

Key Points
Ticagrelor did not reduce the composite end point of vaso-occlusive crises; there was no evidence of efficacy across secondary end points.
Ticagrelor should not be used for the prevention of vaso-occlusive crises in pediatric patients with SCD.

Abstract
The phase 3 HESTIA3 study assessed the efficacy and safety of the reversible P2Y12 inhibitor ticagrelor vs placebo in preventing vaso-occlusive crises in pediatric patients with sickle cell disease (SCD). Patients aged 2 to 17 years were randomly assigned 1:1 to receive weight-based doses of ticagrelor or matching placebo. The primary end point was the rate of vaso-occlusive crises, a composite of painful crises and/or acute chest syndrome (ACS). Key secondary end points included number and duration of painful crises, number of ACS events, and number of vaso-occlusive crises requiring hospitalization or emergency department visits. Exploratory end points included the effect of ticagrelor on platelet activation. In total, 193 patients (ticagrelor, n = 101; placebo, n = 92) underwent randomization at 53 sites across 16 countries. The study was terminated 4 months before planned completion for lack of efficacy. Median ticagrelor exposure duration was 296.5 days. The primary end point was not met: estimated yearly incidence of vaso-occlusive crises was 2.74 in the ticagrelor group and 2.60 in the placebo group (rate ratio, 1.06; 95% confidence interval, 0.75-1.50; P = .7597). There was no evidence of efficacy for ticagrelor vs placebo across secondary end points. Median platelet inhibition with ticagrelor at 6 months was 34.9% predose and 55.7% at 2 hours’ postdose. Nine patients (9%) in the ticagrelor group and eight patients (9%) in the placebo group had at least one bleeding event. In conclusion, no reduction of vaso-occlusive crises was seen with ticagrelor vs placebo in these pediatric patients with SCD. This trial was registered at www.clinicaltrials.gov as #NCT03615924.
Introduction
Sickle cell disease (SCD) is an autosomal recessive inherited hematologic disorder in which β globin is mutated, leading to hemoglobin S (HbS) polymerization when deoxygenated.1,2 HbS polymer distorts the erythrocyte shape into the sickled form and damages the membrane, thereby contributing to both primary SCD pathophysiologies of vaso-occlusive crises and hemolytic anemia.1-3 An estimated 300 000 to 400 000 infants globally are born with SCD each year.4 Individuals with SCD experience high disease-related morbidity and reduced life expectancy due to acute and chronic complications.3
Erythrocytes from patients with SCD, containing sickle hemoglobin, have a shorter life span, and the chronic hemolysis results in hemolytic anemia, promotion of oxidative stress, inflammation, endothelial dysfunction, and vascular injury via complex downstream pathways.5-7 Vaso-occlusive crises, which are caused by obstruction of the microvasculature and associated ischemic tissue/organ injury, can be intensely painful and, when pulmonary vasculature is affected, result in acute chest syndrome (ACS).8 There is evidence of a role for thrombin and tissue factor “upstream” of the actual vaso-occlusion leading to the phenotype of ACS. Specifically, Jimenez et al9 found that neutrophil–platelet aggregation enables vaso-occlusion in SCD. Sparkenbaugh et al10 showed that tissue factor inhibition upstream of platelet-to-platelet interactions can reduce neutrophil–platelet aggregation in mouse lungs. This study followed earlier work by Bennewitz et al,11 which showed that lung vaso-occlusion in sickle mice was mediated by neutrophil–platelet microemboli.
Four disease-modifying medications are approved for the treatment of SCD.12-15 Hydroxyurea induces beneficial myelosuppression and γ globin (and thereby fetal hemoglobin) stimulation to reduce the frequency of SCD complications.12l-glutamine, an amino acid with antioxidant properties, can reduce acute SCD complications.13 Crizanlizumab, a P-selectin inhibitor, reduces the frequency of vaso-occlusive crises by reducing the intercellular adhesion between blood cells and endothelium.14 Finally, voxelotor shifts the HbS–oxygen dissociation curve, reduces HbS polymerization, and improves hemolytic anemia.15 Although these treatments have shown benefits in SCD, most patients continue to have SCD complications.
Platelet activation is increased at baseline in patients with SCD, and platelets are further activated during vaso-occlusive events, thus making antiplatelet therapy a potential therapeutic option. High circulating levels of monocyte–platelet and neutrophil–platelet aggregates indicate platelet activation in patients with SCD and thus suggest a potential role for antiplatelet therapy.16-20 Adherent platelets interact with neutrophils via platelet glycoprotein IIb/IIIa (GPIIb/IIIa) in a fibrinogen-dependent manner.21 However, the activation and role of platelets in SCD pathophysiology are complex due to ongoing inflammation, hypoxia, free hemoglobin, fibrinogen receptor activation, and decreased nitric oxide bioavailability.22 Inflammation and adenosine diphosphate (ADP)-associated platelet activation and aggregation in individuals with SCD contribute to the vaso-occlusive processes.3 All these findings raise the possibility that targeting a single mode of action may not be effective but that successful pharmacologic treatment may require action at multiple targets.
The irreversible ADP P2Y12 receptor antagonist prasugrel exhibited a trend toward efficacy but no significant decrease in rates of vaso-occlusive crisis vs placebo (the primary end point) in children with SCD in the phase 3 DOVE (Determining Effects of Platelet Inhibition on Vaso-Occlusive Events) trial. However, the actual platelet inhibition by prasugrel in the DOVE trial was only ∼25%, which was less than the targeted inhibition of 30% to 60% and, therefore, left open the important question of whether greater platelet inhibition would have offered therapeutic benefit.23,24
Ticagrelor is an oral, reversible ADP P2Y12 receptor antagonist used for the prevention of thrombotic events such as myocardial infarction and stroke in adults with coronary artery disease or cerebrovascular disease.25-28 Mean platelet inhibition was ∼80% predose and ∼90% at 2 hours’ postdose in patients with a history of myocardial infarction who received ticagrelor 60 mg or 90 mg twice a day for at least 4 weeks in the PEGASUS-TIMI 54 (Prevention of Cardiovascular Events in Patients With Prior Heart Attack Using Ticagrelor Compared to Placebo on a Background of Aspirin) trial.29 The sickle cell program with ticagrelor (HESTIA) was designed to assess the potential therapeutic benefits of ticagrelor in patients with SCD,30-32 using ticagrelor doses selected to target greater platelet inhibition (35%-80%) than was observed in the DOVE trial (∼25%).32 A range of ticagrelor doses were investigated in phase 2 studies in children (3-17 years; N = 45; HESTIA1) and young adults (18-30 years; N = 87; HESTIA2). Although these studies were not designed to evaluate the effect of ticagrelor on the prevention of vaso-occlusive crises, the treatment was shown to be well tolerated, with a low bleeding risk and with adverse events (AEs) consistent with common medical issues in SCD.30,31
The present phase 3 HESTIA3 (A Randomised, Double-Blind, Parallel-Group, Multicentre, Phase III Study to Evaluate the Effect of Ticagrelor Versus Placebo in Reducing the Rate of Vaso-Occlusive Crises in Paediatric Patients With Sickle Cell Disease) trial was designed to evaluate the efficacy, safety, and tolerability of ticagrelor vs placebo in preventing vaso-occlusive crises in pediatric patients with SCD. The selected ticagrelor doses in HESTIA3 were predicted by pharmacokinetic/pharmacodynamic modeling and simulation to provide >35% but <80% platelet inhibition during a dosing interval to achieve a higher platelet inhibition than was achieved in the DOVE study (Appendix 1; available on the Blood Web site)32 and to limit the risk of bleeding. The aim of the ticagrelor treatment in the HESTIA3 trial was to reduce the rate of vaso-occlusive crises while also having an acceptable safety profile.
Methods
Study design and oversight
HESTIA3 was an international, multicenter, double-blind, randomized, parallel-group, placebo-controlled, phase 3 study (#NCT03615924; EudraCT2017-002421-38). The design of the study has been described previously.32 This study was performed in accordance with the principles of the Declaration of Helsinki, the International Conference on Harmonisation for Good Clinical Practice guidelines, all applicable regulatory and ethical requirements, and the AstraZeneca policy on bioethics. The study protocol was approved by the institutional review board/independent ethics committee for each participating site and the national regulatory authorities according to local regulations. Written informed consent to participate was obtained from the patient’s parent or legal guardian, and age-appropriate assents were obtained from the patients. Patients who turned 18 years of age during the study signed a new informed consent form. The steering committee comprised international academic leaders and representatives of the sponsor. An independent data and safety monitoring committee conducted regular safety assessments of unblinded study results during the study and reviewed the overall study conduct.
Study enrollment began on September 26, 2018, and was completed on October 18, 2019, with 193 randomized patients. On June 15, 2020, the data and safety monitoring committee recommended study termination because the potential risk to patients of continuing the study outweighed any possible beneficial effect that ticagrelor may show if the study were completed. The study was terminated on June 18, 2020, just 4 months before the planned completion date. Full inclusion and exclusion criteria are available in the study design paper32 and at www.clinicaltrials.gov.
Patients
Pediatric patients were eligible for inclusion if they were 2 to 17 years old, weighed at least 12 kg, had homozygous sickle cell anemia (hemoglobin SS [HbSS]) or sickle β-zero thalassemia (HbS/β0) and had experienced at least 2 vaso-occlusive crises in the past 12 months (defined as acute painful crisis and/or ACS). Patients who were being treated with hydroxyurea were eligible if their weight-adjusted dose had been stable for the past 3 months.
Exclusion criteria included: history of transient ischemic attack or ischemic or hemorrhagic stroke, severe head trauma, intracranial hemorrhage, intracranial neoplasm, arteriovenous malformation, aneurysm, or proliferative retinopathy; conditional or abnormal time-averaged mean of the maximum velocity values (≥153 cm per second using transcranial Doppler imaging, which corresponds to ≥170 cm per second by the nonimaging technique); active pathologic bleeding or increased risk of bleeding complications; hemoglobin <6 g/dL at screening; platelet count <100 × 109/L at screening; undergoing treatment with chronic red blood cell transfusion therapy; continuous use of nonsteroidal anti-inflammatory drugs on >3 days per week; chronic treatment with anticoagulant or antiplatelet drugs; and active untreated malaria.
Study treatments and follow-up
After a screening period of 7 to 28 days, participants were randomly assigned, in a 1:1 ratio, to receive double-blind treatment with ticagrelor or matching placebo, using an interactive voice/Web response system. The doses in the study were selected based on ticagrelor pharmacokinetic/pharmacodynamic modeling and simulation work using data from earlier studies in patients with SCD (HESTIA1, HESTIA230,31).33 A description of the pharmacokinetic/pharmacodynamic model development and the rationale for the proposed doses is provided in supplemental Appendix 1 (available on the Blood Web site). Three different doses of ticagrelor (or matching placebo) were used, depending on body weight (12 to 24 kg body weight, 15 mg [1 tablet] twice daily; >24 kg to 48 kg body weight, 30 mg [2 tablets] twice daily; and >48 kg body weight, 45 mg [3 tablets] twice daily). These doses were predicted to result in a platelet activity corresponding to >35% but not exceeding 80% platelet inhibition compared with baseline. Given the reversible mechanism of action for ticagrelor, the level of P2Y12 inhibition during ticagrelor treatment was expected to vary within a dosing interval and peak ∼2 hours after dosing. Ticagrelor and matching placebo were taken as tablets in the morning and evening at ∼12-hour intervals, with or without food. Physical study visits were scheduled at 2 weeks, 4 weeks, and 3 months, and then every 3 months until the end of study, with safety follow-up 2 weeks after the end of study. Telephone visits were scheduled for the months during which no physical visits were scheduled. Patients were to be followed up for up to 24 months or until a common study end date was reached, defined as 12 months after the last patient was randomized.
Pharmacokinetic and pharmacodynamic assessments
Blood sample collections for pharmacokinetic and pharmacodynamic assessments were scheduled at randomization (day 0), 4 weeks, and 6, 12, 18, and 24 months. Time points for collection were predose (except day 0) and 2 hours’ postdose. Plasma concentrations of ticagrelor and its active metabolite AR-C124910XX were assessed by using a validated, highly sensitive and specific liquid chromatography/mass spectrometry/mass spectrometry method. The lower limit of quantification was 1.00 ng/mL for ticagrelor and 2.50 ng/mL for the metabolite. Platelet reactivity was measured by vasodilator-stimulated phosphoprotein assay using enzyme-linked immunosorbent assay methodology (BioCytex, Marseille, France) according to the manufacturer’s instructions.
Study end points
Efficacy
The primary objective of HESTIA3 was to compare the efficacy of ticagrelor vs placebo in reducing the number of vaso-occlusive crises, which were defined as a composite of painful crisis and/or ACS, in children with SCD. A painful crisis was defined as an onset or worsening of pain that lasted at least 2 hours, for which there was no explanation other than vaso-occlusion and that required therapy with oral or parenteral opioids, parenteral nonsteroidal anti-inflammatory drugs, or other analgesics prescribed by a health care provider in a medical setting (eg, hospital, clinic, or emergency department visit) or at home. An ACS event was defined, per Ballas et al,34 as an acute illness characterized by fever and/or respiratory symptoms, accompanied by a new pulmonary infiltrate on a chest radiograph.
Secondary end points included the number and duration of painful crises, number of ACS events, number of vaso-occlusive crises requiring hospitalization or emergency department visits, days hospitalized for vaso-occlusive crises, number of acute SCD complications, days hospitalized for acute SCD complications, and number of sickle cell–related red blood cell transfusions. Exploratory end points included pharmacokinetic features of ticagrelor and its active metabolite AR-C124910XX and pharmacodynamic evaluation of the effect of ticagrelor on platelet activation.
Patients or their caregivers were trained in an age-adapted way in their local language on how to use a handheld electronic device to record vaso-occlusive crisis-related pain (according to the Revised Faces Pain Scale and the Face, Legs, Activity, Cry, Consolability scale).
Safety
The long-term safety and tolerability of therapy with ticagrelor vs placebo were assessed. AEs and serious AEs, including bleeding, vital signs, and laboratory safety variables, were captured.
Statistical analysis
Sample size calculations were based on simulations, assuming that the number of vaso-occlusive crises would have a negative binomial distribution with a shape parameter of 0.8 and a mean number of crises per year of 2.0 in the placebo group, with a reduction of 50% in the ticagrelor group. With a minimum follow-up of 12 months and a mean follow-up of 18 months, a sample size of 182 patients randomized in a 1:1 ratio to receive ticagrelor or placebo was estimated to provide ∼90% power, with a two-sided test at a significance level of 5%, allowing for a 15% dropout rate. Ticagrelor data were pooled and analyzed irrespective of dose.
The primary and secondary efficacy analyses were based on the intention-to-treat principle using negative binomial regression adjusted for the treatment group (placebo as the reference group) and baseline hydroxyurea therapy (yes; no), with the log-transformed patient follow-up time as offset. Any vaso-occlusive crisis with onset date within 7 days of a prior vaso-occlusive crisis onset date was not counted as a new event. Prespecified subgroup analyses were conducted based on age (<12 years; ≥12 years), number of vaso-occlusive crises within the previous 12 months (2-4; >4), baseline hydroxyurea use (yes; no), sickle cell genotype (HbSS; HbS/β0), geographic region (Africa and Asia; Europe; North and South America), sex (male; female), and race (Black or African American; Asian; White; other). Secondary efficacy variables were analyzed by using the same analysis method as for the primary end point.
The pharmacodynamic analysis included data from all patients who received at least 1 dose of randomized study treatment and who provided at least baseline and one postbaseline (predose and/or postdose) analyzable plasma sample. The pharmacokinetic analysis included data from all patients who received at least 1 dose of randomized study treatment and who provided at least 1 postdose analyzable plasma sample.
Post hoc descriptive analyses were conducted of vaso-occlusive crises rates for 4 different age groups (2-5 years, 6-11 years, 12-14 years, and 15-17 years). The relationship between ticagrelor plasma concentrations and number of vaso-occlusive crises was also assessed post hoc.
All randomized patients, regardless of treatment received, were included in the full analysis set. All patients who received at least 1 dose of randomized study treatment (ticagrelor or placebo), and for whom any postdose data were available, were included in the safety analysis set.
Results
Patients
A total of 193 patients were randomized to treatment and were included in the full analysis set. Patients underwent randomization at 53 sites across 16 countries in Africa, the Americas, Asia, Europe, and the Middle East, with 101 patients assigned to ticagrelor and 92 to placebo. The disposition of the study population is shown in Figure 1. The baseline demographic characteristics of the 2 treatment groups are summarized in Table 1. Baseline laboratory values for hemoglobin, leukocytes, platelets, and neutrophils are shown in supplemental Table 2. The 2 groups were similar in terms of distributions of age, sex, race, SCD genotype, and number of vaso-occlusive crises in the past 12 months. The most common surgical history procedure was splenectomy (ticagrelor group, n = 15 [15%]; placebo group, n = 4 [4%]). The proportion of patients who were at least 80% adherent to study treatment was 87% in the ticagrelor group and 82% in the placebo group. As a result of the premature termination, the end-of-study visit was defined by a common study end date (June 18, 2020). All patients had their randomized study treatment stopped by June 23, 2020, and the last visit of the last patient was on August 13, 2020.
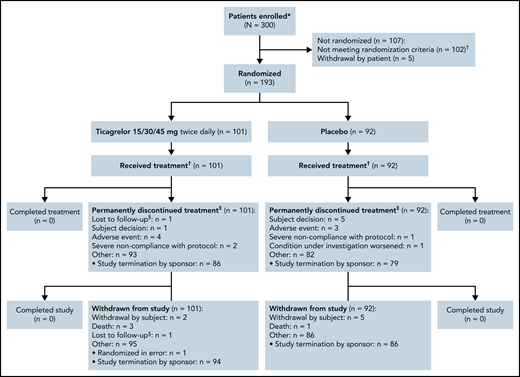
Consolidated Standards of Reporting Trials flowchart. *Informed consent/assent received. †The most common reasons for exclusion were: absent or inadequate/incomplete transcranial Doppler imaging (n = 23), abnormal liver function test results (n = 20), hemoglobin <6 g/dL (n = 18), judged unsuitable by the principal investigator (n = 12), and not having experienced at least 2 vaso-occlusive crises in the past 12 months before visit 1 (n = 12). ‡Randomized to double-blind study treatment and received at least 1 dose of double-blind, randomized study treatment. §Includes patients who prematurely discontinued study treatment. ¶Lost to follow-up: patients were only considered lost to follow-up after 3 documented failed attempts to reach the patient, and all other options of reaching the patient had been exhausted.
Consolidated Standards of Reporting Trials flowchart. *Informed consent/assent received. †The most common reasons for exclusion were: absent or inadequate/incomplete transcranial Doppler imaging (n = 23), abnormal liver function test results (n = 20), hemoglobin <6 g/dL (n = 18), judged unsuitable by the principal investigator (n = 12), and not having experienced at least 2 vaso-occlusive crises in the past 12 months before visit 1 (n = 12). ‡Randomized to double-blind study treatment and received at least 1 dose of double-blind, randomized study treatment. §Includes patients who prematurely discontinued study treatment. ¶Lost to follow-up: patients were only considered lost to follow-up after 3 documented failed attempts to reach the patient, and all other options of reaching the patient had been exhausted.
Baseline characteristics (full analysis set)
| Characteristic | Ticagrelor group (n = 101) | Placebo group (n = 92) |
|---|---|---|
| Age, y | ||
| Mean ± SD | 10.4 ± 4.1 | 10.1 ± 3.8 |
| Median (minimum, maximum) | 10.0 (3.0, 17.0) | 10.5 (3.0, 17.0) |
| Age group, n (%) | ||
| <12 y | 61 (60) | 54 (59) |
| ≥12 y | 40 (40) | 38 (41) |
| Female sex, n (%) | 48 (48) | 43 (47) |
| Race, n (%) | ||
| Black or African American | 60 (59) | 51 (55) |
| White | 25 (25) | 21 (23) |
| Asian | 15 (15) | 15 (16) |
| Other | 1 (1.0) | 5 (5) |
| Geographic region, n (%) | ||
| Ghana, Kenya, South Africa, Tanzania, Uganda | 45 (45) | 44 (48) |
| Egypt, Lebanon, Turkey | 21 (21) | 18 (20) |
| India | 15 (15) | 15 (16) |
| Belgium, Greece, Italy, Spain, United Kingdom | 11 (11) | 8 (9) |
| United States | 4 (4) | 4 (4) |
| Brazil | 5 (5) | 3 (3) |
| SCD genotype, n (%) | ||
| HbSS | 87 (86) | 83 (90) |
| HbS/β0 | 13 (13) | 9 (10) |
| Missing* | 1 (1) | 0 |
| No. of vaso-occlusive crises in past 12 mo, n (%) | ||
| ≤1† | 0 | 1 (1) |
| 2 to 4 | 99 (98) | 89 (97) |
| >4 | 2 (2) | 2 (2) |
| Patients using hydroxyurea at baseline, n (%) | 65 (64) | 58 (63) |
| Characteristic | Ticagrelor group (n = 101) | Placebo group (n = 92) |
|---|---|---|
| Age, y | ||
| Mean ± SD | 10.4 ± 4.1 | 10.1 ± 3.8 |
| Median (minimum, maximum) | 10.0 (3.0, 17.0) | 10.5 (3.0, 17.0) |
| Age group, n (%) | ||
| <12 y | 61 (60) | 54 (59) |
| ≥12 y | 40 (40) | 38 (41) |
| Female sex, n (%) | 48 (48) | 43 (47) |
| Race, n (%) | ||
| Black or African American | 60 (59) | 51 (55) |
| White | 25 (25) | 21 (23) |
| Asian | 15 (15) | 15 (16) |
| Other | 1 (1.0) | 5 (5) |
| Geographic region, n (%) | ||
| Ghana, Kenya, South Africa, Tanzania, Uganda | 45 (45) | 44 (48) |
| Egypt, Lebanon, Turkey | 21 (21) | 18 (20) |
| India | 15 (15) | 15 (16) |
| Belgium, Greece, Italy, Spain, United Kingdom | 11 (11) | 8 (9) |
| United States | 4 (4) | 4 (4) |
| Brazil | 5 (5) | 3 (3) |
| SCD genotype, n (%) | ||
| HbSS | 87 (86) | 83 (90) |
| HbS/β0 | 13 (13) | 9 (10) |
| Missing* | 1 (1) | 0 |
| No. of vaso-occlusive crises in past 12 mo, n (%) | ||
| ≤1† | 0 | 1 (1) |
| 2 to 4 | 99 (98) | 89 (97) |
| >4 | 2 (2) | 2 (2) |
| Patients using hydroxyurea at baseline, n (%) | 65 (64) | 58 (63) |
SD, standard deviation.
This patient had sickle cell–hemoglobin C; the patient was kept in the study but was marked as a protocol deviation.
The patient with ≤1 prior vaso-occlusive crisis is a protocol deviation.
Study end points
Efficacy
The primary end point of the study, the efficacy of ticagrelor vs placebo in reducing the number of vaso-occlusive crises, was not met (Table 2). The estimated yearly incidence rate of vaso-occlusive crises was 2.74 (standard error, 0.334; 95% confidence interval [CI], 2.16-3.48) in the ticagrelor group and 2.60 (standard error, 0.336; 95% CI, 2.01-3.34) in the placebo group. The incidence rate ratio was 1.06 (95% CI, 0.75-1.50; P = .7597). Primary efficacy results were consistent across predefined subgroups, including those based on age (<12 years; ≥12 years) and baseline hydroxyurea use (yes; no) (supplemental Table 1). Post hoc analysis of absolute neutrophil count on study according to the stratification factor hydroxyurea use indicated that the hydroxyurea subgroup had greater myelosuppression than the non-hydroxyurea subgroup, suggesting a hydroxyurea-related myelosuppression (supplemental Table 3); however, there was no apparent added benefit of adding ticagrelor to hydroxyurea use. Further post hoc descriptive results across 4 age groups showed similar rates of vaso-occlusive crises between treatment groups across the age groups (supplemental Figure 1). Time to first vaso-occlusive crisis was consistent with the primary analysis (Figure 2).
Primary and secondary efficacy end points (full analysis set)
| End point | Ticagrelor group (n = 101) | Placebo group (n = 92) | Rate ratio (95% CI) | P |
|---|---|---|---|---|
| Vaso-occlusive crises (primary end point)*† | ||||
| Patients with events, n (%) | 70 (69) | 58 (63) | ||
| Total no. of events | 249 | 202 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (per year) | 2.74 | 2.60 | 1.06 (0.75-1.50) | .7597 |
| Painful crises†‡ | ||||
| Patients with events, n (%) | 69 (68) | 58 (63) | ||
| Total no. of events | 248 | 209 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (per year) | 2.73 | 2.67 | 1.02 (0.72-1.45) | .9037 |
| ACS†‡ | ||||
| Patients with events, n (%) | 5 (5) | 4 (4) | ||
| Total no. of events | 6 | 6 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (per year) | 0.05 | 0.06 | 0.76 (0.17-3.30) | .7136 |
| Duration of painful crises (days)§ | ||||
| Patients with events, n (%) | 69 (68) | 58 (63) | ||
| Total no. of days | 1476 | 1441 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (number of days per year) | 16.09 | 19.20 | 0.84 (0.50-1.40) | .4970 |
| Hospitalization or emergency department visit for vaso-occlusive crises†‡ | ||||
| Patients with events, n (%) | 42 (42) | 27 (29) | ||
| Total no. of events | 87 | 51 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (per year) | 0.87 | 0.61 | 1.43 (0.87-2.36) | .1636 |
| Days hospitalized for vaso-occlusive crises§ | ||||
| Patients with events, n (%) | 39 (39) | 23 (25) | ||
| Total no. of days | 526‖ | 256‖ | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (number of days per year) | 5.07 | 3.01 | 1.68 (0.76-3.75) | .2011 |
| Acute SCD complications‡ | ||||
| Patients with events, n (%) | 3 (3) | 2 (2) | ||
| Total no. of events | 6 | 3 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (per year) | NP¶ | NP¶ | ||
| Days hospitalized for acute SCD complications§ | ||||
| Patients with events, n (%) | 0 (0) | 3 (3) | ||
| Total no. of days | 0 | 6 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (number of days per year) | NP¶ | NP¶ | ||
| Red blood cell transfusions due to SCD‡# | ||||
| Patients with events, n (%) | 21 (21) | 19 (21) | ||
| Total no. of events | 39 | 49 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (per year) | 0.41 | 0.53 | 0.77 (0.38-1.58) | .4822 |
| End point | Ticagrelor group (n = 101) | Placebo group (n = 92) | Rate ratio (95% CI) | P |
|---|---|---|---|---|
| Vaso-occlusive crises (primary end point)*† | ||||
| Patients with events, n (%) | 70 (69) | 58 (63) | ||
| Total no. of events | 249 | 202 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (per year) | 2.74 | 2.60 | 1.06 (0.75-1.50) | .7597 |
| Painful crises†‡ | ||||
| Patients with events, n (%) | 69 (68) | 58 (63) | ||
| Total no. of events | 248 | 209 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (per year) | 2.73 | 2.67 | 1.02 (0.72-1.45) | .9037 |
| ACS†‡ | ||||
| Patients with events, n (%) | 5 (5) | 4 (4) | ||
| Total no. of events | 6 | 6 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (per year) | 0.05 | 0.06 | 0.76 (0.17-3.30) | .7136 |
| Duration of painful crises (days)§ | ||||
| Patients with events, n (%) | 69 (68) | 58 (63) | ||
| Total no. of days | 1476 | 1441 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (number of days per year) | 16.09 | 19.20 | 0.84 (0.50-1.40) | .4970 |
| Hospitalization or emergency department visit for vaso-occlusive crises†‡ | ||||
| Patients with events, n (%) | 42 (42) | 27 (29) | ||
| Total no. of events | 87 | 51 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (per year) | 0.87 | 0.61 | 1.43 (0.87-2.36) | .1636 |
| Days hospitalized for vaso-occlusive crises§ | ||||
| Patients with events, n (%) | 39 (39) | 23 (25) | ||
| Total no. of days | 526‖ | 256‖ | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (number of days per year) | 5.07 | 3.01 | 1.68 (0.76-3.75) | .2011 |
| Acute SCD complications‡ | ||||
| Patients with events, n (%) | 3 (3) | 2 (2) | ||
| Total no. of events | 6 | 3 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (per year) | NP¶ | NP¶ | ||
| Days hospitalized for acute SCD complications§ | ||||
| Patients with events, n (%) | 0 (0) | 3 (3) | ||
| Total no. of days | 0 | 6 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (number of days per year) | NP¶ | NP¶ | ||
| Red blood cell transfusions due to SCD‡# | ||||
| Patients with events, n (%) | 21 (21) | 19 (21) | ||
| Total no. of events | 39 | 49 | ||
| Total follow-up, patient-years | 89.0 | 80.0 | ||
| Incidence rate (per year) | 0.41 | 0.53 | 0.77 (0.38-1.58) | .4822 |
Vaso-occlusive crisis was defined as a composite of painful crisis and/or ACS. Number of vaso-occlusive crises is defined as the count of events of vaso-occlusive crisis assessed throughout the treatment period from randomization to end-of-study visit or date of premature study discontinuation.
Events with an onset date within 7 days of the previous event onset date are not counted as new events.
Number of secondary end point events as assessed throughout the treatment period from randomization to end-of-study visit or date of premature study discontinuation (observed follow-up).
For event-free patients, the duration is set to 0.
The difference in number of days hospitalized for vaso-occlusive crises was due to a few patients with many days of hospitalization.
Incidence rate not presented (NP) owing to the low number of events.
Sickle cell–related red blood cell transfusions as identified by study physician review.
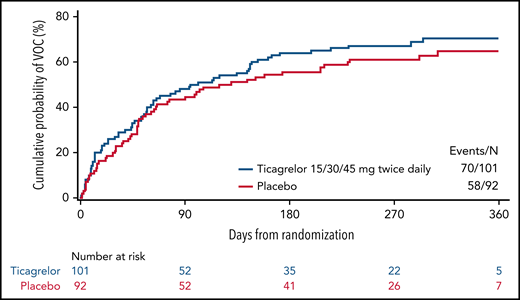
Time to first vaso-occlusive crisis (VOC). Kaplan-Meier plot (full analysis set).
Time to first vaso-occlusive crisis (VOC). Kaplan-Meier plot (full analysis set).
Results of the analyses of secondary efficacy end points are included in Table 2. There was no evidence of efficacy for ticagrelor across any of the secondary end points. Forty-two patients in the ticagrelor group and 27 patients in the placebo group were hospitalized for vaso-occlusive crises. The total number of days hospitalized for vaso-occlusive crises was 526 for the ticagrelor group and 256 for the placebo group.
Ticagrelor plasma exposure overall successfully reached the level predicted in the pharmacokinetic/pharmacodynamic modeling and simulation of HESTIA1 and HESTIA2 study data (Figure 3A). Geometric mean predose plasma concentrations of ticagrelor and its active metabolite AR-C124910XX over time are presented in supplemental Table 4. The number of patients contributing data decreased beyond 6 months. No relationship was observed between ticagrelor plasma concentrations and number of vaso-occlusive crises (negative binomial regression model, slope = 0.0129; P = .95) (Figure 3B).
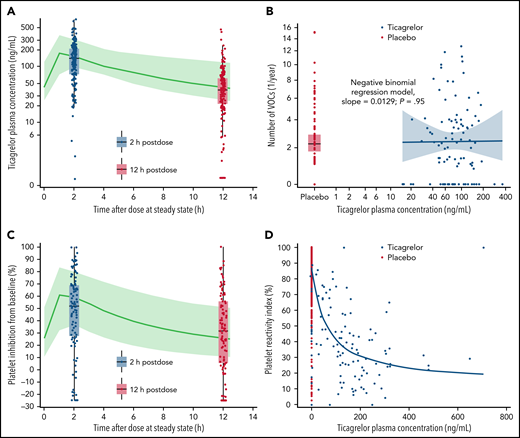
Ticagrelor plasma concentration. Observed and predicted plasma concentrations (A) and relationship with number of vaso-occlusive crises (VOCs) (B). Observed and predicted platelet inhibition (C) and relationship with platelet reactivity index (D). As shown in panels A and C, predicted plasma concentrations based on HESTIA1 and HESTIA2: green line is the median, and the green area is the 5% to 95% quantiles. Because ticagrelor is given every 12 hours, the 12 hours’ postdose time point is also the immediate predose (or trough) time point. As shown in panel B, individual geometric mean ticagrelor plasma concentration at visits 2, 4, and 9: blue line is the negative binomial regression model. As shown in panel D, time-matched platelet reactivity index and ticagrelor plasma concentrations, all visits: blue line is the exposure–response model of the maximum effect the drug can have.
Ticagrelor plasma concentration. Observed and predicted plasma concentrations (A) and relationship with number of vaso-occlusive crises (VOCs) (B). Observed and predicted platelet inhibition (C) and relationship with platelet reactivity index (D). As shown in panels A and C, predicted plasma concentrations based on HESTIA1 and HESTIA2: green line is the median, and the green area is the 5% to 95% quantiles. Because ticagrelor is given every 12 hours, the 12 hours’ postdose time point is also the immediate predose (or trough) time point. As shown in panel B, individual geometric mean ticagrelor plasma concentration at visits 2, 4, and 9: blue line is the negative binomial regression model. As shown in panel D, time-matched platelet reactivity index and ticagrelor plasma concentrations, all visits: blue line is the exposure–response model of the maximum effect the drug can have.
Platelet reactivity index values over time are listed in Table 3. Two hours after the first dose of ticagrelor, the median platelet inhibition was 43.9% compared with baseline. At week 4, the median platelet inhibition was 25.1% predose and 54.1% at 2 hours’ postdose, compared with baseline. Predose and postdose median platelet inhibition was 34.9% and 55.7%, respectively, compared with baseline, at month 6. Platelet inhibition successfully reached predicted levels (Figure 3C). Figure 3D shows the exposure–response relationship for platelet inhibition levels (maximum effect exposure–response model, estimated concentration of half-maximum effect = 61.7 ng/mL).
Platelet reactivity index (%) (pharmacodynamics analysis set)
| Visit | Time point | Ticagrelor | Placebo | |||
|---|---|---|---|---|---|---|
| n | Median (minimum, maximum) | Platelet inhibition, %* | n | Median (minimum, maximum) | ||
| Baseline | Predose | 68 | 84.8 (–5.9, 100.0) | 59 | 89.7 (–1.8, 100.0) | |
| Day 0 | 2 h postdose | 67 | 41.1 (1.7, 100.0) | 43.9 | 56 | 91.7 (2.7, 100.0) |
| Week 4 | Predose | 50 | 56.3 (–5.4, 96.7) | 25.1 | 43 | 91.6 (24.6, 100.0) |
| 2 h postdose | 50 | 40.0 (–0.8, 100.0) | 54.1 | 43 | 93.9 (10.5, 100.0) | |
| Month 6 | Predose | 45 | 47.2 (12.6, 100.0) | 34.9 | 43 | 90.2 (–7.6, 97.9) |
| 2 h postdose | 45 | 29.2 (0.0, 85.9) | 55.7 | 43 | 91.4 (7.5, 97.1) | |
| Visit | Time point | Ticagrelor | Placebo | |||
|---|---|---|---|---|---|---|
| n | Median (minimum, maximum) | Platelet inhibition, %* | n | Median (minimum, maximum) | ||
| Baseline | Predose | 68 | 84.8 (–5.9, 100.0) | 59 | 89.7 (–1.8, 100.0) | |
| Day 0 | 2 h postdose | 67 | 41.1 (1.7, 100.0) | 43.9 | 56 | 91.7 (2.7, 100.0) |
| Week 4 | Predose | 50 | 56.3 (–5.4, 96.7) | 25.1 | 43 | 91.6 (24.6, 100.0) |
| 2 h postdose | 50 | 40.0 (–0.8, 100.0) | 54.1 | 43 | 93.9 (10.5, 100.0) | |
| Month 6 | Predose | 45 | 47.2 (12.6, 100.0) | 34.9 | 43 | 90.2 (–7.6, 97.9) |
| 2 h postdose | 45 | 29.2 (0.0, 85.9) | 55.7 | 43 | 91.4 (7.5, 97.1) | |
Platelet reactivity index assessed by vasodilator-stimulated phosphoprotein assay. Data from month 12 and month 18 are not included owing to the small number of observations (ticagrelor month 12: predose, n = 8; postdose, n = 9; ticagrelor month 18: predose and postdose, n = 1. Placebo month 12: predose and postdose, n = 5; placebo month 18: predose and postdose, n = 0).
Relative to baseline median. Manually derived as: median change from baseline (%)/median platelet reactivity index (%) at baseline × 100, where median platelet reactivity index (%) at baseline is 84.8.
Safety
The median duration of exposure to the study drug in the ticagrelor group was 296.5 days (range, 5-598 days) and in the placebo group was 288.0 days (range, 34-548 days). The number of patients with AEs in any category on treatment and the most common AEs (frequency ≥10%) are shown in Table 4. Post hoc descriptive results of safety data broken down by region (Africa and Asia, Europe, and North and South America) are presented in supplemental Table 5. AEs were reported in 95 patients (95%) in the ticagrelor group and 84 patients (91%) in the placebo group.
Number of patients with AEs in any category on treatment and most common AEs by preferred term (safety analysis set)
| AE category* | Ticagrelor group (n = 100) | Placebo group (n = 92) |
|---|---|---|
| Any AE | 95 (95) | 84 (91) |
| AE with maximum intensity* | ||
| Mild | 21 (21) | 16 (17) |
| Moderate | 32 (32) | 39 (42) |
| Severe | 42 (42) | 29 (32) |
| Patients with bleeding events | 9 (9) | 8 (9) |
| Major† | 1 (1) | 0 |
| Clinically relevant nonmajor‡ | 0 | 0 |
| Minor§ | 8 (8) | 8 (9) |
| Any AE with outcome death | 3 (3) | 1 (1) |
| Any serious AE | 44 (44) | 29 (32) |
| Any AE leading to study drug discontinuation | 4 (4) | 4 (4) |
| AEs with ≥10% frequency | ||
| Sickle cell anemia with crisis | 71 (71) | 57 (62) |
| Pain in extremity | 28 (28) | 23 (25) |
| Headache | 24 (24) | 18 (20) |
| Back pain | 21 (21) | 12 (13) |
| Upper respiratory tract infection | 20 (20) | 24 (26) |
| Cough | 17 (17) | 7 (8) |
| Arthralgia | 14 (14) | 14 (15) |
| Abdominal pain | 15 (15) | 10 (11) |
| Malaria | 15 (15) | 8 (9) |
| Pyrexia | 13 (13) | 13 (14) |
| Anemia | 10 (10) | 11 (12) |
| AE category* | Ticagrelor group (n = 100) | Placebo group (n = 92) |
|---|---|---|
| Any AE | 95 (95) | 84 (91) |
| AE with maximum intensity* | ||
| Mild | 21 (21) | 16 (17) |
| Moderate | 32 (32) | 39 (42) |
| Severe | 42 (42) | 29 (32) |
| Patients with bleeding events | 9 (9) | 8 (9) |
| Major† | 1 (1) | 0 |
| Clinically relevant nonmajor‡ | 0 | 0 |
| Minor§ | 8 (8) | 8 (9) |
| Any AE with outcome death | 3 (3) | 1 (1) |
| Any serious AE | 44 (44) | 29 (32) |
| Any AE leading to study drug discontinuation | 4 (4) | 4 (4) |
| AEs with ≥10% frequency | ||
| Sickle cell anemia with crisis | 71 (71) | 57 (62) |
| Pain in extremity | 28 (28) | 23 (25) |
| Headache | 24 (24) | 18 (20) |
| Back pain | 21 (21) | 12 (13) |
| Upper respiratory tract infection | 20 (20) | 24 (26) |
| Cough | 17 (17) | 7 (8) |
| Arthralgia | 14 (14) | 14 (15) |
| Abdominal pain | 15 (15) | 10 (11) |
| Malaria | 15 (15) | 8 (9) |
| Pyrexia | 13 (13) | 13 (14) |
| Anemia | 10 (10) | 11 (12) |
Data are expressed as n (%). AEs on treatment: AEs with onset date on or after the date of first dose of study treatment and on or before the date of last dose of study treatment plus 7 days. AEs were recorded by using the Medical Dictionary for Regulatory Activities version 23.0.
Patients with multiple events in the same category are counted only once in the maximum intensity category. Patients with events in >1 category are counted once in each of those categories.
Major bleeding was defined as any fatal bleeding; clinically overt bleeding associated with a decrease in hemoglobin levels of at least 20 g/L (2 g/dL); bleeding that is retroperitoneal, pulmonary, intracranial, or otherwise involves the central nervous system; or bleeding that requires surgical intervention in an operating suite.
Clinically relevant nonmajor bleeding was defined as overt bleeding for which a blood product is administered and which is not directly attributable to the patient’s underlying medical condition, and bleeding that requires medical or surgical intervention to restore hemostasis, other than in an operating room.
Minor bleeding was defined as any overt or macroscopic evidence of bleeding that does not fulfill the criteria for either major bleeding or clinically relevant, nonmajor bleeding.
Nine patients (9%) in the ticagrelor group and eight patients (9%) in the placebo group had at least one bleeding event (Table 4). One major bleeding event was reported as a fatal intracranial hemorrhage in a patient in the ticagrelor group. This relates to the patient in the ticagrelor group who had an AE of cerebrovascular accident with outcome of death (as discussed later in this section). All other bleeding events were classified as minor bleeding. No patients had bleeding events classified as clinically relevant nonmajor events during the study treatment period. All bleeding events were spontaneous except for one trauma-related bleeding (gingival bleeding after brushing). There were no procedural bleeds.
Serious AEs were reported in 44 patients (44%) in the ticagrelor group and 29 patients (32%) in the placebo group. The most commonly reported serious AE by preferred term was sickle cell anemia with crisis, which was reported in 39 patients (39%) in the ticagrelor group and 24 patients (26%) in the placebo group.
Three patients in the ticagrelor group and one patient in the placebo group had a fatal AE. One patient in the ticagrelor group had a fatal cerebrovascular accident, with computed tomography scan findings indicating subarachnoid bleed in the right hemisphere together with signs of meningitis. A second patient in the ticagrelor group had fatal septicemia, and the cause of death was confirmed by autopsy as meningitis and pneumonia, although no pathogen was identified. The third patient in the ticagrelor group was reported by the investigator as having had a sudden death of unclear etiology. This patient lost consciousness during exercise and was dead on arrival at hospital. No imaging or autopsy was performed, and the diagnosis is thus unclear. This patient had a normal transcranial Doppler and a normal cranial magnetic resonance imaging result before randomization. In the placebo group, one patient had a fatal event of sickle cell anemia with crisis after being hospitalized with severe general body pain and fever.
Discussion
This international, phase 3, randomized controlled trial was designed to evaluate the efficacy of ticagrelor vs placebo to reduce vaso-occlusive crises in pediatric patients with SCD. Study enrollment across 4 continents was intended to provide equitable trial access for individuals with SCD. Following a recommendation from the independent data and safety monitoring committee, the sponsor terminated the study early. The planned enrollment had completed with 193 randomized patients, the recruited population was consistent with the intended study population, and the planned age distribution was achieved. In total, 451 vaso-occlusive crises were recorded during the study treatment period, and the sample size assumption of a mean of 2.0 crises per year in the placebo group was exceeded. The early termination, only 4 months ahead of the anticipated completion date, did not affect the ability to perform the predefined analysis and was not considered to have had a meaningful impact on the robustness of the efficacy end points.
The study did not meet its primary objective of efficacy for ticagrelor over placebo in reducing the number of vaso-occlusive crises. The results of the primary efficacy analysis were consistent across all predefined subgroups, including those based on age and baseline hydroxyurea use. No evidence of efficacy with ticagrelor was observed in the secondary endpoint results. The observed ticagrelor plasma concentrations reached the levels predicted based on the modeling of data from HESTIA1 and HESTIA2.30,31 Median platelet inhibition with ticagrelor at 6 months was 34.9% predose and 55.7% at 2 hours’ postdose, compared with baseline; thus, as prospectively planned, this exceeded the level of platelet inhibition (∼25%) observed with prasugrel in the DOVE trial.24 Notwithstanding the exposure–response relationship for platelet inhibition levels, no relationship was observed between ticagrelor plasma concentrations and number of vaso-occlusive crises. Even though ticagrelor’s P2Y12 inhibition is reversible, the antiplatelet effect over the dosing interval is well documented and shown to have a clinical effect in earlier studies and in other populations.25,28-31 Despite the pathophysiological rationale, HESTIA3 casts further doubt on the utility and safety of platelet inhibition for the reduction of vaso-occlusive crises in children with SCD.
The observed median total exposure of 296.5 days on ticagrelor and 288.0 days on placebo was sufficient to allow for robust evaluation of the safety profile. The overall AE profile was as expected in a pediatric population with SCD. However, the proportion of patients reporting serious AEs was higher in the ticagrelor group than in the placebo group, driven by the reports of sickle cell anemia with crisis. In addition, there were 4 deaths reported among the 193 study patients during ∼10 months of treatment, suggesting an estimated mortality of 2.5 per 100 person-years, in line with reasonable expectations in this patient group, the majority of whom were recruited in Sub-Saharan Africa.35,36 Three patients in the ticagrelor group and one patient in the placebo group died during the study. The deaths in the ticagrelor group were from an intracranial hemorrhage, septicemia, and unclear etiology. The observed higher frequency of splenectomy in the medical and surgical history at baseline in the ticagrelor group, compared with placebo, did not seem to have any impact on the numerical differences observed in the safety results.
The preclinical data for increased platelet activation in patients with SCD are referred to in the “Introduction.” In addition, there is growing evidence of von Willebrand factor interaction via GPIbα, tissue factor, and thrombin to promote neutrophil–platelet aggregation.37 The lack of effect of ticagrelor on the incidence of vaso-occlusive crises in this study may further implicate the primacy of neutrophil involvement.9-11 It may be that the propagation of cellular aggregation and initiation of vaso-occlusive crises is more dependent on endothelial interaction via von Willebrand factor, selectins, GPIbα, and/or GPIIb/IIIa.37
Despite a clear biological rationale for antiplatelet therapy in SCD, and the greater platelet inhibition in HESTIA3 than in the previous DOVE study, there was no benefit of ticagrelor in this patient group. In the DOVE study, there was a trend toward efficacy but no significant reduction in rates of vaso-occlusive crisesversus placebo.23 Thus, there are now 2 studies using P2Y12 inhibitors that have failed to show efficacy in children, indicating that platelet inhibition (at least via P2Y12) is not a mechanism to reduce vaso-occlusive crises in pediatric patients with SCD. We can only speculate on why inhibition of platelet activation and aggregation by P2Y12 inhibitors is not a successful mechanism for the prevention of vaso-occlusive crises compared with, for example, that of crizanlizumab.14 One possible reason is that the degree of platelet activation and the cumulative endothelial damage is not as substantial in children as it is in adults living with SCD; thus, any possible benefit of platelet inhibition may be less evident.
Conclusions from this study should not be applied to adults living with SCD or pediatric patients with genotypes other than the HbSS and HbS/β0 studied in HESTIA3 (eg, hemoglobin C). P2Y12 inhibition also affects only one aspect of the complex molecular and cellular pathophysiology of SCD and, although subgroup analyses showed no synergy of ticagrelor with hydroxyurea, perhaps antiplatelet therapy will have an adjunctive role in future multiagent therapy. At present, we conclude that ticagrelor is not effective for the prevention of vaso-occlusive crises in pediatric patients with the SCD genetic traits HbSS and HbS/β0. Unfortunately, the negative result of the HESTIA3 trial of ticagrelor joins a list of negative results for other investigational agents in SCD, including senicapoc,38 prasugrel,23 regadenoson,39 sevuparin,40 poloxamer 188,41 olinciguat,42 and rivipansel.43 There continues to be a significant unmet need for prevention and treatment of vaso-occlusive crises in SCD.
Acknowledgments
The authors thank all study participants and their families.
The HESTIA3 study was funded by AstraZeneca. Medical writing support was provided by Anja Becher of Oxford PharmaGenesis and was funded by AstraZeneca.
Authorship
Contribution: M.M.H. contributed to design, acquisition, and interpretation of data, and drafting and critical revision of the manuscript; M.R.A. contributed to conception, design, and acquisition of data, and critical revision of the manuscript; J.G. contributed to design, acquisition, and interpretation of data, and critical revision of the manuscript; B.P.D.I. contributed to the design and data acquisition, and critical revision of the manuscript; J.K. contributed to the to HESTIA3 study concept and design, acquisition, analysis and interpretation of data, and critical revision of the manuscript; A.D.M. contributed to HESTIA3 study concept and design, interpretation of data, and critical revision of the manuscript; V.N. contributed to data acquisition and critical revision of the manuscript; V.M. contributed to participant recruitment and data collection, and review of all manuscript versions; M. Apte contributed to acquisition of data and critical revision of the manuscript; A.I. contributed to patient recruitment, data acquisition, and manuscript revision; A.M.T. contributed to acquisition and interpretation of data, and revision of the manuscript; M.A. contributed to acquisition and interpretation of data, and revision of the manuscript; M.Å. and M.N. contributed to HESTIA3 study design, acquisition, and interpretation of pharmacokinetic and pharmacodynamic data, and critical revision of the manuscript; N.M. contributed to the HESTIA3 study concept and design, acquisition, analysis and interpretation of data, and critical revision of the manuscript; and A.H. and A.R.B. contributed to the HESTIA3 study concept and design, acquisition and interpretation of data, and critical revision of the manuscript.
Conflict-of-interest disclosure: M.M.H. is a consultant to AstraZeneca, bluebird bio, Cyclerion, Dova Pharmaceuticals, Forma Therapeutics, Keros Therapeutics, Micelle BioPharma, and Novartis. M.R.A. is a consultant to Novartis; received research funding from Novartis, Global Blood Therapeutics, and AstraZeneca; received speaker fees from Novartis and Novo Nordisk; is on an advisory board for Global Blood Therapeutics; is on a data safety committee for Vertex Pharmaceuticals; and is on a steering committee for AstraZeneca. B.P.D.I. received educational funding from AstraZeneca, Celgene, and Novartis; and honoraria from AstraZeneca, Cyclerion, and Novartis. J.K. is a consultant to Agios, AstraZeneca, Beam, Forma Therapeutics, Graphie, and Novartis; and on a Data and Safety Monitoring Board for Novo Nordisk. A.D.M. is a consultant to AstraZeneca, Janssen, Medtronic, Pfizer, and Stasys; and received research funding from Pfizer. A.I. receives research funding from AstraZeneca, Cyclerion, GBT, Imara, Novartis, and OctaPharma; and is a consultant for GBT and Novartis. M.A., M.Å., N.M., M.N., A.H., and A.R.B. are employees of AstraZeneca. The remaining authors declare no competing financial interests.
A complete list of the principal investigators of the HESTIA3 study appears in supplemental Appendix 2 (available on the Blood Web site).
Correspondence: Matthew M. Heeney, Division of Hematology/Oncology, Boston Children’s Hospital, Harvard Medical School, 300 Longwood Ave, Boston, MA 02115; e-mail: matthew.heeney@childrens.harvard.edu.
Data underlying the findings described in the manuscript may be obtained in accordance with AstraZeneca’s data-sharing policy described at: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal