

Abstract
Hematopoietic stem cells (HSCs) are the blood-forming stem cells thought to be responsible for supporting the blood system throughout life. Transplantability has long been the flagship assay used to define and characterize HSCs throughout ontogeny. However, it has recently become clear that many cells emerge during ontogeny that lack transplantability yet nevertheless are fated to ultimately contribute to the adult HSC pool. Here, we explore recent advances in understanding the numbers and kinetics of cells that emerge during development to support lifelong hematopoiesis; these advances are made possible by new technologies allowing interrogation of lifelong blood potential without embryo perturbation or transplantation. Illuminating the dynamics of these cells during normal development informs efforts to better understand the origins of hematologic disease and engineer HSCs from differentiating pluripotent stem cells.
Introduction
Hematopoietic stem cells (HSCs) are the blood-forming stem cells thought to be responsible for maintaining and replenishing blood throughout life and during stress.1,2 As the first stem cell population identified, HSCs serve as a paradigm for stem cell biology.1,2 In adult vertebrates, they are found in the bone marrow (mammals) or kidney marrow (fish).3-5 Classically, they have been most rigorously defined by their functional ability to reconstitute all major blood lineages for long periods of time when transplanted. Transplantability has been exploited as a functional readout to identify their unique molecular and other phenotypic markers. These markers have been used in turn to define their unique transcriptional profiles, interactions with the bone marrow (BM) niche, and functional potential and assess how perturbations to homeostasis (eg, infection, disease, and genetic manipulation) influence numbers and function. Limiting dilution transplantation and imaging of mammalian whole BM have established that transplantable HSCs are extremely rare in adults, although variable with age.3,6-11 However, if we think of HSCs as the cells that maintain blood for life, a reasonable question is whether transplantability is the best approach to interrogate the size of the entire HSC pool, especially given the many stress hurdles that HSCs must overcome to effectively repopulate and reconstitute the blood of ablated recipients (eg, homing, normoxia, encountering a niche damaged by ablation, and ex vivo manipulation). HSCs capable of durable self-renewal,12-16 as evidenced by repeated serial transplantation, are experimentally subjected to repeated external stressors and regenerative insult. Indeed, serial transplantation dramatically reduces the clonal complexity of reconstituted marrow.17 Thus, durably self-renewing HSCs represent rare cells that can survive repeated exposure to intense stress and might not equate perfectly to physiologic HSCs that quietly support blood homeostasis in an unperturbed setting throughout life.
Here, we explore recent advances in understanding the number and dynamics of cells that emerge during development to support lifelong hematopoiesis. Many of these advances are made possible by new technologies and approaches that allow interrogation of lifelong blood potential without embryo perturbation or transplantation. Illuminating the dynamics of these cells during normal development informs efforts to better understand the origins of hematologic disease and engineer HSCs from differentiating pluripotent stem cells.
Historical perspective on transplantability as the gold standard of HSC activity
The history of how HSCs were conceived, identified, and characterized helps to explain the original estimated numbers of clonal specification events and how those numbers expand over the course of ontogeny. At the end of the 1800s, a stem cell foundation for blood had been proposed,18 and in 1916, Danchakoff19 articulated a reasonably sophisticated model of stem cells and maturation supporting diverse lineage output and malignancy. Studies in the 1960s by McCulloch and Till and associates20-24 established assays with quantifiable output, such as splenic colony formation to define clonality and lineage output, which were complemented by in vitro assays pioneered by Bradley and Metcalf25 and expanded over the years to test hematopoietic potential.26 In vitro assays, however, were not able to determine self-renewal potential, and ultimately, the gold standard of HSC potential became the ability to completely reconstitute for a long period of time (months or longer) the blood lineages of an animal lethally irradiated to ablate its hematopoietic system (so-called long-term reconstitution [LTR]) and more stringently to serially reconstitute through multiple subsequent transplantations.27 Using these assays, multiple groups phenotypically defined the cell populations containing HSCs and other lineage maturation intermediates (eg, size and density, adhesive properties, and cell-surface molecular markers).28 Refined molecular phenotypes were iteratively developed that highly enriched for both murine (ie, a cocktail of antibodies to eliminate markers of lineage differentiation [Lin−] in combination with Sca-1, c-Kit, CD150, and EPCR and the absence of CD48) and human HSCs (ie, CD34+CD38−CD90+CD45A−CD49f+).9,10,28-39
Developmental ontogeny of HSCs
Classical embryology studies implicated endothelial cells as the immediate precursors of the earliest blood cells in vertebrate species, including pig, rabbit, guinea pig, cat, mouse, chick, and human,18,40-44 and hemogenic endothelium was confirmed in the modern era using techniques including dye-based and genetic in vivo lineage-tracing studies,45,46 in vitro assays,47,48 functional genetic studies,49 and direct imaging in live zebrafish and mouse embryos.50-52 However, precisely where this hemogenic endothelium was located has been heavily investigated and disputed. The earliest emergence of blood is observed in extraembryonic tissues (the mammalian yolk sac and chick area vasculosa18,40,43,53-55), and early studies seemed to provide evidence of diverse lineage potential and LTR by progenitors from that region.56-59 However, in the 1970s, Dieterlen-Lièvre and associates60,61 showed, using avian embryos, that all long-term hematopoietic activity is derived intraembryonically from mesodermal tissue containing the dorsal aorta, whereas yolk sac–derived lineages disappeared in adulthood, and this observation was confirmed in amphibian embryos.62-65 In the 1990s, it was established in mouse that an intraembryonic trunk region termed the aorta-gonado-mesonephros (AGM), containing the primitive dorsal aorta, is a potent source of colony-forming units and HSCs with LTR activity between ∼9 days postcoitum (E9.0) and E10.5.66-72 These studies raised the possibility that definitive HSC potential observed in the yolk sac might derive from circulating progenitors that originate in the AGM, because circulation initiates at ∼E8.0.73
A clear objective was to identify and geographically locate the putative source of intraembryonic HSCs, but it was not clear that the collection of phenotypic markers known to identify adult HSCs would be expressed in developing HSCs. Markers such as c-Kit, Sca-1, CD31, and CDH574-76 and genes newly discovered to be functionally important in the specification of HSCs (eg, Runx177-79) were found to label a population in the dorsal aorta of vertebrates with HSC potential. We now know that newly emerging HSCs and hematopoietic progenitors form clusters of c-Kit+ cells in the dorsal aorta (intra-aortic cell clusters) in most vertebrates, often localized near the midcaudal region of the aorta close to the junction with the vitelline artery (VA).3,66,80,81 Limited numbers of c-Kit+ cells are seen in the aorta at E9.5 (∼0-5 cells).80 In contrast, large numbers are evident by E10.5 (∼609 ± 84 c-Kit+ cells),80 with some forming clusters of >10 cells (∼7 large clusters visible at E10.5). c-Kit+ cell numbers begin declining by E11.5 (439 ± 87 c-Kit+ cells; ∼1 large cluster)80 as newly specified HSCs and progenitors move on to the fetal liver (FL). By E14.5, only 30 to 40 single c-Kit+ cells are detected in the aortic endothelium.80 Similarly, the numbers of c-Kit+ clusters and large c-Kit+ clusters localized to the VA and umbilical artery (UA) peak at E10.5 (250-300 c-Kit+ per vessel at E10.5 and 100-200 at E11.5; 5 large clusters at E10.5 and 1 at E11.5 per vessel). Interestingly, the largest c-Kit+ clusters are found in the VA (76 c-Kit+ cells per cluster) and UA (48 c-Kit+ cells per cluster) rather than in the aorta proper (19 c-Kit+ cells per cluster).80 As investigators sought to better define the phenotype and function of nascent mammalian HSCs, they turned to the functional definition that had been established for adult HSCs: LTR potential in adults or neonates undergoing transplantation.8,68,82-85
Original estimates of the numbers of newly emerging HSCs during mammalian development were based on the transplantation of freshly isolated murine embryonic tissues.7,8,82,86 When these transplantations are performed at limiting dilution, the number of transplantable HSCs (also known as hematopoietic repopulating units [RUs]) based on Poisson statistics can be estimated.3,7,8,50,68,70,82,84-89 Via this approach, only 1 to 2 RUs are detected in E10.5 to E11.5 murine embryos.8,68,70 Thus, nascent HSCs as defined by transplantation are rare at midgestation in the developing mammalian embryo.90,91 Although these elegant studies shed light on the embryonic origin of HSCs and their migration to successive anatomic locations (AGM, FL, placenta, and BM), transplantation provides only an instantaneous snapshot of the number of RUs present at a particular time and place.6,92 Moreover, nascent HSCs are not programmed to migrate to or engage with the adult BM niche, as would be required in most transplantation studies. Indeed, one can detect many more RUs when AGM-derived cells are transplanted at limiting dilution into neonates compared with adults.93 Thus, transplantation-based estimates of nascent HSC numbers overlook cells too immature to repopulate the hematopoietic system of an ablated recipient, but that would ultimately realize their potential to contribute to lifelong hematopoiesis if left developmentally unperturbed. These observations explain why transplantation-based estimates of nascent HSC potential underestimate the actual number of physiologically specified HSCs. The limitations of transplantation-based techniques set the stage for new approaches to estimate clonal specification of HSCs: ex vivo reaggregate culture systems and fate mapping in unmanipulated embryos.
Specification is more frequent than previously thought: new insights from novel approaches
Beginning in the early 2000s, the Medvinsky laboratory94-97 pioneered a new culture system that allowed for functional detection of immature HSC precursors with LTR potential. Dissected tissues from E8.5 to E11.5 murine embryos were kept intact or dissociated and then reaggregated in the presence or absence of supportive OP9 stromal cells and subsequently cultured as explants at the air-liquid interface in the presence of cytokines and serum.71,94-98 These studies show that both expansion of existing nascent HSCs and de novo HSC specification are ongoing in cultured embryo explants.92,97 This system has been used to investigate the hierarchy of HSC precursors between E9.5 and E11.5 by assessing prospectively isolated candidate populations for HSC potential after culture in reaggregated explants.94,96,97 Combined with subsequent limiting dilution transplants, 1 to 2 E9.5, ∼50 E10.5, and ∼70 E11.5 immature HSC precursors are detectable in the AGM and major arteries,95 revealing far greater numbers of cells that ultimately develop transplantable HSC potential than estimated by previous approaches.7,8,82,86 Moreover, these studies reveal that the number of HSC precursors at E11.5 in the AGM matches the functional HSC number in the E12.5 FL, suggesting that the early FL HSC pool derives from a mixture of maturing and transplantable HSC precursors rather than from extensive division of rare previously specified HSCs.95 Thus, the E12.5 FL may comprise a niche supportive of HSC maturation, although this model requires further exploration.6
Although reaggregated explant cultures have proven extremely useful to uncover and define HSC precursor phenotypes at multiple stages of mammalian ontogeny, they probably do not fully recapitulate the in vivo specification and maturation environment (eg, ex vivo cultures involve supraphysiologic cytokine concentrations). Nonphysiologic RUs may emerge in cultures, and the potential of all legitimate HSC precursors may not be realized.6,71,94-98 Moreover, transplantation of fresh or cultured embryonic tissues reveals only the functional potential of a given population rather than its actual in vivo fate (ie, contribution to lifelong blood production; Figure 1). Therefore, estimated numbers of HSCs and HSC precursors that derive from these studies may not reflect in vivo reality. Indeed, the numerous c-Kit+ intra-aortic cells observed between E10.5 and E11.5 suggest that endothelial-to-hematopoietic transition (EHT) is not a rare event80 (Figure 1). Determining exactly how many cells in the aorta and UA and VA actually contribute to the adult HSC pool requires noninvasive lineage tracing.
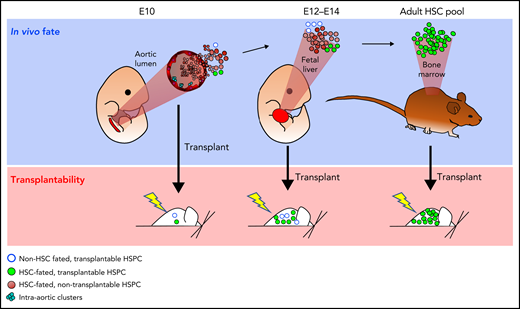
Possible differences in unperturbed in vivo fate and transplantability. At key developmental time points, noninvasive fate mapping studies and phenotypic analyses indicate the presence of far more HSC-fated precursors than are capable of engraftment in transplantation assays. At E10 in the aortic lumen and E12 to E14 in the FL, a few rare HSC-committed hematopoietic stem and progenitor cells (HSPCs) are already sufficiently mature to be transplantable (green), but most HSC-fated HSPCs are not (brown). A small transplantable pool of HSPCs may also be present that would never realize an adult HSC fate without transplantation (blue circles). Differences in unperturbed in vivo fate vs transplantation potential may arise from immaturity of HSPCs at the time of transplantation, stresses associated with ex vivo manipulations, and niche availability.
Possible differences in unperturbed in vivo fate and transplantability. At key developmental time points, noninvasive fate mapping studies and phenotypic analyses indicate the presence of far more HSC-fated precursors than are capable of engraftment in transplantation assays. At E10 in the aortic lumen and E12 to E14 in the FL, a few rare HSC-committed hematopoietic stem and progenitor cells (HSPCs) are already sufficiently mature to be transplantable (green), but most HSC-fated HSPCs are not (brown). A small transplantable pool of HSPCs may also be present that would never realize an adult HSC fate without transplantation (blue circles). Differences in unperturbed in vivo fate vs transplantation potential may arise from immaturity of HSPCs at the time of transplantation, stresses associated with ex vivo manipulations, and niche availability.
Recent novel noninvasive lineage-tracing approaches allow for tracking and quantification of numbers of hematopoietic precursors with lifelong hematopoietic potential in an unperturbed setting. For example, some fluorescence-labeling methods used to study the clonal origins of blood are based on the Brainbow allele, which was initially developed to study neuronal circuitry.99 The Brainbow allele allows inducible random expression of combinations of multiple copies of 4 different fluorescent proteins (CFP, GFP, RFP, and YFP) and thereby produces ∼100 detectable colors in neurons.99 The system was adapted for use in zebrafish as a CRE-inducible zebrabow allele (which harbors ∼20 transgene insertions and produces 40 distinguishable fluorescent colors).11 Tamoxifen-inducible draculin (drl:creERT2; active during early hematopoiesis) in combination with the zebrabow allele revealed that zebrafish embryos harbor ∼30 HSC precursors (21 pre-HSCs before HSC emergence at 24 hours postfertilization [hpf] and 34 HSCs at 72 hpf).11 In agreement with this estimate, hsp70l LASER-induced labeling of individual HSC precursors in Tg(cd41:eGFP; bactin2:switch; hsp70l; mCherry-T2A-creERT2) fish estimates 25 to 36 HSCs in the ventral dorsal aorta.11,100 Together, these data suggest a larger number of independently specified clonal HSC precursors than predicted by transplantation approaches in mice (1-2 HSCs at E11.5) and support a model in which HSC specification is more frequent, especially considering that mice are larger than zebrafish.
In mice, Fatima et al101 sought to label a single E7.5 HSC progenitor via low marking of embryonic blood precursors by exposing Abcg2-CreERT2Cre-ERT2/Cre-ERT2; Rosa26Lox-STOP-Lox-YFPYFP/YFP embryos to a single dose of 4-hydroxytamoxifen at E7.5. ABCG2 is a member of the ATP-binding cassette transporter family expressed by all HSCs that allows for embryonic labeling of some HSC precursors.102 This strategy resulted in 0.3% to 0.8% labeling of adult myeloid and lymphoid blood lineages, confirming labeling of HSC precursors. Assuming that 0.3% labeling of the blood corresponds to a single labeled HSC, the authors estimated that at least 333 HSC precursors exist in E7.5 to E8.5 murine embryos.101,102 The inability of this system to determine how many HSC precursors were actually labeled at E7.5 precludes the model from placing an upper bound on the actual number of HSC precursors present at E7.5.
We previously used a CRE-inducible Confetti allele,103 also derived from the Brainbow system, to estimate independent clonal HSC specification frequency during embryogenesis at key time points. The Confetti allele can be recombined to 1 of 4 colors (GFP, YFP, RFP, or CFP), and expression persists in subsequent cellular progeny.17,92,103 Although only 4 colors are available, the distribution of colors in the adult HSC-derived peripheral blood can be mathematically correlated to the number of discrete clones present at the time of recombination; if few clones are present at recombination, color distributions show high mouse-to-mouse variance (MtMV), whereas if numerous clones are initially labeled, the variance is small.17,92 This approach allowed estimates of the number of murine mesodermal blood precursors (Flk1-Cre) as ∼700 (∼E7), endothelial precursors (VE-cadherin-Cre) as ∼600 (∼E8.5), and mature HSC precursors (Vav1-Cre) as ∼600 (∼E11-E14).
The MtMV in the Confetti color distribution yields the most accurate estimate of initial labeling events in the range of 50 to 2500 cells; outside this range, estimates are qualitative rather than quantitative (ie, very few vs very large numbers of initially labeled events).92 Additionally, Confetti labeling efficiency needs to be >3%, and >500 cells must be analyzed.92 Therefore, each of these technical nuances must be considered when evaluating the likely fidelity of estimates of clonal complexity using MtMV of Confetti labeling. Furthermore, the system does not allow for the tracking of individual clones. Nonetheless, the evolution of the global clonal complexity of a cellular system as a whole can be followed, allowing one to interrogate the effects of aging and other insults on the global clonal complexity of the blood.17
Recently, a number of genetic barcoding technologies have emerged and been applied to adult hematopoiesis. The Sleeping Beauty system, based on a doxycycline-inducible transposase,104 suggested that native hematopoiesis is maintained by thousands of multipotent progenitors, rather than HSCs, and that HSCs predominantly contribute to megakaryopoiesis.104,105 This finding remains controversial, supported by studies where hematopoiesis continued when HSCs were ablated in adults106-108 but also refuted by lineage-tracing approaches that support a traditional view of HSC contribution to steady-state hematopoiesis.109-111 Additional innate barcoding systems include PolyLox, which generates random barcodes via induced recombination of a cassette containing many loxP sites,112 and CRISPR-based molecular recording or scarring, which revealed skewed contribution of FL HSC precursors and reduced clonal diversity in blood after 5-fluorouracil treatment.113-115 To date, these technologies have not been reported to estimate frequency of developmental HSC specification, but we are likely to have insights in the near future, including tracking of individual cells and study of lineage output and proliferative behavior. However, if future work establishes definitively that multipotent progenitors contribute substantially to steady-state hematopoiesis, key questions will need to be revisited to clarify our understanding of the establishment of the adult hematopoietic system, such as whether multipotent progenitors are specified independently of classically defined HSCs.116
Insights from human studies
Gaining insights into the frequency of HSC specification during human ontogeny has been more challenging, but recent technologic innovations are yielding fresh understanding. Early studies seeking to investigate human clonal dynamics took advantage of X inactivation and the evolution of telomere lengths with age. These and other studies hinted at the highly polyclonal nature of human adult blood and suggested that most HSC expansion takes place during childhood and adolescence and stabilizes in adulthood.117-120 Observations of shifting patterns of X inactivation with age in women provided early glimpses of age-associated clonal hematopoiesis that have subsequently been laid bare in studies of large cohorts of patients whose blood was subjected to whole-exome sequencing.119,121-124
In 2014, Behjati et al125 reported a proof of principle that somatic mutations acquired naturally over time and detected via whole-genome sequencing (WGS) of many independently expanded clones (murine gut organoids in this case) could be exploited to trace the developmental history of a given tissue. Using estimates of the rate of mutation accrual and the distribution of mutations among independently examined clones, these naturally acquired barcodes can be organized into a hierarchy and placed in ontologic time all the way back to the 2-cell embryo. WGS is now revolutionizing our understanding of the clonal dynamics of human blood development during ontogeny, as well as the clonal evolution of mutations associated with hematologic disease.121,122,124
In a groundbreaking study, Lee-Six et al118 used natural accumulation of somatic mutations in clones of expanded human HSPCs to examine the origins, lineage relationships, and clonal complexity of the human hematopoietic system via WGS. They and others estimated that adult human blood emerges from numerous embryonic clones, with ∼50 000 to 200 000 HSCs actively contributing to hematopoiesis in a healthy adult at any given moment and most expansion of adult blood precursors occurring during adolescence.118,126 This approach has now been applied to expanded HSPC clones isolated from the liver and BM of human fetuses. Here, investigators demonstrated explicitly that even early in development, HSPCs have acquired dozens of somatic mutations.127 They also concluded that the cells of a 2-cell embryo do not contribute equally to downstream tissues.127 Similarly, Ludwig et al128 recently demonstrated that acquired somatic mutations in the mitochondrial genome, which is almost entirely transcribed, can be exploited in single-cell RNA-sequencing data sets as a useful barcode to examine clonal relationships among sequenced human HSPCs. Most recently, WGS of expanded clones has been applied to illuminate the developmental timing of the acquisition of driver mutations in patients with myeloproliferative neoplasms (MPNs).129 Here, JAK2V617F and DNMT3A mutations were estimated to arise in early fetal life and childhood, expanding for years (sometimes many decades) before clinical presentation of an MPN. This finding has major implications for thinking about when and how screening should be performed for MPNs. However, because each of these studies depends on the sequencing of clones expanded in vitro, the data are inherently biased for cells capable of in vitro expansion, which may or may not be influenced by disease mutations. Furthermore, it is difficult to exclude mutation acquisition during colony expansion. Finally, the high cost of WGS imposes a limitation on the number of sequenced clones and potentially the ability to effectively extrapolate the data.
Adding complexity: transient and tissue-specific populations
Multiple investigations have revealed the existence of transient embryonic HSPCs with restricted lineage potential, generally erythroid and erythromyeloid oligopotency.130,131 More recently, a suite of studies has defined long-lasting or permanent precursors with highly restricted lineage potential, including tissue-resident macrophage and specific subsets of adult lymphoid cells, which may not derive from HSCs.66,132-134 Some of these HSPCs may have a role in the emergence of HSCs (eg, proinflammatory AGM-associated macrophages).135 Here, we briefly highlight additional layers of complexity that derive from these newly discovered transient and restricted lineage HSPCs, as well as how they might confound interpretations of the dynamics and magnitude of lifelong HSC specification during embryogenesis.
During embryonic development, transient hematopoietic waves have been detected that emerge from the yolk sac, contribute with little or no frequency to the adult HSC pool, and harbor erythroid or erythromyeloid potential.3,71,72,136-139 Primitive erythrocytes arise earliest in blood islands of the yolk sac, and by E8.5 in mouse or 32 hpf in zebrafish, a subsequent oligopotent precursor termed the erythromyeloid progenitor (EMP) is specified.66,130,136,138 Recently, lineage tracing was performed via activation of Tg(drl:creERT2;ubi:lox-GFP-lox-mCherry) reporter fish with TAM at 30 vs 54 hpf, suggesting that definitive HSPCs minimally contribute to embryonic lymphomyelopoiesis in zebrafish.140 These findings are supported by additional experiments that deplete HSCs with nitroreductase/metronidazole treatments,140 validating previous observations in mice.136
Until recently, it was thought that EMP-derived myeloid cells did not perdure into adulthood, but multiple recent studies have now shown that certain types of adult tissue-resident macrophages, including microglia in the brain, alveolar macrophages of the lung, Kupffer’s cells in the liver, and Langerhans cells in the skin, are partly or wholly EMP derived and migrate to the embryo proper, where they differentiate during organogenesis, beginning at ∼E10.5.132-134 In mammals, these cells are able to self-renew and persist into adulthood, unless faced with unusual challenges, such as organismal irradiation. Interestingly, it seems that in lower vertebrates, such as teleost fish, there are 2 successive waves of microglial specification, where a second HSC-derived population of microglia (development of which relies on colony-stimulating factor 1 receptor b [csfr1b]) replaces the earlier EMP-derived wave, so these specification processes may not be completely conserved across vertebrate phyla.141 Additionally, a transient wave of tissue-resident macrophages derived from the heart endocardium and essential for cardiac valve development has also been discovered via lineage tracing that exploited the endocardial-specific Nfatc1-Cre mouse line.142
Similarly, it now seems that some embryonic lymphoid populations are also specified independently of HSCs and persist into adulthood.132,134 Fetal T-lymphoid progenitors have been reported in circulation before the establishment of the thymus.143-145 Multiple studies have reported development of a restricted lymphoid or T-lineage precursor in the mouse yolk sac or directly from hemogenic endothelium of mouse and zebrafish embryos, and in some cases, these precursors arise earlier than AGM HSCs and in the absence of blood flow (ie, yolk sac in origin), although recent depletion and lineage-tracing studies have revealed an HSC origin for specific innate lymphoid subsets.146-150 It seems that embryonically derived γδ T cells persist and self-renew in the skin of mice.151
A subset of B cells, known as B1a cells, residing in the mucosa and body cavities of adult tissues has also been proposed to have an HSC-independent origin. In vitro lineage potential and in vivo transplantation studies have revealed FL and neonatal HSPCs to be more potent than adult BM HSCs at generating B1a cells.152 Indeed, adult HSCs do not cell autonomously contain high-level B1a potential (as reviewed by Ghosn et al153). Furthermore, B1a potential can be detected in yolk sac and AGM tissues before HSC emergence,154 and B1a progenitors are detected in Cbfβ−/−; Tek:GFP-Cbfβ mice, which lack transplantable HSCs.155 However, B1a progenitors observed in Cbfβ−/−; Tek:GFP-Cbfβ mice also display maturation defects, raising questions as to whether this model accurately reflects steady-state biology. Furthermore, fate mapping studies have revealed that B1a progenitors are absent in the developing embryo until HSC emergence.150 Barcoding studies have also revealed the presence of FL HSPCs that give rise to both B2 and B1a progeny when transplanted.156 These studies were complemented by single-cell transplantations in which phenotypic FL long-term HSCs (LT-HSCs) gave rise to both B2 and B1a cells. In contrast, Ghosn et al157 failed to observe B1a potential after transplantation of bulk FL LT-HSC. This disparate result might be explained by phenotypic differences in the populations interrogated, because Ghosn et al depleted Mac-1+ cells from LT-HSCs; previous work has shown that FL LT-HSCs express Mac-1.82 In an interesting twist, a fate mapping study in which cells that transit through an Flk2+ state are irreversibly switched from TdTomato+ to GFP+ showed that an embryonic Flk2+ precursor, termed transient HSCs, has multilineage potential with bias toward B1a B and γδ T lymphocytes but does not persist in the adult HSC compartment.158 It was recently reported that B2 vs B1a identity can be directed by the nature of the B-cell receptor (BCR) being expressed and that this potential is somewhat plastic (ie, B2 cells can be made to revert to a B1a phenotype simply by switching their BCR to a typical B1a BCR).159 This raises the question of whether appropriate antigen exposure could be key to the development of B1a cells. Might differential exposure to antigen during in vitro culture assays, transplantation, or steady-state development explain some of the disparate findings described? Further investigation is needed to decipher these puzzles.
Together, these results demonstrate how limited phenotypic markers may be misleading and how functional studies and high-resolution lineage-tracing studies must continue to inform and complement our attempts to understand fate potentials and the clonal establishment of hematopoietic lineage potency. In addition, recent quantitative modeling of normal and malignant HSPC dynamics has highlighted the importance of a unifying mathematic model that could reconcile different observations by considering cell heterogeneity and an extended nonsteady state, in which cells do not evenly contribute to downstream cellular compartments.160 Early experiments relied on the quantification of transplantable repopulating units as a measurement of HSC numbers and thus only captured a fraction of blood precursors at any given developmental stage. Furthermore, the contribution of each blood precursor to the adult HSC pool may be distinct, hence the importance of unbiased labeling methods to accurately measure blood precursor dynamics and of appropriate mathematic modeling able to integrate cell heterogeneity as defined by murine Cre lines displaying overlapping and nonoverlapping labeling of distinct blood precursors.
Conclusions
Together, lineage-tracing data and genomic mutational studies reveal that HSC specification is not a rare event; 10s of zebrafish HSPCs, 100s of murine HSPCs, and 1000s of human HSPCs contribute to the adult HSC pool11,92,101,118 (Figure 1). Each of these approaches has its own advantages and limitations (Table 1). Additionally, the presence of transient waves of progeny derived from embryonic HSPCs and long-lived non–HSC-derived cells complicates interpretations of the origins of hematopoietic clonal complexity. Indeed, key for future studies will be to definitively resolve the role of classically transplantable HSCs in steady-state hematopoiesis. This will likely require new tools that allow for clean fate mapping of multipotent progenitors vs HSCs, because all existing published tools show some lack of discrimination in labeling between these two populations.109-111 Another important question to address is whether multipotent progenitors are specified independently from HSCs during embryogenesis and if they contribute progeny into adult life. Alternatively, these cells might simply be organized into a hierarchy of transplantation and lifelong blood potential, consistent with the classic view. The novel approaches described here and others to come should eventually bring some clarity on these outstanding issues.
Summary of multiple approaches to study the frequency and dynamics of HSC specification in mice, zebrafish, and humans
| Method | Organism | N of HSPC/HSPC precursors specified* | Advantages | Limitations | Equipment/technical requirements | References |
|---|---|---|---|---|---|---|
| IAC cluster cartography | Mouse | 609 ± 84 c-Kit+ cells/E10.5 embryo 439 ± 87 c-Kit+ cells/E11.5 embryo | Direct morphologic evaluation; flexibility on use of additional markers | Lack of functional validation; single time point snapshot | Histology equipment; antibodies; confocal microscopy | 80 |
| Transplantation of fresh tissues | Mouse | 1-2 RUs/embryo at E10.5-E11.5; 60 RUs/ee at E12.5 | Well established; statistically robust and simple (Poisson statistics); commercial software available for LDA analysis | Does not score immature HSC precursors | Requires large cohorts of mice as transplant recipients; flow cytometry | 7,8,68,82,86 |
| Transplantation of cultured explants | Mouse | 60 RUs/ee between E9.5 and E12.5 | Allows for quantification of immature HSC precursor potential at early developmental time points (E8.5-E10.5) in murine embryos | Native development perturbed | Requires large cohorts of mice as transplant recipients; flow cytometry; explant culture | 94-98 |
| Marking of single blood precursors | Mouse | 333 E7.5-E8.5 blood precursors (Abcg2-CreERT2; Rosa26Lox-STOP-Lox-YFP) | Native development is preserved; simple math that assumes observed blood labeling is output of single precursor from which total n of precursors is extrapolated | Difficult to verify single precursor labeling; cannot determine upper bound for n of precursors | Abcg2-CreERT2; Rosa26Lox-STOP-Lox-YFP mouse model; single group of mice (≥5 mice); flow cytometry | 101 |
| MtMV of Confetti-reporter labeling | Mouse | 600 blood precursors (E7.5-E12.5) | Native development is preserved; quantitative; simple genetic mouse models; statistically robust and simple; based on Poisson statistics | Individual clones cannot be tracked; requires >3% of Confetti labeling and >500 cells examined; quantitative range is limited; commercial software for analyses not available | Cre recombinase mouse models; single group of mice (≥5 mice); flow cytometry with 405, 445, 488, 562, and 640 nm and specific band-pass filter configurations to discriminate Confetti colors | 17,92 |
| Zebrabow | Zebrafish | 20-26 lifelong precursors | Native development is preserved | Reliant on hue and saturation to identify individual clones | Driver and effector transgenic lines; color-clustering computational analysis platform; flow cytometry; confocal and inverted microscope | 11 |
| Hsp701 LASER induction | Zebrafish | 30 lifelong precursors | Native development is preserved | Reliant on labeling 1 cell, otherwise individual clones cannot be tracked | Micropoint microscope-targeted 440-nm laser; driver and effector transgenic lines; flow cytometry; confocal and inverted microscopes | 11 |
| Transposase-mediated tagging | Mouse | Not yet employed | Native development is preserved; single clones can be tracked; lineage relationships inferred | Complex genetic mouse model; bioinformatics expertise required to interpret data | Targeted sequencing; bioinformatics expertise | 104,105 |
| PolyLox-based barcoding | Mouse | Not yet employed | Native development is preserved; single clones can be tracked; lineage relationships inferred | Complex genetic mouse model; bioinformatics expertise required to interpret data; need to eliminate common rearrangements | Targeted sequencing; bioinformatics expertise | 112 |
| CRISPR-based barcoding | Mouse | Not yet employed | Native development is preserved; single clones can be tracked; lineage relationships inferred | Complex genetic mouse model; bioinformatics expertise required to interpret data | Targeted sequencing; bioinformatics expertise | 113-115 |
| Somatic mutations | Human | Native development is preserved | Costs limit n of analyzed clones; potential for confounding mutations during cell expansion; biased for sequencing clones able to expand in vitro; complex bioinformatics analyses | WGS platforms; bioinformatics expertise | 118 |
| Method | Organism | N of HSPC/HSPC precursors specified* | Advantages | Limitations | Equipment/technical requirements | References |
|---|---|---|---|---|---|---|
| IAC cluster cartography | Mouse | 609 ± 84 c-Kit+ cells/E10.5 embryo 439 ± 87 c-Kit+ cells/E11.5 embryo | Direct morphologic evaluation; flexibility on use of additional markers | Lack of functional validation; single time point snapshot | Histology equipment; antibodies; confocal microscopy | 80 |
| Transplantation of fresh tissues | Mouse | 1-2 RUs/embryo at E10.5-E11.5; 60 RUs/ee at E12.5 | Well established; statistically robust and simple (Poisson statistics); commercial software available for LDA analysis | Does not score immature HSC precursors | Requires large cohorts of mice as transplant recipients; flow cytometry | 7,8,68,82,86 |
| Transplantation of cultured explants | Mouse | 60 RUs/ee between E9.5 and E12.5 | Allows for quantification of immature HSC precursor potential at early developmental time points (E8.5-E10.5) in murine embryos | Native development perturbed | Requires large cohorts of mice as transplant recipients; flow cytometry; explant culture | 94-98 |
| Marking of single blood precursors | Mouse | 333 E7.5-E8.5 blood precursors (Abcg2-CreERT2; Rosa26Lox-STOP-Lox-YFP) | Native development is preserved; simple math that assumes observed blood labeling is output of single precursor from which total n of precursors is extrapolated | Difficult to verify single precursor labeling; cannot determine upper bound for n of precursors | Abcg2-CreERT2; Rosa26Lox-STOP-Lox-YFP mouse model; single group of mice (≥5 mice); flow cytometry | 101 |
| MtMV of Confetti-reporter labeling | Mouse | 600 blood precursors (E7.5-E12.5) | Native development is preserved; quantitative; simple genetic mouse models; statistically robust and simple; based on Poisson statistics | Individual clones cannot be tracked; requires >3% of Confetti labeling and >500 cells examined; quantitative range is limited; commercial software for analyses not available | Cre recombinase mouse models; single group of mice (≥5 mice); flow cytometry with 405, 445, 488, 562, and 640 nm and specific band-pass filter configurations to discriminate Confetti colors | 17,92 |
| Zebrabow | Zebrafish | 20-26 lifelong precursors | Native development is preserved | Reliant on hue and saturation to identify individual clones | Driver and effector transgenic lines; color-clustering computational analysis platform; flow cytometry; confocal and inverted microscope | 11 |
| Hsp701 LASER induction | Zebrafish | 30 lifelong precursors | Native development is preserved | Reliant on labeling 1 cell, otherwise individual clones cannot be tracked | Micropoint microscope-targeted 440-nm laser; driver and effector transgenic lines; flow cytometry; confocal and inverted microscopes | 11 |
| Transposase-mediated tagging | Mouse | Not yet employed | Native development is preserved; single clones can be tracked; lineage relationships inferred | Complex genetic mouse model; bioinformatics expertise required to interpret data | Targeted sequencing; bioinformatics expertise | 104,105 |
| PolyLox-based barcoding | Mouse | Not yet employed | Native development is preserved; single clones can be tracked; lineage relationships inferred | Complex genetic mouse model; bioinformatics expertise required to interpret data; need to eliminate common rearrangements | Targeted sequencing; bioinformatics expertise | 112 |
| CRISPR-based barcoding | Mouse | Not yet employed | Native development is preserved; single clones can be tracked; lineage relationships inferred | Complex genetic mouse model; bioinformatics expertise required to interpret data | Targeted sequencing; bioinformatics expertise | 113-115 |
| Somatic mutations | Human | Native development is preserved | Costs limit n of analyzed clones; potential for confounding mutations during cell expansion; biased for sequencing clones able to expand in vitro; complex bioinformatics analyses | WGS platforms; bioinformatics expertise | 118 |
ee, embryo equivalent; IAC, intra-aortic cluster; LDA, limiting dilution analysis.
N of IAC cells, RUs, or lineage-traced cells.
Acknowledgments
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, grants R01DK116835 and R01DK104028 (S.M.-F.) and R01DK113973 (W.C.), the Leukemia and Lymphoma Society (S.M.-F.), and the American Lebanese Syrian Associated Charities (S.M.-F. and W.C.). M.G. is funded by the American Society of Hematology (Global Research Award), Barts Charity, Leukaemia UK (John Goldman Fellowship 2020/JGF/001), and Medical Research Council (Career Development Award MR/V009222/1).
This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: W.C., M.G., and S.M.-F. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Shannon McKinney-Freeman, 262 Danny Thomas Place, Memphis, TN 38105; e-mail: shannon.mckinney-freeman@stjude.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal