVA followed by Tukey HSD post hoc test for comparison among 3 to 6 groups. *P < .05 vs HbAS mice of the same treatment. #P < .05 vs vehicle-treated mice of the same genotype.
We then sought to explore the mechanisms of heme-mediated cardiomyopathy in SCD. Although Hx prevents heme uptake into cardiac cells,3 other cardiac heme importers like Flvcr2 could take up free heme,21 and consequently result in Hmox1 induction.4 Cardiac Hmox1 was upregulated in HbSS mice (Figure 1H). Consequently, cardiac non-heme iron, a breakdown product of heme by Hmox1, was higher in HbSS mice (supplemental Figure 1F), although cardiac heme levels were unchanged (supplemental Figure 1E). Although these results differ from patients with SCD who do not display cardiac iron loading,22 it should be noted that T2*, the most common technique to determine cardiac iron in humans, only measures iron bound to proteins like hemosiderin or ferritin, and not other redox-active forms of iron (eg, labile or free iron).23 Additionally, there was no change in cardiac heme synthesis, determined by aminolevulinic acid synthase 1, in HbSS mice (supplemental Figure 1G), suggesting that Hmox1 upregulation could primarily occur due to increased heme uptake from circulation rather than cardiac heme.3 Notably, Hx administration corrected cardiac Hmox1 upregulation and iron loading (Figure 1I; supplemental Figure 1H), supporting the idea that cardiac Hmox1 is regulated by systemic heme status.
Next, we altered Hmox1 activity using a known inducer hemin or an inhibitor SnPP, respectively (supplemental Figure 1I). Hemin depleted serum Hx (supplemental Figure 1K) and increased non-Hx bound heme levels (supplemental Figure 1L) in HbAS mice. Also, hemin increased cardiac Hmox1 (Figure 1J) and non-heme iron (supplemental Figure 1N) and induced cardiac damage in HbAS mice and exacerbated cardiomyopathy in HbSS mice (Figure 1K; supplemental Figures 1M and 2G). Conversely, SnPP corrected cardiac damage in HbSS mice, although modest cardiac iron deficiency was observed in HbAS mice (Figure 1J,K; supplemental Figures 1M and 2G). These results identify Hmox1 as a key player in promoting cardiomyopathy in SCD and suggest that modulation of Hmox1 alters the susceptibility to cardiomyopathy. Future studies using SCD × cardiac-specific Hmox1-knockout or overexpression models will delineate the roles of systemic and cardiac Hmox1 in SCD cardiomyopathy.
Although Hmox1 may play a protective role in vascular function,24,25 accumulating evidence suggests the role of Hmox1 in ferroptosis, an iron-dependent cell death.6,26,27 Thus, we examined the downstream effects of elevated cardiac Hmox1 in SCD. Consistent with ferroptotic damage, we found increased levels of malondialdehyde (MDA), a lipid peroxidation marker, and Ptgs2 (a ferroptotic marker) in HbSS hearts (supplemental Figure 2A,B). The antioxidant enzyme Gpx4 was unchanged (supplemental Figure 2B), which is consistent with a previous finding in iron-induced ferroptosis.28 Iron overload could upregulate Slc7a11, a cysteine-glutamate antiporter,29 which may explain unchanged Gpx4 levels. This suggests that iron overload ferroptosis is distinct from erastin (system Xc− inhibitor)-mediated ferroptosis.29 Moreover, Hx administration prevented lipid peroxidation and ferroptosis in HbSS mice (Figure 2A,B), suggesting that attenuation of systemic heme overload decreases cardiac ferroptosis in SCD.
![Upregulated Hmox1 promotes cardiac ferroptosis in mice with sickle cell disease. (A-B) HbAS and HbSS mice were injected with human Hx (4 mg/kg) once a week for 4 weeks. (A) Lipid peroxidation was assessed as levels of malondialdehyde (MDA) by ELISA. (B) Relative mRNA expression of ferroptotic markers was determined using qPCR and normalized to β-actin. (C-E) HbAS and HbSS mice were injected with ferroptosis inhibitors (ferrostatin-1 [Fer-1; 1 mg/kg] or deferoxamine [DFO; 100 mg/kg]) or ferroptosis inducer (erastin; 20 mg/kg) intraperitoneally 3 times per week for 4 weeks. (C) Lipid peroxidation was assessed as levels of MDA. Relative expression of (D) ferroptotic and (E) hypertrophic markers were determined using qPCR and normalized to β-actin. (F-G) HbAS and HbSS mice were injected with hemin (25 mg/kg) or SnPP (12 mg/kg) intraperitoneally 3 times per week for 4 weeks. (F) Lipid peroxidation was assessed as MDA levels. (G) Ferroptotic markers were determined by qPCR and normalized to β-actin. Results are representative of n = 7-8 per group. Data are expressed as mean ± standard error of the mean. Statistical significance was assessed using 1-way ANOVA followed by Tukey HSD post hoc test. *P < .05 vs HbAS mice of the same treatment. #P < .05 vs vehicle-treated mice of the same genotype.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/6/10.1182_blood.2020008455/3/m_bloodbld2020008455f2.png?Expires=1769085487&Signature=fxCGA10i4Gb7iw~aSHvQiwgDOzV2~nT66-yGANvfEtOLoTcl9caJSbB9izLb8YTZAUv7qkoU6UXVMnPzw-OcennYdqmVIgZ3G1JGvyW6~IYrmkCdylStlpEE6qp2MlaNTuYCuYTxJFr5puSvzM9tur1qWOnCg3qEE~i4wjUWu5olABlBluYJBP-ZNQLcWEYc039QJhIcLDY7tBs1sWsLY4x1aELXv-W80QPZYWLZyHfEIgydydCqG0zqhpeqb9OoeFcF8xXnAsrHmvUU67Gxke3-ZSy775~vLsjYI1zespEKIsTAjtQylek77~4rfgS2rK2IIHgaK~8UOnsj7Ptx1g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Upregulated Hmox1 promotes cardiac ferroptosis in mice with sickle cell disease. (A-B) HbAS and HbSS mice were injected with human Hx (4 mg/kg) once a week for 4 weeks. (A) Lipid peroxidation was assessed as levels of malondialdehyde (MDA) by ELISA. (B) Relative mRNA expression of ferroptotic markers was determined using qPCR and normalized to β-actin. (C-E) HbAS and HbSS mice were injected with ferroptosis inhibitors (ferrostatin-1 [Fer-1; 1 mg/kg] or deferoxamine [DFO; 100 mg/kg]) or ferroptosis inducer (erastin; 20 mg/kg) intraperitoneally 3 times per week for 4 weeks. (C) Lipid peroxidation was assessed as levels of MDA. Relative expression of (D) ferroptotic and (E) hypertrophic markers were determined using qPCR and normalized to β-actin. (F-G) HbAS and HbSS mice were injected with hemin (25 mg/kg) or SnPP (12 mg/kg) intraperitoneally 3 times per week for 4 weeks. (F) Lipid peroxidation was assessed as MDA levels. (G) Ferroptotic markers were determined by qPCR and normalized to β-actin. Results are representative of n = 7-8 per group. Data are expressed as mean ± standard error of the mean. Statistical significance was assessed using 1-way ANOVA followed by Tukey HSD post hoc test. *P < .05 vs HbAS mice of the same treatment. #P < .05 vs vehicle-treated mice of the same genotype.
Upregulated Hmox1 promotes cardiac ferroptosis in mice with sickle cell disease. (A-B) HbAS and HbSS mice were injected with human Hx (4 mg/kg) once a week for 4 weeks. (A) Lipid peroxidation was assessed as levels of malondialdehyde (MDA) by ELISA. (B) Relative mRNA expression of ferroptotic markers was determined using qPCR and normalized to β-actin. (C-E) HbAS and HbSS mice were injected with ferroptosis inhibitors (ferrostatin-1 [Fer-1; 1 mg/kg] or deferoxamine [DFO; 100 mg/kg]) or ferroptosis inducer (erastin; 20 mg/kg) intraperitoneally 3 times per week for 4 weeks. (C) Lipid peroxidation was assessed as levels of MDA. Relative expression of (D) ferroptotic and (E) hypertrophic markers were determined using qPCR and normalized to β-actin. (F-G) HbAS and HbSS mice were injected with hemin (25 mg/kg) or SnPP (12 mg/kg) intraperitoneally 3 times per week for 4 weeks. (F) Lipid peroxidation was assessed as MDA levels. (G) Ferroptotic markers were determined by qPCR and normalized to β-actin. Results are representative of n = 7-8 per group. Data are expressed as mean ± standard error of the mean. Statistical significance was assessed using 1-way ANOVA followed by Tukey HSD post hoc test. *P < .05 vs HbAS mice of the same treatment. #P < .05 vs vehicle-treated mice of the same genotype.
To explore the contribution of ferroptosis toward cardiomyopathy in SCD, we used 2 small molecule ferroptosis inhibitors: an iron chelator DFO and a lipid radical scavenger Fer-1 as well as a ferroptosis inducer erastin (supplemental Figure 2C). Inhibition of ferroptosis corrected cardiac damage, whereas erastin exacerbated cardiac ferroptosis in HbSS mice (Figure 2C-E; supplemental Figure 2D,E,G). In HbAS mice, DFO decreased cardiac iron, but increased cardiac damage (supplemental Figure 2D,E,G; Figure 2E). However, no differences in any ferroptotic markers were observed (Figure 2C,D), suggesting that iron deficiency could cause cardiomyopathy by mechanisms independent of ferroptosis. Conversely, Fer-1 treatment did not change any of the parameters measured in HbAS mice (Figure 2C-E; supplemental Figure 2D,E,G). Finally, erastin decreased levels of Gpx4 (Figure 2D) and promoted ferroptosis (Figure 2C,D) and cardiac damage (Figure 2E; supplemental Figure 2E,G) in HbAS mice without influencing cardiac iron levels (supplemental Figure 2D). To determine if cardiomyopathy in SCD could result from cell death mechanisms like apoptosis, we measured caspase-3 and its cleaved form as well as Bax and Bcl-2 and found no differences (supplemental Figure 2F), which was confirmed by TUNEL staining (data not shown). Because myocardial apoptosis is associated with aging,30 it remains to be determined if SCD represents a risk for age-dependent cardiac apoptosis. Finally, we examined if modulation of Hmox1 alters cardiac ferroptosis in SCD. Ferroptotic damage in HbSS mice was corrected by SnPP and exacerbated by hemin (Figure 2F,G). Additionally, hemin administration induced lipid peroxidation and ferroptosis in HbAS mice (Figure 2F,G). Combined, our data confirm that heme-induced Hmox1 drives cardiomyopathy through ferroptosis, and that inhibition or induction of ferroptosis can ameliorate or exacerbate SCD-associated cardiomyopathy, respectively.
Together, our study demonstrates that excess systemic heme in SCD upregulates Hmox1, which promotes cardiac ferroptosis (supplemental Figure 2H). Further studies are required to evaluate if other hemolytic products, like hemoglobin, can contribute to cardiovascular events in SCD through other pathways such as scavenging nitric oxide.31 Alternatively, increased reticulocytes in SCD, associated with iron-loaded mitochondria and reactive oxygen species,32 may be responsible for cardiomyopathy by inducing a pro-inflammatory state.33 Our findings provide the molecular basis for cardiac ferroptosis in SCD and help to identify therapeutic strategies to alleviate SCD-associated cardiomyopathy.
Acknowledgments
The authors thank Flora Sam for her helpful comments regarding cardiotoxic markers and HaYoung Nam, Mitchell Lobo, Lisa Luka, and Uksavatey Nuth for their assistance in maintaining mouse colonies.
This study was in part supported by the National Institutes of Health National Heart, Lung, and Blood Institute (R01 HL143020) (J.K.).
Authorship
Contribution: A.V.M. and J.K. conceived the project, designed research, interpreted the results, and wrote the paper; A.V.M. also performed most research and analyzed the data; H.P.T., L.Z., and S.Y. conducted biochemical experiments and assisted with mouse experiments; and J.L. and A.A. performed echocardiography and analyzed the echo data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Aarti Asnani, Center for Life Sciences, 9th Floor, Beth Israel Deaconess Medical Center, 330 Brookline Ave, Boston, MA 02115; e-mail: aasnani@bidmc.harvard.edu; and Jonghan Kim, 3 Solomont Way, Suite 4, Lowell, MA 01854; e-mail: jonghan_kim@uml.edu.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal