

Key Points
Systemic IL-15 promotes allogeneic cell rejection by host T cells, limiting clinical responses to allogeneic adoptive cellular therapy.
The cytokines delivered affect the competitive balance of host immunity and in vivo persistence of adoptive NK cell therapy.

Abstract
Natural killer (NK) cells are a promising alternative to T cells for cancer immunotherapy. Adoptive therapies with allogeneic, cytokine-activated NK cells are being investigated in clinical trials. However, the optimal cytokine support after adoptive transfer to promote NK cell expansion, and persistence remains unclear. Correlative studies from 2 independent clinical trial cohorts treated with major histocompatibility complex-haploidentical NK cell therapy for relapsed/refractory acute myeloid leukemia revealed that cytokine support by systemic interleukin-15 (IL-15; N-803) resulted in reduced clinical activity, compared with IL-2. We hypothesized that the mechanism responsible was IL-15/N-803 promoting recipient CD8 T-cell activation that in turn accelerated donor NK cell rejection. This idea was supported by increased proliferating CD8+ T-cell numbers in patients treated with IL-15/N-803, compared with IL-2. Moreover, mixed lymphocyte reactions showed that IL-15/N-803 enhanced responder CD8 T-cell activation and proliferation, compared with IL-2 alone. Additionally, IL-15/N-803 accelerated the ability of responding T cells to kill stimulator-derived memory-like NK cells, demonstrating that additional IL-15 can hasten donor NK cell elimination. Thus, systemic IL-15 used to support allogeneic cell therapy may paradoxically limit their therapeutic window of opportunity and clinical activity. This study indicates that stimulating patient CD8 T-cell allo-rejection responses may critically limit allogeneic cellular therapy supported with IL-15. This trial was registered at www.clinicaltrials.gov as #NCT03050216 and #NCT01898793.
Introduction
Natural killer (NK) cells are a promising alternative to T cells for allogeneic cellular immunotherapy because they have been administered safely, naturally eliminate malignant cells, and are amendable to cellular engineering.1 All NK products require signaling through interleukin-2 (IL-2)/IL-15 cytokine receptor (IL-15R) to promote their survival, expansion, and persistence.2 Enriched conventional NK cell therapy has been tested in clinical trials for patients with acute myeloid leukemia (AML), and have been supported mainly by low-dose IL-2.3,4 Memory-like (ML) NK cells, which are induced after brief activation with the cytokines IL-12, IL-15, and IL-18, are currently being advanced in the clinic and supported by low-dose IL-2.5-8 Studies suggest that a surge of endogenous IL-15 following lymphodepleting chemotherapy can support transferred NK cells, and exogenous cytokines may be further expand them in vivo.3,9,10 However, the optimal cytokine, dose, and schedule to support transferred NK cells remain unclear. Based on the importance of IL-15 for NK cell homeostasis and function, coupled with a distinct IL-15:IL-15Rα transpresentation receptor biology, IL-15R agonists have been advanced as an alternative to IL-2.11-14 Here, we investigated replacing IL-2 with the longer acting IL-15R agonist N-803 (formerly known as ALT-803) to promote NK cell persistence after transfer into patients. Unexpectedly, every 5-day systemic IL-15/N-803 administration limited allogeneic NK cell therapy by expediting host T cell-mediated rejection, resulting in reduced clinical activity.
Study design
Patient samples
Samples from patients treated on an open-label, nonrandomized, phase 1 dose escalation trial are included in this study (NCT-01898793; Figure 1A; supplemental Tables 1 and 2, available on the Blood Web site).7,8 Written informed consent was obtained from all patients under a Washington University School of Medicine institutional review board–approved clinical protocol. Additional samples were tested from trials at the University of Minnesota (UMN) institutional review board (NCT01106950, NCT03050216; Figure 1A; supplemental Tables 1 and 2). Informed consent was given by all patients and donors for treatment and prospective data collection in accordance with Declaration of Helsinki.
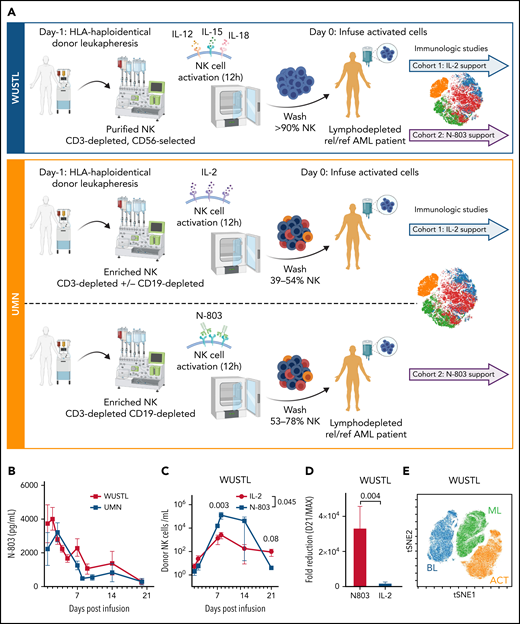
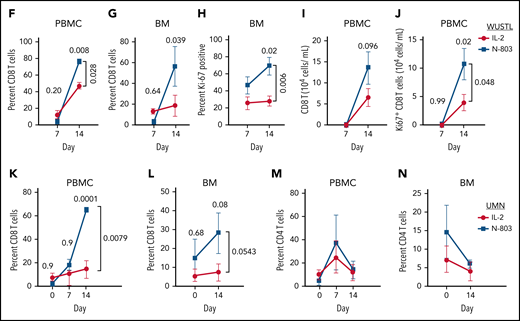
N-803 promotes donor NK and recipient CD8 T-cell expansion in vivo. (A) WUSTL and UMN trial schemas. Briefly, relapsed/refractory AML patients were lymphodepleted with fludarabine (25 mg/m3 × 5) on study days -6 to -2 and cyclophosphamide (60 mg/kg × 2) on study days -5 and -4. On study day -1, related, haploidentical donors were apheresed, NK cells were purified (WUSTL) or enriched (UMN) and activated with IL-12, IL-15, and IL-18 (WUSTL), IL-2 (UMN cohort 1), or N-803 (UMN cohort 2). Products were washed and infused into patients on study day 0 (NK purity for each cohort indicated as a percentage). Infused products were supported with IL-2 (WUSTL/UMN cohort 1) or N-803 (WUSTL/UMN cohort 2). (B) N-803 concentration in the PB from patients at the indicated times. (C) Donor NK cell expansion over time, as determined by flow cytometry between IL-2- (blue) and N-803 (purple) supported WUSTL patients (IL-2 n = 6; N-803 n = 7). (D) Fold reduction in cells from day 21 compared with maximal measure NK cells, typically days 8 through 14. (E) Representative overlay viSNE plot of purified donor NK cells (baseline, BL), infusion product (activated, ACT), and in vivo differentiated donor ML NK cells assessed by mass cytometry. (F-J) WUSTL patient CD8 T cells from PBMC and bone marrow (BM) were assessed by mass cytometry at the indicated days after NK cell infusion. (F-G) Summary data depicting recipient CD8 T-cell frequency (of CD45+ lymphocytes) in the (F) PBMC (IL-2; day 7 n = 8, day 14 n = 3; N-802 n = 6) and (G) BM (day 7 n = 4, day 14 n = 3). (H) Summary data showing percent Ki-67+ CD8 T cells in recipient BM at 7 and 14 days, after NK cell infusion (day 7 n = 4, day 14 n = 3). (I-J) Summary data showing absolute CD8+ (I) and Ki67+ CD8 (J) T-cell numbers in the PBMC at the indicated days after infusion (IL-2 day 7 n = 8, day 14 n = 3; N-802 n = 6). (K-N) Patients treated on UMN trials using IL-2 activated NK cells supported in vivo with IL-2 (gold) or N-803 (red) were also assessed by mass cytometry (see schema, supplemental Figure 2). Summary data depicting percent CD8 T cells from the (K) PBMC before (day 0) and the indicated days after NK cell infusion (IL-2 day 0 n = 6, day 7 n = 3, day 14 n = 6; N-802 day 0 n = 3, day 7 n = 2, day 14 n = 3), (L) BM (IL-2 n = 6; N-802 day 0 n = 3, day 14 n = 5). Summary data depicting percent CD4 T cells from the (M) PBMC and (N) BM. Summary data were analyzed using 2-way analysis of variance. Mean is depicted with error represented as standard error of the mean. P values are indicated within the graphs; no significant differences were detected in panels K-N.
N-803 promotes donor NK and recipient CD8 T-cell expansion in vivo. (A) WUSTL and UMN trial schemas. Briefly, relapsed/refractory AML patients were lymphodepleted with fludarabine (25 mg/m3 × 5) on study days -6 to -2 and cyclophosphamide (60 mg/kg × 2) on study days -5 and -4. On study day -1, related, haploidentical donors were apheresed, NK cells were purified (WUSTL) or enriched (UMN) and activated with IL-12, IL-15, and IL-18 (WUSTL), IL-2 (UMN cohort 1), or N-803 (UMN cohort 2). Products were washed and infused into patients on study day 0 (NK purity for each cohort indicated as a percentage). Infused products were supported with IL-2 (WUSTL/UMN cohort 1) or N-803 (WUSTL/UMN cohort 2). (B) N-803 concentration in the PB from patients at the indicated times. (C) Donor NK cell expansion over time, as determined by flow cytometry between IL-2- (blue) and N-803 (purple) supported WUSTL patients (IL-2 n = 6; N-803 n = 7). (D) Fold reduction in cells from day 21 compared with maximal measure NK cells, typically days 8 through 14. (E) Representative overlay viSNE plot of purified donor NK cells (baseline, BL), infusion product (activated, ACT), and in vivo differentiated donor ML NK cells assessed by mass cytometry. (F-J) WUSTL patient CD8 T cells from PBMC and bone marrow (BM) were assessed by mass cytometry at the indicated days after NK cell infusion. (F-G) Summary data depicting recipient CD8 T-cell frequency (of CD45+ lymphocytes) in the (F) PBMC (IL-2; day 7 n = 8, day 14 n = 3; N-802 n = 6) and (G) BM (day 7 n = 4, day 14 n = 3). (H) Summary data showing percent Ki-67+ CD8 T cells in recipient BM at 7 and 14 days, after NK cell infusion (day 7 n = 4, day 14 n = 3). (I-J) Summary data showing absolute CD8+ (I) and Ki67+ CD8 (J) T-cell numbers in the PBMC at the indicated days after infusion (IL-2 day 7 n = 8, day 14 n = 3; N-802 n = 6). (K-N) Patients treated on UMN trials using IL-2 activated NK cells supported in vivo with IL-2 (gold) or N-803 (red) were also assessed by mass cytometry (see schema, supplemental Figure 2). Summary data depicting percent CD8 T cells from the (K) PBMC before (day 0) and the indicated days after NK cell infusion (IL-2 day 0 n = 6, day 7 n = 3, day 14 n = 6; N-802 day 0 n = 3, day 7 n = 2, day 14 n = 3), (L) BM (IL-2 n = 6; N-802 day 0 n = 3, day 14 n = 5). Summary data depicting percent CD4 T cells from the (M) PBMC and (N) BM. Summary data were analyzed using 2-way analysis of variance. Mean is depicted with error represented as standard error of the mean. P values are indicated within the graphs; no significant differences were detected in panels K-N.
N-803 serum concentration
N-803 serum concentration was determined as previously described (supplemental Methods).15
Mass cytometry
Mass cytometry staining, acquisition and analyses were performed as previously described (supplemental Methods; supplemental Table 3).8,13,16
MLRs
Mixed lymphocyte reactions (MLRs) were performed as previously described (supplemental Methods).17
51Cr release killing assay
Cytotoxicity assays were performed as previously described (supplemental Methods).18
Results and discussion
Allogeneic cellular therapies are enthusiastically being explored in clinical trials. However, the best cytokine to support these transferred cells is unclear. Here, patients with relapsed/refractory AML, including MRD+ patients,19 were treated with major histocompatibility complex (MHC)-haploidentical, related donor-derived ML NK cell infusions that were supported with IL-2 (N = 15) or N-803 (N = 8; NCT01898793; supplemental Tables 1 and 2; Figure 1A) at Washington University in St Louis (WUSTL cohorts).8 In separate trials at UMN (UMN cohort), comparable patients were treated with CD3-depleted/enriched NK cells activated and supported in vivo with IL-2 (NCT01106950; N = 32) or N-803 (NCT03050216; Figure 1A; N = 7; supplemental Tables 1 and 2).4 Clinical response rates by International Working Group criteria20 were significantly different between the 2 treatment cohorts at WUSTL with 47% (7/15)8 achieving complete remission (CR)/complete remission with incomplete count recovery (CRi) with IL-2 support and 0% (0/8) patients achieving CR/CRi with N-803 support given every 5 days (P < .05; Figure 1A). For UMN, patients with IL-2 support had a 28% CR/CRi (9/32),4 whereas patients supported with N-803 had 14% CRi (1/7; Figure 1A; supplemental Tables 1 and 2). Despite AML heterogeneity and the limitations in comparing these separate studies, these findings suggest that supporting allogeneic/MHC-haploidentical NK cells with N-803 reduced the expected outcomes for patients and warranted further investigation.
With this dose and schedule of N-803, high IL-15 levels could be detected for weeks (Figure 1B), consistent with prior studies.13 Donor ML NK cell expansion was monitored using donor- or recipient-specific anti-HLA monoclonal antibodies by flow cytometry (Figure 1C).7,8 Despite similar NK cell doses (supplemental Figure 1A,B), donor NK cells supported with N-803 had a higher number of cells present at peak expansion (days 7-14) in line with prior reports,13,14 but were rapidly reduced (Figure 1C,D). Although total NK cell numbers are not significantly different at day 21 (mean ± standard deviation; 4.2 ± 2.6 N-803 vs 87.4 ± 118.0; P = .086), these data suggest that N-803 promoted increased numbers of NK cells in the peripheral blood (PB) at early timepoints but not persistence, compared with IL-2. Mass cytometry revealed that NK cells supported with N-803 in vivo were distinct from purified donor NK cells and infusion product (Figure 1E). N-803-supported ML NK cells demonstrated the expected multidimensional ML NK cell phenotype (supplemental Figure 1C).8 These data indicated that NK cells were similar in the IL-2 and N-803 cohorts, suggesting other factors within the recipient contributed to poor responses and NK cell loss following N-803 support.
CD8 T cells also respond to IL-15 (N-803) in vivo, although to a lesser extent than NK cells.13 Mass cytometry revealed that recipient T cells in the PB and bone marrow were significantly increased in frequency with N-803 support compared with IL-2 (Figure 1F,G), corresponding to increased proliferation (Ki-67) in the recipient bone marrow CD8 T cells (Figure 1H), a trend in increased total PB CD8 T cells (Figure 1I), and increased total Ki-67+ CD8 T cells in the PB (Figure 1H). Although the IL-2 UMN cohort samples were limited, similar results were observed in comparing the UMN studies (Figure 1K,L; supplemental Tables 1 and 2), and CD4 T cells were not differentially regulated (Figure 1M,N). Together, these data suggest that N-803 activated recipient CD8 T cells to a greater extent than low-dose IL-2. We hypothesized this resulted in accelerated NK cell rejection, thereby shortening the opportunity for NK cell anti-AML responses.
To determine if IL-15 could impact allogeneic rejection by T cells in vivo, MLRs were performed (Figure 2A). PB mononuclear cells (PBMCs) were incubated with irradiated allogenic stimulator cells with IL-2 (10 U/mL), N-803, or with IL-2 and N-803 concentrations.21 T-cell proliferation and activation were assessed by flow cytometry (Figure 2B-D; supplemental Figure 3). N-803/IL-15 supported earlier proliferation (carboxyfluorescein diacetate succinimidyl ester dilution) and upregulation of activation markers CD25 and CD38 on both T-cell subsets. However, by day 11, there were no significant differences between the IL-2 and N-803 incubated conditions. These data indicated that N-803/IL-15 promote accelerated activation against allogeneic target cells. Next, ML NK cells differentiated from the allogeneic stimulator cells were used as targets in short-term cytotoxicity assays, where PBMCs from the MLR at day 10 were used as the effectors (Figure 2E). Effector T cells activated in the presence of N-803 exhibited significantly increased ML NK cell killing, compared with IL-2 (Figure 2F). The increased killing was mediated by CD8 T cells because cytotoxicity was abrogated when MHC-I was blocked (Figure 2G; supplemental Figure 3).22 Modest contributions from FAS/FAS-L to target cell killing were also detected (Figure 2H,I; supplemental Figure 3). Together, these data indicate that N-803 promotes a faster, more robust T-cell response against allogenic NK cells, mediated in part by CD8 T cells.
![N-803 hastens T-cell activation and allogenic rejection in mixed lymphocyte reactions. (A) MLR experimental design. Briefly, PBMCs were carboxyfluorescein diacetate succinimidyl ester (CFSE) labeled and incubated with unmatched, irradiated PBMCs with or without IL-2 and increasing concentrations of N-803. NK cells from stimulator donor were 12/15/18-activated and allowed to differentiate into ML-NK in parallel. (B-C) CD8 T cells were examined by flow cytometry from days 4 through 11 after incubation with allogenic stimulators. (B) Representative histograms depicting proliferation (CFSE dilution) and activation markers CD25 and CD38. (C) Summary data from panel B. (D) Summary data from CD4 T cells as examined in panels B and C. (E-I) 51CR-release killing assays using MLR-stimulated PBMCs (R; responder) as effectors against allogeneic in vitro differentiated ML NK cells as targets. (E) Killing assay schema. PBMCs were harvested from MLR on day 10 and cocultured with 51Cr-pulsed ML NK cells (matched to original allogenic stimulator [S] cells; S-ML NK) and killing assessed. (F) Summary data from panel E. (G-I) PBMCs incubated with IL-2 and 35 ng/mL N-803 were incubated with anti-MHC-I (G), anti-FAS-L (H), or anti-TRAIL (I) before addition of labeled ML-NK targets and allogenic killing assessed in the presence of blocking antibodies. N = 6 normal donor responders, 4 normal donor stimulator/targets in 2 independent experiments. Data were analyzed using 2-way analysis of variance, *P < .05, **P < .01, ***P < .001. Unless indicated (purple asterisk), statistics are for each condition, compared with IL-2 only condition. Purple asterisk indicates significance for 35 ng/mL N-803 + IL-2 compared with IL-2 only. Mean is depicted with error represented as standard error of the mean.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/8/10.1182_blood.2021011532/5/m_bloodbld2021011532f2.png?Expires=1769112302&Signature=mECc1NQE2Tim3ZyqXY5w13-FuyUz1fV3Jm84bavtHYuHqSsVDdOMekbZC9vhcM5K2TfJ74f0nDx-AgnkAlbm7h5egsQuX0L~UwMX6Uc~RkSOzolgmniJcuv9h0fYu2HVHADgXw~Ntj2dzkwdsZ7pVDZdxjBurYTuiAUmIL1gzh3f9ScZ2-HjtoA~VAN2fPeE9U3Q5xbf29hgpr5l-0Qfj53IF9mTpVZWDUzY2kJBlyanksXdZnGGq9d0PGim4eFJw13EwG6K-zcQR9ueA1gxu3NDeYvBPxwGHPHPv8oIUoUGnUOiwzM~iurqXEtJLX6YoRQzaOoesNAOhr2L2YbMtQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
N-803 hastens T-cell activation and allogenic rejection in mixed lymphocyte reactions. (A) MLR experimental design. Briefly, PBMCs were carboxyfluorescein diacetate succinimidyl ester (CFSE) labeled and incubated with unmatched, irradiated PBMCs with or without IL-2 and increasing concentrations of N-803. NK cells from stimulator donor were 12/15/18-activated and allowed to differentiate into ML-NK in parallel. (B-C) CD8 T cells were examined by flow cytometry from days 4 through 11 after incubation with allogenic stimulators. (B) Representative histograms depicting proliferation (CFSE dilution) and activation markers CD25 and CD38. (C) Summary data from panel B. (D) Summary data from CD4 T cells as examined in panels B and C. (E-I) 51CR-release killing assays using MLR-stimulated PBMCs (R; responder) as effectors against allogeneic in vitro differentiated ML NK cells as targets. (E) Killing assay schema. PBMCs were harvested from MLR on day 10 and cocultured with 51Cr-pulsed ML NK cells (matched to original allogenic stimulator [S] cells; S-ML NK) and killing assessed. (F) Summary data from panel E. (G-I) PBMCs incubated with IL-2 and 35 ng/mL N-803 were incubated with anti-MHC-I (G), anti-FAS-L (H), or anti-TRAIL (I) before addition of labeled ML-NK targets and allogenic killing assessed in the presence of blocking antibodies. N = 6 normal donor responders, 4 normal donor stimulator/targets in 2 independent experiments. Data were analyzed using 2-way analysis of variance, *P < .05, **P < .01, ***P < .001. Unless indicated (purple asterisk), statistics are for each condition, compared with IL-2 only condition. Purple asterisk indicates significance for 35 ng/mL N-803 + IL-2 compared with IL-2 only. Mean is depicted with error represented as standard error of the mean.
N-803 hastens T-cell activation and allogenic rejection in mixed lymphocyte reactions. (A) MLR experimental design. Briefly, PBMCs were carboxyfluorescein diacetate succinimidyl ester (CFSE) labeled and incubated with unmatched, irradiated PBMCs with or without IL-2 and increasing concentrations of N-803. NK cells from stimulator donor were 12/15/18-activated and allowed to differentiate into ML-NK in parallel. (B-C) CD8 T cells were examined by flow cytometry from days 4 through 11 after incubation with allogenic stimulators. (B) Representative histograms depicting proliferation (CFSE dilution) and activation markers CD25 and CD38. (C) Summary data from panel B. (D) Summary data from CD4 T cells as examined in panels B and C. (E-I) 51CR-release killing assays using MLR-stimulated PBMCs (R; responder) as effectors against allogeneic in vitro differentiated ML NK cells as targets. (E) Killing assay schema. PBMCs were harvested from MLR on day 10 and cocultured with 51Cr-pulsed ML NK cells (matched to original allogenic stimulator [S] cells; S-ML NK) and killing assessed. (F) Summary data from panel E. (G-I) PBMCs incubated with IL-2 and 35 ng/mL N-803 were incubated with anti-MHC-I (G), anti-FAS-L (H), or anti-TRAIL (I) before addition of labeled ML-NK targets and allogenic killing assessed in the presence of blocking antibodies. N = 6 normal donor responders, 4 normal donor stimulator/targets in 2 independent experiments. Data were analyzed using 2-way analysis of variance, *P < .05, **P < .01, ***P < .001. Unless indicated (purple asterisk), statistics are for each condition, compared with IL-2 only condition. Purple asterisk indicates significance for 35 ng/mL N-803 + IL-2 compared with IL-2 only. Mean is depicted with error represented as standard error of the mean.
Here we show that every 5-day IL-15/N-803 support resulted in sustained levels of IL-15 in serum and had the unintended consequence of shortening the window of opportunity allogeneic NK cells had to mediate their antitumor responses. Although N-803 administration resulted in higher short-term NK cell levels in the PB at peak expansion, as expected from the first-in-human trial,13 this did not result in improved outcomes, suggesting that NK cell persistence is an important parameter for clinical efficacy. In other studies at Washington University, weekly N-803 has been successfully used to support NK cell expansion and durability for >2 months, when the NK cells and the T cells are immune compatible (from the same donor).10,23 Additionally, Cooley et al observed that the route and type of IL-15 administration could be important. Subcutaneous recombinant human IL-15 dosing lead to higher, more prolonged IL-15 concentrations and increased the incidence of cytokine release syndrome and immune effector cell-association neurotoxicity syndrome, compared with IV application, without improving clinical responses, likely from enhanced T-cell activation.14 Miller et al described an association between increased IL-15 serum levels following lymphodepletion and allogeneic NK cell expansion.3 However, the endogenous IL-15 levels observed after lymphodepletion are much lower than those achieved with subcutaneous N-803 administration. This suggests that low doses of IL-15 may not negatively impact allogeneic NK cell persistence, but this remains to be tested. As enthusiasm for NK cellular therapies rises, strategies that promote NK cell persistence in vivo are of great interest.24 Multiple groups are now engineering IL-15 into their NK cellular products.24,25 The effective local IL-15 concentration generated by these engineered products may promote NK cell persistence, as well as recipient T-cell activation and thus NK cell allo-rejection. The IL-15 mechanism reported here will assist in interpreting the allogeneic NK cell persistence within these trials as data become available. These data provide a warning about systemic IL-15 combined with allogeneic effector cells because it clearly impacts the balance between recipient T cells and donor cells, and suggests the γc cytokine, route, dose/interval, and formulation are important factors to consider for each therapy.
Acknowledgments
The authors acknowledge support from Bill Eades, Siteman Flow Cytometry Core; Stephen Oh, Immune Monitoring Laboratory; and Biological Therapy Core Facility of the Siteman Cancer Center supported by the National Institutes of Health National Cancer Institute Cancer Center Support Grant (P30CA91842). T.A.F. reports grants from National Institutes of Health National Cancer Institute (R01CA205239), Leukemia and Lymphoma Society, V Foundation for Cancer Research, and Siteman Cancer Center at Washington University School of Medicine and Children’s Discovery Institute. M.M.B.-E. was supported by funding from National Institutes of Health National Cancer Institute (K12CA167540). University of Minnesota studies were supported by funding to J.S.M. from National Institutes of Health National Cancer Institute (P01CA111412, P01CA65493, and R35CA197292). M.M.B.-E., T.A.F., and A.F.C. were also funded by National Institutes of Health National Cancer Institute (grant P50CA171063). P. Wong was supported by a grant from the National Institutes of Health National Heart, Lung, and Blood Institute (T32HL007088). Schemas were created using Biorender.com.
Authorship
Contribution: M.M.B.-E., M.B.-H., J.S.M., and T.A.F. designed the research; M.M.B.-E., M.B.-H., M.A.J., P. Wong, M.F., E.M., S.D., and F.G. performed the research; M.M.B.-E., M.B.H., and M.F. analyzed data; A.F.C., C.B., S.C., C.N.A., G.L.U., P. Westervelt, M.A.J., I.P., K.E.S.-G., M.A.S., J.F.D., and J.S.M. provided clinical care and contributed samples to this study; P.S.-S. provided clinical reagents; M.M.B.-E. and T.A.F. wrote the manuscript; and all authors edited and approved the final draft.
Conflict-of-interest disclosure: M.M.B.-E. and T.A.F. consult for Wugen (equity) and are inventors of technology that Washington University has licensed to Wugen (royalties). T.A.F. has received research support from Immunity Bio, Compass Therapeutics, HCW Biologics, and Wugen and advises Kiadis, Nkarta, Indapta, and Orca Biosystems. C.B. has received research funds from Gamida Cell and Fate Therapeutics and consults for Allovir (data and safety monitoring board). I.P. is on the advisory board for Incyte, Kadmon, and Syndax. J.S.M. reports consultancy, patents, royalties, and research funding from Fate Therapeutics and GT Biopharma; consultancy for Vycellix; and honoraria and membership on the advisory committees of ONK Therapeutics and Sanofi. P.S.-S. is a majority shareholder of ImmunityBio, Inc., and Altor BioScience, LLC, related to N-803. The remaining authors declare no competing financial interests.
The current affiliation for S.C. is Fate Therapeutics, San Diego, CA.
Correspondence: Jeffrey S. Miller, Washington University School of Medicine, 660 South Euclid Ave, Campus Box 8007, St. Louis, MO 63110; e-mail: mille011@umn.edu; and Todd A. Fehniger, Washington University School of Medicine, 660 South Euclid Ave, Campus Box 8007, St. Louis, MO 63110; e-mail: tfehnige@wustl.edu.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal