

In this issue of Blood, ,1 by a whole-genome analysis of 150 cases of adult T-cell leukemia/lymphoma (ATL), identify new oncogenic drivers, noncoding and structural mutations, and a novel molecular signature that distinguishes the leukemic from the lymphomatous forms.
ATL remains one of the most aggressive hematological malignancies and one of the most refractory to therapy. Although some cases have been successfully treated, in particular, with a combination of zidovudine and type 1 interferon,2 there has been no substantial increase in average life expectancy in the last 30 years.3
In a landmark paper in 2015, Kataoka et al4 identified recurrent putative oncogenic driver mutations in a large series of ATL cases, using whole-exome sequencing: the commonest mutations were found in genes concerned with T-cell function or proliferation. The paper by Kogure et al extends these observations with an analysis of the frequency, distribution, and type of mutations throughout the whole genome, providing significant insights into the etiology of the condition.
ATL is a malignancy of CD4+ T cells, caused by human T-cell leukemia virus type 1 (HTLV-1).5 A typical patient with ATL possesses a single malignant clone that carries 1 copy of the HTLV-1 provirus integrated in the T-cell genome. Proviral integration sites vary widely between cases, although integration upstream of a known cancer-associated gene was observed in 6% of cases.6
HTLV-1 expresses 2 genes, tax and HBZ, that regulate the expression of numerous host genes as well as HTLV-1 itself. Products of these genes have been implicated in the pathogenesis of ATL. HBZ has the stronger association: whereas Tax protein expression is lost in 50% of cases, HBZ appears to be invariably maintained intact. Furthermore, ATL can arise in a clone that never expressed Tax.7 However, neither tax nor HBZ is an acutely transforming gene like myc or src: rather, the 2 genes cooperate to maintain persistent replication of HTLV-1+ T-cell clones, which are effectively immortalized. A simple hypothesis of the oncogenesis of ATL is that replicative mutations accumulate in these long-lived, persistently replicating clones.8,9
By whole-genome sequencing of 150 cases of ATL, Kogure et al found 10 previously unidentified oncogenic drivers among 56 altered genes, 32 of which were altered in >10% of cases. The median number of altered driver genes in each case was 9, and 149 of the 150 cases had 1 or more driver alterations. The frequency of point mutations was highest in intergenic regions, whereas structural variants were commoner in transcribed genomic regions. This difference might result from a difference either in the mechanisms of generation of the mutation, as suggested by the authors, or in selection, because mutations in transcribed regions are more likely to affect T-cell function.
Remarkably, alterations were found in the genes encoding the transcriptional repressive complex CIC-ATXN1 in 53% of the cases. The mutations in CIC were not previously recognized, because the CIC-L exon encoding the long isoform of the protein, in which most mutations were present, was not well understood. The functional importance of this complex in T cells was shown by knocking out the CIC-L exon in mice, which doubled the number of CD4+ CD25+ CD127− FoxP3+ T cells in the circulation. The authors interpreted these cells as regulatory T cells (Tregs); however, no single set of markers is uniquely associated with Tregs, which are properly defined by function rather than by phenotype, and many ATL clones lack the most characteristic Treg markers.
Kogure et al also found alterations in the noncoding genome, which could not be identified with the previous whole-exome sequencing approach. Particularly, frequent alterations were found in the enhancer of the immunoglobulin gene IGH (9% of cases) and the 3′ untranslated region of NFKBIZ.
Frequent alterations were observed in several genes directly involved in T-cell activation or immune recognition. Clustered hypermutations were found in one-third of cases, mainly in IG or TCR genes. Splice-site mutations were observed in HLA-A, HLA-B, CD58 (LFA-3), and FAS. These splice-site alterations were strongly associated with potential neoantigen mutations as well as structural variants, suggesting that immune escape had occurred. The implication that the immune response plays an important part in preventing ATL is also consistent with the previous observation6 that the ATL clone is often accompanied by several abnormally abundant but nonmalignant HTLV-1–infected T-cell clones.
Kogure et al found mutations in certain transcription factors or their binding sites. Of particular interest were mutations in binding sites for the HTLV-1 protein HBZ that contained recognition sequences for the transcription factors AP-1 and ETS. This observation again points to a critical role of HBZ in ATL oncogenesis.
Each cause of genetic mutations can produce a characteristic pattern of changes, called a mutational signature.10 Of the 7 mutational signatures identified by Kogure et al, the most prominent was the aging-related signature. This observation is consistent with the idea that ATL results from the accumulation of replicative mutations in the long-lived, HTLV-1–infected T-cell clones.8,9
By a cluster analysis of 56 driver genes, the authors identified 2 molecular subgroups. Group 1, common in leukemic cases, was associated with alterations in TCR signaling molecules, such as PLCG1, whereas genes involved in antigen recognition (HLA-A, HLA-B, CD58) were enriched in group 2, in which most lymphoma cases were classified. This observation suggests that impairment of immune surveillance plays a greater role in the emergence of lymphoma, whereas constitutive activation of T cells, caused, for example, by activating mutations in PLCG1, is associated with leukemia. This unexpected association is of particular interest, because it is still poorly understood why some individuals develop the lymphomatous form of ATL, whereas others develop the leukemia. ATL lymphoma has a particularly poor prognosis; the group 2 molecular signature also carried a worse prognosis than the group 1 signature, independent of the clinical subtype of ATL.
The results reported by Kogure et al provide strong evidence of the importance of certain key factors in the oncogenesis of ATL, namely the accumulation of replicative mutations with age, the T-cell immune response, and the HTLV-1 gene HBZ (see figure). The novel identification of different clusters of altered genes associated with the phenotype of ATL suggests an intriguing difference in the dominant mechanisms of pathogenesis of the leukemic and lymphomatous subtypes of this problematic and devastating disease.
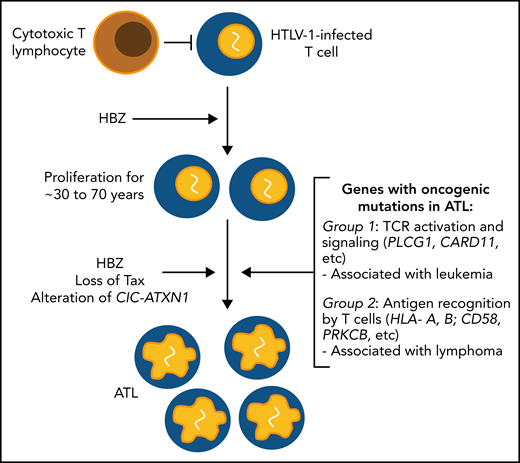
Schematic depiction of key factors in the oncogenesis of ATL identified or implicated in the report by Kogure et al. Many other factors are known to contribute to the pathogenesis of the condition, notably, genomic instability, insertional mutagenesis, and inhibition of DNA repair.8
Schematic depiction of key factors in the oncogenesis of ATL identified or implicated in the report by Kogure et al. Many other factors are known to contribute to the pathogenesis of the condition, notably, genomic instability, insertional mutagenesis, and inhibition of DNA repair.8
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal