

Key Points
GPIIb-dependent outside-in signaling through Gα13, c-Src, and 14-3-3ζ is essential for the initiation and maintenance of platelet migration.
Blockade of this signaling cascade abrogates platelet migration in vivo and in dasatinib-treated patients with leukemia.

Abstract
Platelets are not only the first responders in thrombosis and hemostasis but also central players in inflammation. Compared with platelets recruited to thrombi, immune-responsive platelets use distinct effector functions including actin-related protein complex 2/3-dependent migration along adhesive substrate gradients (haptotaxis), which prevents inflammatory bleeding and contributes to host defense. How platelet migration in this context is regulated on a cellular level is incompletely understood. Here, we use time-resolved morphodynamic profiling of individual platelets to show that migration, in contrast to clot retraction, requires anisotropic myosin IIa-activity at the platelet rear which is preceded by polarized actin polymerization at the front to initiate and maintain migration. Integrin GPIIb-dependent outside-in signaling via Gα13 coordinates polarization of migrating platelets to trigger tyrosine kinase c-Src/14-3-3ζ–dependent lamellipodium formation and functions independent of soluble agonists or chemotactic signals. Inhibitors of this signaling cascade, including the clinically used ABL/c-Src inhibitor dasatinib, interfere predominantly with the migratory capacity of platelets, without major impairment of classical platelet functions. In murine inflammation models, this translates to reduced migration of platelets visualized by 4D intravital microscopy, resulting in increased inflammation-associated hemorrhage in acute lung injury. Finally, platelets isolated from patients with leukemia treated with dasatinib who are prone to clinically relevant hemorrhage exhibit prominent migration defects, whereas other platelet functions are only partially affected. In summary, we define a distinct signaling pathway essential for migration and provide novel mechanistic insights explaining dasatinib-related platelet dysfunction and bleeding.
Introduction
Platelets are central cellular effectors of clot formation after traumatic vessel injury. Upon exposure of subendothelial matrix proteins, platelets are rapidly recruited to the site of injury to form a first barrier and prevent excessive blood loss.1 Activation and recruitment are orchestrated in a tightly regulated sequence. After engagement of glycoprotein receptors, activation signals are propagated in platelets through a process termed as inside-out signaling, leading to conformational changes of integrin receptors, most notably the fibrin(ogen) receptor GPIIBIIIA, which allows for stable adherence. Moreover, integrin-mediated engagement of ligands can also trigger platelet responses, termed outside-in signaling,1-5 with concomitant activation of downstream effectors, such as sarcoma (Src) family kinases (SFKs) and SFK-binding proteins, such as 14-3-3ζ.6-8 The release of auto- and paracrine mediators, thromboxane A2, and adenosine diphosphate (ADP), further drives platelet recruitment and activation. The resulting rapid response by the cytoskeleton leads to shape change, enforced platelet-platelet interaction, and finally retractile activity, stabilizing the growing thrombus.4,9
In addition to their crucial contribution to hemostasis and coagulation, platelets are vital first responders in the setting of inflammation and infection, mediating pathogen clearance and innate immune cell recruitment and even shaping adaptive immune responses.10-15 We have previously shown that immune-responsive platelets migrate along substrate gradients to identify sites of microvascular injury and that this process not only aids in the prevention of inflammatory bleeding and bacterial dissemination but can also contribute to hyperinflammation in sepsis.16,17 In addition, we provided evidence that both myosin- and actin-associated proteins, including myosin heavy chain 9 (Myh9) and actin-related protein complex 2/3 (Arp2/3), are essential regulators of migration, rendering Myh9- and Arp2/3-deficient platelets unable to migrate.16,18 Interestingly, inhibition of Arp2/3 does not affect thrombus formation or hemostasis in vivo, suggesting that its role as an actin nucleator in platelets is specific for migration and immunity. Understanding how the cytoskeleton is regulated in this context could help to specifically target platelets in inflammation or thrombosis. However, the intracellular signaling cascades that initiate and propagate platelet migration up- and downstream of these 2 key mechanisms remain unclear.
Here, we use high-throughput in vitro assays of platelet recruitment, retraction, and migration to decipher the sequence of events that initiate and propagate migratory behavior in human and murine platelets. Using high-resolution epifluorescence and confocal microscopy, we identify key morphological and behavioral elements mediated by the actin- and myosin-associated platelet cytoskeleton. Interference with myosin IIa–dependent contraction or actin-dependent lamellipodia formation abrogates migratory capacity. Through systematic interference with downstream effectors, we identify that concomitant c-Src kinase and 14-3-3ζ signaling downstream of GPIIBIIIA-coupled Gα13 are crucial for platelet polarization and migration. Consequently, Gα13 inhibition, 14-3-3ζ blockade, or c-Src inhibition with the clinically approved antineoplastic tyrosine kinase inhibitor (TKI), dasatinib, block platelet migration even at nanomolar doses, which do not interfere with other basic platelet functions. In mouse models of lipopolysaccharide (LPS)-induced inflammation, low-dose dasatinib blocks platelet migration and aggravates inflammatory bleeding without affecting traumatic hemostasis or arterial thrombus formation.
Dasatinib—frequently used in chronic myeloid leukemia (CML) and Philadelphia chromosome–positive (Ph+) acute lymphoblastic leukemia—has been associated with severe adverse effects on the platelet lineage, including thrombocytopenia and a striking hemorrhage risk affecting ∼40% of patients across the spectrum of chronic, accelerating, and blastic leukemia, including gastrointestinal, mucosal, and intracranial bleeding19-27: 23% of the 138 patients included in the phase 1 or 2 studies were reported to have suffered from drug-associated hemorrhage, with 81% of these being attributed to gastrointestinal or mucosal bleeding events, and 69% of these bleeding events occurring during the first 3 months of treatment.19,21,26,28,29 Although this clinical phenotype can partly be explained by thrombocytopenia and defects in procoagulant function and aggregation, ∼40% of patients receiving dasatinib who suffer from hemorrhage are not thrombocytopenic, and the observed effect of the drug on classical effector functions is inconsistent and does not correlate with bleeding events.19,20,26 Here, we show that platelets from patients with leukemia treated with dasatinib consistently show abrogated platelet migration, whereas other core platelet functions are only partially affected. Our findings comprehensively assess the underlying signaling events that precede and drive platelet migration and provide evidence that platelet migratory capacity while sharing intracellular signaling hubs with other platelet functions, is more sensitive to pharmacological interference than these classical functions. Thus, impaired platelet migration may serve as a potential explanation for the exponential increase of mucosal bleeding events in patients treated with dasatinib.
Methods
Detailed methodology is provided in the supplemental Materials and methods, available on the Blood website.
Generation of fibrinogen/albumin and fibrin/albumin surfaces
Microchambers for coating with fibrin, fibrinogen, and/or albumin were generated as described previously.16,17,30 In brief, coverslips (no. 1.5, D263T; Nexterion) were consecutively washed in nitric acid and water for 1 hour each, air-dried and silanized with hexamethyldisilazane (HMDS, Sigma) by spin-coating for 30 seconds at 80 revolutions/sec. Sticky slide plastic channels (Ibidi VI0.4, #80608) were attached to the silanized coverslip. Chambers were then coated with 37.5 μg/mL AlexaFluor-conjugated or unconjugated fibrinogen and 0.2% human serum albumin resuspended in modified Tyrode’s buffer (pH 7.2, 15 minutes, room temperature [RT]). To generate crosslinked fibrin surfaces, thrombin (1 U/mL), calcium (1 mM), and platelet-poor plasma were added to fibrin(ogen)/albumin mixes.
Migration and retraction assays
In vitro assays investigating platelet migration and retraction were performed as described previously.16,17,30 Isolated mouse and human platelets were diluted to a concentration of 150 000 to 200 000/μL using Tyrode’s buffer. A total of 4 × 106 platelets were transferred to a new tube and activated by the addition of 4 μM ADP, 2 μM U46619, and 200 μM calcium chloride. In some experiments, antagonists targeting surface receptors or intracellular signaling hubs (supplemental Table 2) were added before platelet seeding. Next, platelets were pipetted into precoated custom chambers and incubated for 15 minutes at 37°C. Suspended platelets were removed by 3 washing steps with cell-free wash buffer containing 200 μM calcium chloride and antibodies for detection of platelets and respective activation markers and/or inhibitory compounds targeting surface receptors or intracellular signaling hubs. After 30 to 60 minutes, cells were fixated with fixation mix containing 2% paraformaldehyde (PFA) and 0.05% glutaraldehyde (both Sigma). In some cases, immunofluorescence stainings were performed as described in the supplemental Materials and methods. Samples were imaged using an epifluorescence microscope (Olympus IX83) or a Zeiss LSM 880 confocal microscope. For quantification, 5 to 6 random images were acquired per biological replicate. The absolute cleared area (in μm2), a proxy for migratory capacity, was automatically measured using a custom macro in ImageJ Fiji.31
Immunofluorescence staining
Immunofluorescence staining of adherent, retracting, and migrating platelets were performed as described previously.16,17,30 After fixating with 2% PFA and 0.05% glutaraldehyde (both Sigma) and washing steps with phosphate-buffered saline (PBS), samples were blocked with 2% bovine serum albumin (BSA) for 1 hour at RT. In some experiments, platelets were fixated with 1% PFA followed by permeabilization with Triton X-100 0.5% in PBS. Primary antibodies targeting integrin α3, 14-3-3ζ, and c-Src kinase as well as actin, phospho-myosin light chain 2, and tubulin (supplemental Table 1 for concentrations and ID) were diluted 1:100 in 1% BSA and incubated for 1 hour at RT or overnight at 4°C. After 3 washing steps with PBS, secondary antibodies (supplemental Table 1) diluted in 1% BSA were added (1:200, 1 hour at RT). Samples were imaged on a Zeiss LSM 880 confocal microscope in AiryScan Superresolution mode.
Acute lung injury
We performed subacute lung injury models as described previously.16,30 Briefly, mice were anesthetized before 20 μg of Escherichia coli–derived LPS (O111:B4; Sigma) was swiftly applied to the nostrils of each animal. After LPS application, anesthesia was immediately antagonized. Mice were clinically scored to assess severity of pulmonary inflammation. In some experiments, mice were injected intraperitoneally with either vehicle (2.5% dimethyl sulfoxide in sterile NaCl 0.9%) or 1 mg/kg body weight (BW) dasatinib (solved in 2.5% dimethyl sulfoxide in sterile NaCl 0.9%) 12 hours before, immediately before, and 12 hours after LPS application. Twenty-four hours after LPS treatment, mice were killed by cervical dislocation and bronchoalveolar lavage fluid (BALF) or whole lungs were collected. For BAL fluid collection, intratracheal flushing with 1% BSA and 2 mM EDTA was performed twice, and aliquots of the collected BALF were either stained with antibodies and analyzed by flow cytometry or sonicated to assess hemoglobin content using a Tecan Infinite F200 plate reader (absorption at 405 nm, buffer-only containing wells serving as controls for normalization of experimental samples). For all BALF samples, >1 mL of the injected fluid was recovered. For immunofluorescence stainings, lungs were surgically removed without collecting BAL fluid, fixated in 4% PFA for 1 hour at RT, dehydrated overnight in 30% sucrose, and subsequently cryo-embedded. Detailed immunofluorescence stainings of organs are described in the supplemental Materials and methods.
Clinical pilot study
To investigate the impact of systemic dasatinib application in humans, we recruited patients with CML treated with BCR-ABL/c-Src/c-Kit TKIs dasatinib (n = 3) and bosutinib (n = 1) or the non–c-Src targeting TKI and BCR-ABL antagonist, imatinib (n = 3; see Table 1 for patient data). Informed consent of the patients was obtained in accordance with the Declaration of Helsinki. Pseudonymized data were used for analysis, and the study was approved by the ethics committee of LMU Munich (no. 20-1067). Blood was drawn by cubital venipuncture at a mean of 9.5 hours after intake of the respective inhibitors. Healthy, age- and sex-matched volunteers (n = 5) served as negative controls (total of n = 12 participants in this study). Immediately after drawing blood, platelets were isolated (supplemental Materials and methods) and used for analysis of platelet migration, spread, and activation in in vitro assays. Platelet numbers were normalized to include equal platelet counts in all assays. Migratory and spreading capacities as well as levels of platelet activation and surface expression were normalized to healthy controls measured on the same day.
Results
Directional lamellipodium formation initiates migration
The cellular mechanisms and morphological characteristics that initiate and maintain platelet migration are incompletely understood. We analyzed 3-dimensional reconstructions of polarized platelets migrating on a fibrinogen/albumin matrix and immunostained for proteins essential for platelet migration, including the fibrinogen receptor GPIIBIIIA (and its activated conformation), the myosin II–associated myosin light chain (MLC) as well as the actin-related complex protein 2/3 necessary for lamellipodium formation (Arp2/3, supplemental Video 1; Figure 1A-B).16,17 Immunofluorescence staining for phosphorylated myosin light chain (pMLC), a marker of myosin II activity, and Arp2/3 revealed a polarized cell shape with enrichment of Arp2/3 and costained F-actin at the leading edge, consisting of a sheet-like lamellipodium, whereas pMLC was enriched at the trailing edge, where a pseudonucleus (PN) formed (Figure 1B-C). Next, we studied the morphodynamic sequence preceding this polarized state. Contour tracing of PN movement and of lamellipodia revealed a dynamic protrusion and formation of the leading edge while the PN initially remained stationary (Figure 1D). Tracking both the lamellipodium and PN over time revealed distinct phases of migration initiation, with initial and full spreading (phases 1 and 2, respectively) followed by protrusion of the leading edge (phase 3) and subsequent initiation of migration, characterized by PN movement (phase 4, Figure 1E-F; supplemental Video 2). We confirmed these findings using kymograph analyses (Figure 1G). Live microscopy of platelets isolated from LifeAct-eGFP mice showed prominent F-actin polymerization and nucleation waves, which correlated with protrusion formation (Figure 1H-I; supplemental Video 3). In contrast, targeting assembly or disassembly of tubulin microtubules through colchicine or paclitaxel, respectively, did not affect platelet migration (supplemental Figure 1A-C).

Directional lamellipodium formation initiates migration. (A) 3D reconstruction of a polarized migrating human platelet stained for the fibrinogen receptor GPIIBIIIA and its activated form (stained with antibody clone PAC-1). (B) p-MLC, Arp2/3, and F-actin immunofluorescence staining of migrating human platelets. Asterisks (∗) indicate the formation of pseudonuclei (PN) at the trailing edge. (C) Histogram of colocalization of Myh9 (upper panel, green), Arp2/3 (lower panel, green), and F-actin (pink) expression, along the green lines in Figure 1B. (D) Time-resolved morphology tracking of the PN (green) and outer shape of a human platelet (pink) in the initiation phase of migration. (E) Representative phase contrast (PH) micrographs of live imaging of a human platelet starting to spread and eventually migrating. Green: PN, pink: lamellipodial leading edge. Scale bar, 5 μm. (F) Longitudinal depiction of PN and leading-edge tracking analysis. (G) Representative micrographs and kymograph analysis (upper panel) of a spread platelet initiating migratory behavior. Kymograph analyses graphically display the spatial position of pixels (and therefore platelets) on the indicated white line (y-axis) across time (x-axis); consequently, the retracted, black PN of the platelet moves down the white line (y-axis) once migration is initiated (white arrowhead). Scale bar, 5 μm. (H) Micrographs of live imaging of a murine LifeAct-eGFP platelet spreading and initiating migration reveals dynamic actin waves at the leading edge (pink arrows); the green circle marks the PN of platelet. (I) Spatiotemporal tracking of dynamic actin waves (black arrowheads) along the leading edge (green spotted line) of a murine LifeAct-eGFP. Bottom panel: histogram of LifeAct eGFP mean fluorescence intensity (MFI) (normalized to maximum intensity) measured along the green spotted line marking the leading edge (upper panel) at the indicated time points. The black arrowheads represent the peak of the actin wave moving along the leading edge for the indicated time points. Panels A-I: assays were performed on fibrinogen/albumin matrices. Scale bars, 5 μm.
Directional lamellipodium formation initiates migration. (A) 3D reconstruction of a polarized migrating human platelet stained for the fibrinogen receptor GPIIBIIIA and its activated form (stained with antibody clone PAC-1). (B) p-MLC, Arp2/3, and F-actin immunofluorescence staining of migrating human platelets. Asterisks (∗) indicate the formation of pseudonuclei (PN) at the trailing edge. (C) Histogram of colocalization of Myh9 (upper panel, green), Arp2/3 (lower panel, green), and F-actin (pink) expression, along the green lines in Figure 1B. (D) Time-resolved morphology tracking of the PN (green) and outer shape of a human platelet (pink) in the initiation phase of migration. (E) Representative phase contrast (PH) micrographs of live imaging of a human platelet starting to spread and eventually migrating. Green: PN, pink: lamellipodial leading edge. Scale bar, 5 μm. (F) Longitudinal depiction of PN and leading-edge tracking analysis. (G) Representative micrographs and kymograph analysis (upper panel) of a spread platelet initiating migratory behavior. Kymograph analyses graphically display the spatial position of pixels (and therefore platelets) on the indicated white line (y-axis) across time (x-axis); consequently, the retracted, black PN of the platelet moves down the white line (y-axis) once migration is initiated (white arrowhead). Scale bar, 5 μm. (H) Micrographs of live imaging of a murine LifeAct-eGFP platelet spreading and initiating migration reveals dynamic actin waves at the leading edge (pink arrows); the green circle marks the PN of platelet. (I) Spatiotemporal tracking of dynamic actin waves (black arrowheads) along the leading edge (green spotted line) of a murine LifeAct-eGFP. Bottom panel: histogram of LifeAct eGFP mean fluorescence intensity (MFI) (normalized to maximum intensity) measured along the green spotted line marking the leading edge (upper panel) at the indicated time points. The black arrowheads represent the peak of the actin wave moving along the leading edge for the indicated time points. Panels A-I: assays were performed on fibrinogen/albumin matrices. Scale bars, 5 μm.
Arp2/3-dependent actin nucleation mediates polarization of platelets to initiate migration
Arp2/3 is essential for platelet migration and haptotaxis.16 Platelets from PF4cre-Arpc2fl/fl mice not only failed to migrate but also exhibited a nonpolarized morphology with increased numbers of long filopodia (Figure 2A-H). Live pharmacological inhibition of Arp2/3 through CK666 abrogated the formation of actin polymerization waves in LifeAct-eGFP platelets and resulted in loss of both polarized phenotype and migratory ability on albumin/fibrin(ogen) hybrid matrices with no effect on substrate retraction (Figure 2C-D; supplemental Figure 1D-E; supplemental Video 4). CK666 immediately arrested platelets and reduced integrin GPIIBIIIA-matrix interaction as revealed by colocalization plots (Figure 2E-F; supplemental Figure 1F-G). This points toward loss of activated integrin-fibrinogen contacts as mechanism that (1) leads to loss of polarization and (2) impairs migratory potential (supplemental Video 4). Notably, 3D clot retraction was unaffected by pharmacological blockade of Arp2/3 function, whereas it was abolished when myosin II was inhibited with blebbistatin (p-Blb, Figure 2G). Cell morphometry revealed an immediate loss of polarized platelet phenotype accompanied by a striking reduction in platelet circularity, driven by the development of long filopodia (Figure 2H). In summary, these findings reveal that Arp2/3 is essential both for initiation of migration and maintenance of cell polarization.
Arp2/3 mediates polarization of platelets to initiate migration, whereas myosin II function regulated via ROCK and MLCK is not involved in platelet polarization. (A) 3D reconstruction of human platelets on fibrinogen/albumin matrix, either control or after inhibition of Arp2/3 with CK666. (B) Longitudinal brightfield images and kymograph analysis of a murine Arp2/3-deficient platelet, depicting the inability to migrate. (C) Representative micrographs of a murine LifeAct eGFP platelet at the indicated timepoints before (0 and 12 seconds) and after (78-426 seconds) treatment with 200 μM CK666 (black arrowhead). Pink arrowheads depict actin nucleation waves at the leading edge. Right panel: quantification of # actin waves at leading edge per minute. Student t test, 2-tailed, unpaired. (D) Representative micrographs of live imaging of a human migrating platelet with sham treatment (upper row) and a human migrating platelet before (−60 seconds) and after treatment with 200 μM CK666 (245-1770 seconds). (E) Colocalization plots of fibrinogen and human platelets treated with CK689 (control) or CK666, with red indicating high and blue indicating low levels of overlap. Signal in the right upper quadrant, depicting high detection of both CD41 (x-axis) and fibrinogen (y-axis), was used as a proxy for receptor-ligand interaction. Colocalization was assessed at the leading-edge lamellipodium. (F) Representative immunofluorescence staining of a human platelet treated with CK666 before seeding; the green dotted line represents measurement along the leading edge. Right panel: histogram of MFIs of the indicated proteins for CK689- (ctrl) and CK666-treated human platelets along the green dotted line, with black arrowheads indicating colocalization. See supplemental Figure 1G for corresponding control. (G) Quantification of 3D clot retraction of human platelet–rich plasma (PRP) incubated with the indicated inhibitors. One-way analysis of variance (ANOVA) with post hoc Dunnett's testing. (H) Immunofluorescence staining of human platelet treated with CK666 (200 μM), cellular outline and quantification of shape analysis. Student t test, unpaired, two-tailed. (I) 3D rendered image of a p-Blb–treated human platelet after initiation of migration. The respective control image is depicted in Figure 2A. (J) Longitudinal micrographs of differential interference contrast and fluorescence images of a migrating platelet 90 seconds before and after addition of p-Blb (50 μM). (K) Colocalization plots of fibrinogen and human platelets treated with p-Blb or vehicle. (L) Representative immunofluorescence staining of a human platelet treated with p-Blb before seeding; the green dotted line represents measurement along the leading edge. Right panel: histogram of MFIs of the indicated proteins along the green dotted line measured for both sham- and p-Blb–treated human platelets, with black arrowheads indicating colocalization. See supplemental Figure 1G for corresponding control. (M) Longitudinal differential interference contrast images and kymograph analysis of a murine Myh9-deficient platelet (PF4cre-Myh9fl/fl). (N) Quantification of migration assay with human platelet treated with the indicated inhibitors. Concentrations: Y27632 50 μM, ML-7 10 μM. (O) Colocalization plot upon indicated treatments (Ctrl vs ML-7 + Y27632), revealing persistent contact between platelet and substrate. (P) Longitudinal brightfield and epifluorescence images of a migrating human platelet upon treatment with MLCK and ROCK inhibitors (same concentrations as panel N). (Q) Representative immunofluorescence images and quantification of human platelets retracting on a silanized glass plate coated with crosslinked fibrin and with or without treatment with MLCK and ROCK inhibitors, ML-7 and Y27632 (same concentrations as panel P). Student t test, unpaired, 2-tailed. (R) Experimental strategy to generate a pliable, fluorescent hybrid matrix consisting of a biotinylated PLL-PEG backbone and a neutravidin-bound RGD motif, allowing rupture of the biotin and neutravidin bond through platelet pulling forces and enabling migration. (S) Representative brightfield (PH) and immunofluorescence images of a migration assay on pliable PLL-PEG-RGD substrate with human platelets for the indicated treatment. (T) Quantification of platelet spreading and migration for untreated, Bleb (+, inactive enantiomer)- and Bleb (−, active enantiomer)-treated platelets. Two-way ANOVA with post hoc Tukey testing. Unless indicated with asterisks, post hoc testing revealed nonsignificant results (P ≥ .05). P values corresponding to asterisks: ∗P < .05, ∗∗P < .01, ∗∗∗P < .01, ∗∗∗∗P < .001. Scale bars, 5 μm.
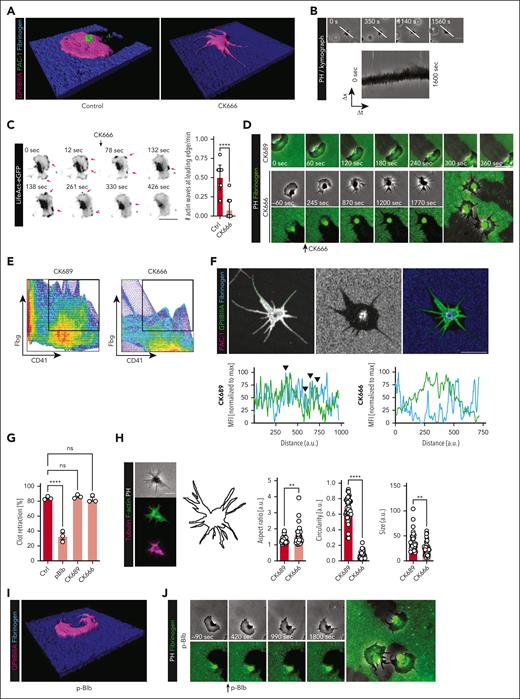

Myosin II function regulated via rho kinase (ROCK) and myosin light chain kinase (MLCK) is not involved in platelet polarization
We have previously shown that Myh9-deficient platelets are unable to migrate.17 When myosin II was inhibited in platelets during active migration using the specific inhibitor, blebbistatin, the cells were arrested in their polarized state without further retraction of the substrate (Figure 2I-J). Colocalization plots showed maintained interaction between fibrinogen and GPIIBIIIA despite potent myosin II inhibition (Figure 2K-L; supplemental Figure 1G). In addition, Myh9-deficient platelets showed no substrate collection at the rear (supplemental Video 5). However, the majority of Myh9-deficient platelets revealed a strong polarization, with a protruding lamellipodium at the front, which even led to cellular disintegration because they were unable to retract their rear (Figure 2M; supplemental Video 5), underlining the inability to migrate. This highlights that coordinated myosin II function per se is required for motility but not for platelet polarization.
Myosin II activity is regulated by ROCK and MLCK in platelets.2,17 Inhibition of either of these kinases using Y27632 or ML7, respectively, reduced migration, whereas the combined inhibition of both kinases almost entirely abrogated migratory capacity and recapitulated the blebbistatin phenotype (Figure 2N-P). In line, analysis of retractile function at single-cell level in a 3D fibrin-rich clot was markedly reduced by combined inhibition of MLCK and ROCK (Figure 2Q). Collectively, these findings indicate that ROCK- and MLCK-mediated myosin II function is dispensable for platelet polarization, whereas coordinated actin polymerization and myosin-dependent retraction are necessary for maintenance of migration.
We next investigated whether platelet polarization and migration are restricted to fibrinogen-albumin substrates on silanized glass surfaces or whether these platelet functions underlie a more general response to specific properties of the adhesive substrate (supplemental Figure 2). On uncoated glass slides, platelets readily spread but were unable to migrate (supplemental Figure 2A). Coating with biologically inert poly (ethylene glycol) (PLL-g-PEG) abolished both spreading and migration on glass (supplemental Figure 2A). When adding integrin recognition motfis (Arg-Gly-Asp, RGD) to the PLL-g-PEG backbone, platelet spreading was restored, however platelets were still unable to migrate (supplemental Figure 2B). Interestingly, although platelet spreading was significantly reduced on lowering RGD densities, platelets remained immotile and did not begin to migrate at any of the RGD densities tested, suggesting that minimization of adhesive interactions between platelets and their substrate is not sufficient to trigger platelet migration (supplemental Figure 2B). In addition, decreasing the stiffness of the underlying substrate did not trigger platelet migration (supplemental Figure 2C). We next altered ligand stability by introducing a fluorescent fragile linker (biotin-avidin bond) to PLL-g-PEG. Strikingly, platelets started to migrate on these fragile RGD peptides, with myosin II–dependent traction necessary to break the adhesive substrate bond (Figure 2R-T; supplemental Figure 2D). These data show that platelets exert single integrin traction forces that overcome the strength of biotin-avidin bonds (∼160 pN). Notably, at a high density of fragile integrin ligands, the tensile force that a platelet can generate per ligand was no longer sufficient to break the individual integrin bonds, rendering the platelets immobile (supplemental Figure 2D).32
Morphodynamic analysis reveals c-Src as a central regulator of Arp2/3 in platelet migration
To further define regulatory mechanisms that drive platelet polarization, we performed an inhibitor screen of platelet retraction and platelet migration in combination with morphometric analyses of platelet contours (Figure 3A). We reasoned that an isolated decrease in migratory behavior combined with a drop in circularity would indicate loss of polarization and define upstream regulators of Arp2/3-dependent actin nucleation. Inhibition of the central signaling regulator phospholipase C through U73122 mitigated both spread and migration, wheres interference with focal adhesion kinase or PI3K signaling through PF-573228 or Ly-294002, respectively, had no significant effects (Figure 3B). Interestingly, broad spectrum SFK inhibition with PP1 or pyrozolopyrimidine (PP2) profoundly decreased migratory capacity, impaired actin polymerization at the leading edge and reduced platelet circularity without affecting 3D and 2D clot retraction (Figure 3B-F). Live imaging of LifeAct-eGFP platelets upon PP2 exposure revealed loss of actin nucleation waves at the leading edge and loss of substrate/GPIIb colocalization as potential culprits for the lack of platelet polarization (Figure 3G-H; supplemental Video 6). Even in the presence of strong agonists, such as ADP and TRAP this phenotype could not be overcome, pointing toward a crucial contribution of SFKs in platelet polarization and migration (Figure 3I). Platelets express a variety of SFKs, with Syk, Fyn, Lyn, Fgr, Yes, and c-Src prominently implicated in platelet activation.33,34 In migration assays, specific inhibition of c-Src through KB SRC4,35 but not of Syk, Fyn, Lyn/Fgr, or Yes, abrogated migratory capacity when using the respective inhibitors near their previously published KI50 values (Figure 3J; supplemental Table 2). Increasing concentrations of SU6656 or BI-1002494, inhibitors of Yes and Syk, respectively, also blocked migration, presumably through unselective SFK inhibition at higher doses (supplemental Figure 3A).

Morphodynamic analysis reveals c-Src as central regulator of Arp2/3 in platelet migration. (A) Experimental scheme of morphodynamic inhibitor screening platform: inhibitor screening and quantification of substrate clearance on substrates leading to retraction (cross-linked fibrin layers) or migration (fibrin monomer layers) in combination with morphological analysis of platelet shape. (B) Quantification of cleared area per cell (μm2) of both migrating (left) and retracting human platelets (right panel) in the presence of the indicated inhibitors, with values being normalized to control in %. One-way ANOVA with post hoc Dunnett's testing. (C) Representative morphologies of human platelets treated with the indicated inhibitors. (D) Representative micrographs of human platelets costained for F-actin and myosin activity (pMLC) following incubation with the indicated treatments. (E) Shape analysis of platelet circularity (a.u., left) and platelet size (μm2, right panel) of migrating human platelets treated with the indicated antagonists. One-way ANOVA with post hoc Dunnett’s testing. (F) Quantification of 3D clot retraction of human PRP incubated with the indicated inhibitors. One-way ANOVA with post hoc Dunnett’s testing. (G) Longitudinal micrographs of live imaging of isolated murine LifeAct-eGFP platelets before (panels 0, 12 seconds) and after addition of the Src inhibitor PP2 (panels 162-492 seconds). Pink arrows mark actin nucleation at the leading edge; pink asterisks show filopodia formation. Right panel: Quantification of # of actin waves (per minute) moving along the leading edge. Student t test, 2-tailed, unpaired. (H) Colocalization plot of fibrinogen and human platelet of sham treatment (left) or after treatment with 10 μM PP2 (right panel). (I) Quantification of cleared area in μm2 upon treatment with indicated inhibitors/agonists (sham vs 10 μM PP2 in the presence or absence of 50 mM ADP and 1 mM TRAP). One-way ANOVA with post hoc Dunnett's testing. (J) Quantification of migrating platelets (%) and cleared area per platelet (μm2) (left panels) as well as the absolute number of recruited and relative amount of spreading platelets on fibrinogen (right panels) for human platelets treated with sham or the indicated inhibitors at the indicated concentrations. One-way ANOVA with post hoc Dunnett’s testing. Unless indicated with asterisks, post hoc testing revealed nonsignificant results (P ≥ .05). P values corresponding to asterisks: ∗P < .05, ∗∗P < .01, ∗∗∗P < .01, ∗∗∗∗P < .001. Scale bars, 5 μm.
Morphodynamic analysis reveals c-Src as central regulator of Arp2/3 in platelet migration. (A) Experimental scheme of morphodynamic inhibitor screening platform: inhibitor screening and quantification of substrate clearance on substrates leading to retraction (cross-linked fibrin layers) or migration (fibrin monomer layers) in combination with morphological analysis of platelet shape. (B) Quantification of cleared area per cell (μm2) of both migrating (left) and retracting human platelets (right panel) in the presence of the indicated inhibitors, with values being normalized to control in %. One-way ANOVA with post hoc Dunnett's testing. (C) Representative morphologies of human platelets treated with the indicated inhibitors. (D) Representative micrographs of human platelets costained for F-actin and myosin activity (pMLC) following incubation with the indicated treatments. (E) Shape analysis of platelet circularity (a.u., left) and platelet size (μm2, right panel) of migrating human platelets treated with the indicated antagonists. One-way ANOVA with post hoc Dunnett’s testing. (F) Quantification of 3D clot retraction of human PRP incubated with the indicated inhibitors. One-way ANOVA with post hoc Dunnett’s testing. (G) Longitudinal micrographs of live imaging of isolated murine LifeAct-eGFP platelets before (panels 0, 12 seconds) and after addition of the Src inhibitor PP2 (panels 162-492 seconds). Pink arrows mark actin nucleation at the leading edge; pink asterisks show filopodia formation. Right panel: Quantification of # of actin waves (per minute) moving along the leading edge. Student t test, 2-tailed, unpaired. (H) Colocalization plot of fibrinogen and human platelet of sham treatment (left) or after treatment with 10 μM PP2 (right panel). (I) Quantification of cleared area in μm2 upon treatment with indicated inhibitors/agonists (sham vs 10 μM PP2 in the presence or absence of 50 mM ADP and 1 mM TRAP). One-way ANOVA with post hoc Dunnett's testing. (J) Quantification of migrating platelets (%) and cleared area per platelet (μm2) (left panels) as well as the absolute number of recruited and relative amount of spreading platelets on fibrinogen (right panels) for human platelets treated with sham or the indicated inhibitors at the indicated concentrations. One-way ANOVA with post hoc Dunnett’s testing. Unless indicated with asterisks, post hoc testing revealed nonsignificant results (P ≥ .05). P values corresponding to asterisks: ∗P < .05, ∗∗P < .01, ∗∗∗P < .01, ∗∗∗∗P < .001. Scale bars, 5 μm.
Platelet SFK function can be modulated by immunoreceptor tyrosine-based inhibition motif (ITIM) receptors, such as G6b-b and CD31 as well as their downstream effectors, including Src homology 2 domain–containing protein-tyrosine phosphatases, such as Shp1 and Shp2.36-39 Interference with ITIM signaling using blocking antibodies to ITIM receptors as well as small molecule inhibitors targeting downstream effectors, such as Shp1/2 did not affect migratory capacity (supplemental Figure 3B-C).
Soluble agonists show no effect on platelet migratory behavior once migration is initiated
Next, we sought to better understand if soluble factors contribute to initiation and maintenance of migration via c-Src signaling. Para- and autocrine platelet activation through soluble agonists is mostly conveyed via ADP binding to purinergic receptors, such as P2Y12 and via thromboxane-mediated TXA2 receptor activation, which serve as clinical targets of antiplatelet therapy.1 Constant flushing of the migration chamber with removal of soluble agonists did not affect migratory capacity, arguing against a role of local agonist accumulation. Inhibition of soluble ADP and TXA2 on the addition of apyrase and indomethacin, respectively, also had no effect once migration had been initiated (Figure 4A). ADP and TXA2 receptor blockade through cangrelor plus terutroban did not affect migration once initiated but significantly reduced the recruitment and adherence of platelets to the substrate when incubated before adhesion (Figure 4B, preincubation). After initiation, the maintenance of platelet migration was not dependent on the presence of ADP or the TXA2 analog, U46619 (Figure 4C-D, superfusion). The absence of both the agonists impaired initial platelet recruitment to the substrate but did not affect the initiation of migration (Figure 4E). Similarly, activation with soluble agonists, such as thrombin, the glycoprotein VI agonist convulxin, or LPS only partially affected platelet recruitment but did not affect migration once initiated (Figure 4F). These findings indicate that auto- and paracrine platelet activation loops, specifically stimulation by soluble agonists ADP and TXA2, are required for initiating but are dispensable for maintenance of platelet migration.

Classical soluble agonists show no effect on platelet migratory behavior once migration is initiated. (A) Quantification of cleared area per platelet (μm2) and representative micrographs of the indicated treatments following platelet migration on albumin/fibrinogen matrices. Continous act. = addition of U46119 and ADP to wash buffer. Wash = regular treatment. Shear = continuous shear stress (15/s). Apyrase and indomethacin were used at 2.9 U/mL and 10 μM, respectively. One-way ANOVA with post hoc Dunnett's testing. (B) Experimental scheme, representative micrograph and quantification of platelets recruited to an albumin/fibrinogen matrix following preincubation with the P2Y12 inhibitor cangrelor (250 nM) and terutroban (1 μg/mL, blocking thromboxane-mediated activation) or sham treatment. Student t test, 2-tailed, unpaired. Scale bar, 5 μm. (C-D) Experimental scheme, representative micrograph, and quantification of cleared area per platelet (μm2) after superfusion of migrating platelets with cangrelor and terutroban or sham treatment. Student t test, 2-tailed, unpaired. Scale bar, 5 μm. (E) Quantification of % migrating cells and the absolute number of adherent cells per field of view (FOV) in a migration assay with human platelets following activation or not with ADP and U46619. Student t test, 2-tailed, unpaired. (F) Quantification of % migrating cells and the absolute number of adherent cells per FOV for the indicated treatments following initiation of platelet migration. One-way ANOVA with post hoc Dunnett's testing. (G) Representative immunofluorescence costainings of migrating human platelets, stained for c-Src, GPIIb and 14-3-3ζ. Lower panels: enlarged depiction of c-Src and 14-3-3ζ colocalization (pink), corresponding to the white rectangle in the merged right upper panel. Scale bars: upper panel 5 μm, lower panel 1 μm. (H) Histogram depicting colocalization of c-Src, CD41/GPIIb and 14-3-3ζ across the leading edge, along the indicated red line in panel G. MFIs were normalized to the maximum intensity of each antigen. (I-J) Representative micrographs and quantification of % migrating platelets and the cleared area per platelet (μm2) of human platelets treated or not with 10 μM 3′,4′,7′-trihydroxyisoflavone (THO). Scale bar, 25 μm. Student t test, 2-tailed, unpaired. Unless indicated with asterisks, post hoc testing revealed nonsignificant results (P ≥ .05). P values corresponding to asterisks: ∗P < .05, ∗∗P < .01, ∗∗∗P < .01, ∗∗∗∗P < .001.
Classical soluble agonists show no effect on platelet migratory behavior once migration is initiated. (A) Quantification of cleared area per platelet (μm2) and representative micrographs of the indicated treatments following platelet migration on albumin/fibrinogen matrices. Continous act. = addition of U46119 and ADP to wash buffer. Wash = regular treatment. Shear = continuous shear stress (15/s). Apyrase and indomethacin were used at 2.9 U/mL and 10 μM, respectively. One-way ANOVA with post hoc Dunnett's testing. (B) Experimental scheme, representative micrograph and quantification of platelets recruited to an albumin/fibrinogen matrix following preincubation with the P2Y12 inhibitor cangrelor (250 nM) and terutroban (1 μg/mL, blocking thromboxane-mediated activation) or sham treatment. Student t test, 2-tailed, unpaired. Scale bar, 5 μm. (C-D) Experimental scheme, representative micrograph, and quantification of cleared area per platelet (μm2) after superfusion of migrating platelets with cangrelor and terutroban or sham treatment. Student t test, 2-tailed, unpaired. Scale bar, 5 μm. (E) Quantification of % migrating cells and the absolute number of adherent cells per field of view (FOV) in a migration assay with human platelets following activation or not with ADP and U46619. Student t test, 2-tailed, unpaired. (F) Quantification of % migrating cells and the absolute number of adherent cells per FOV for the indicated treatments following initiation of platelet migration. One-way ANOVA with post hoc Dunnett's testing. (G) Representative immunofluorescence costainings of migrating human platelets, stained for c-Src, GPIIb and 14-3-3ζ. Lower panels: enlarged depiction of c-Src and 14-3-3ζ colocalization (pink), corresponding to the white rectangle in the merged right upper panel. Scale bars: upper panel 5 μm, lower panel 1 μm. (H) Histogram depicting colocalization of c-Src, CD41/GPIIb and 14-3-3ζ across the leading edge, along the indicated red line in panel G. MFIs were normalized to the maximum intensity of each antigen. (I-J) Representative micrographs and quantification of % migrating platelets and the cleared area per platelet (μm2) of human platelets treated or not with 10 μM 3′,4′,7′-trihydroxyisoflavone (THO). Scale bar, 25 μm. Student t test, 2-tailed, unpaired. Unless indicated with asterisks, post hoc testing revealed nonsignificant results (P ≥ .05). P values corresponding to asterisks: ∗P < .05, ∗∗P < .01, ∗∗∗P < .01, ∗∗∗∗P < .001.
A c-Src/14-3-3ζ axis driving rho GTPase activation in platelet migration
Mechanical substrate characteristics, such as cross-linking of fibrin fibers govern platelet behavior, leading to either migration or fibrin retraction, with GPIIBIIIA being the central adhesion receptor implicated in platelet-fibrin(ogen) interactions.5,16 Given the above findings, we argued that c-Src needs to be activated downstream of this receptor.
Previous studies have shown that GPIIBIIIA-mediated outside-in signaling depends on direct interaction with c-Src and 14-3-3ζ protein.6-8 In migrating platelets, we identified colocalization of GPIIb, c-Src, and 14-3-3ζ at the edge of the lamellipodium (Figure 4G-H). Inhibition of 14-3-3ζ through 3′,4′,7′-trihydroxyisoflavone at previously used concentrations in the micromolar range abrogated migratory capacity (Figure 4I-J).6 Even at nanomolar concentrations of 3′,4′,7′-trihydroxyisoflavone, which have been shown to neither affect platelet aggregation nor in vitro thrombus formation,6,7 platelets migrated at a lower efficiency (supplemental Figure 4A-B). Together, these findings underline the prominent role of the GPIIb/c-Src/14-3-3ζ complex in platelet migration. Screening for downstream effectors of this signaling axis7 revealed a notable decrease of circularity and migratory potential in platelets treated with Rac-1 inhibitor NSC23766 and Cdc42 inhibitor ML141 with additive effects, whereas RhoA inhibition only had negligible effects (supplemental Figure 4C-G). This highlights rho GTPases Cdc42 and Rac1 as mediators of actin nucleation in platelet migration downstream of GPIIb/c-Src/14-3-3ζ complex.
Gα13 serves as a critical adapter mediating GPIIBIIIA-dependent outside-in signaling in platelet migration
GPIIBIIIA-dependent outside-in signaling leading to SFK activation is prominently mediated by heterotrimeric guanine nucleotide–binding protein (G protein) subunit, Gα13.2,40-42 Interference with Gα13/GPIIBIIIA interactions has been shown to reduce platelet activation, thrombus formation and, specifically, exposure of phosphatidylserine associated with procoagulant activation.30,43,44 Although we have shown that subthreshold inhibition of Gα13 mainly affects procoagulant platelet activation,30 higher doses of the Gα13 inhibitor mP6 also impaired migratory capacity while showing negligible effects on cell retraction (Figure 5A-C). Importantly, these doses were not cytotoxic to platelets (supplemental Figure 4H-I). mP6 treatment also reduced cell size and circularity and was associated with a characteristic shape change dominated by defective lamellipodia formation and a striking decrease in circularity (Figure 5C-E).30 The same doses of mP6 had no effect on platelet retraction, as shown by both single platelet 2D retraction and 3D clot retraction in vitro (Figure 5C,F). Live imaging of mP6-treated LifeAct-eGFP platelets revealed an abrogation of organized actin polymerization at the leading edge lamellipodium, underlining the necessity of GPIIBIIIA outside-in signaling for mediating Arp2/3 function in migrating platelets (Figure 5G; supplemental Video 7). These findings confirm a critical role for GPIIBIIIA-mediated outside-in signaling via Gα13 in the initiation of platelet migratory behavior, however these signaling pathways appear less relevant for retractile function.
![Gα13 serves as a critical adapter mediating GPIIBIIIA signaling in migration. (A-B) Quantification of spread cells per FOV, cleared area per platelet (μm2) and % of migrating platelets for human platelets treated or not with 100 μM mP6 before (pre) or after (wash) initiation of migration (fibrinogen/albumin, A) or retraction (fibrin, B). One-way ANOVA with post hoc Dunnett’s testing. (C) Representative micrographs of migrating (upper, albumin/fibrinogen) and retracting (lower panels, crosslinked fibrin) human platelets (pre-)treated or not with 100 μM mP6. Scale bar, 5 μm. (D) Representative cell shapes and PH images of migrating or retracting human platelets treated or not with 100 μM mP6. Scale bar, 5 μm. (E) Quantification of cell size (μm2) and circularity [a.u.] of human platelets spreading on fibrin(ogen) matrices for the indicated treatments. One-way ANOVA with post hoc Dunnett's testing, comparing the respective shape changes with sham-treated platelets on the same substrate. (F) Representative images of 3D clot retraction experiments of human PRP treated or not with mP6. (G) Micrographs from live imaging experiments using platelets from LifeAct-eGFP mice before (0, 12 seconds) and after (96-246 seconds) treatment with 100 μM mP6. Pink arrows mark actin nucleation at the leading edge, while asterisks indicate marked filopodia formation. Scale bar, 5 μm. Right panel: quantification of # of actin waves (per minute) moving along the leading edge. Student t test, 2-tailed, unpaired. Unless indicated with asterisks, post hoc testing revealed nonsignificant results (P ≥ .05). P values corresponding to asterisks: ∗P < .05, ∗∗P < .01, ∗∗∗P < .01, ∗∗∗∗P < .001.](/view-large/figure/11734222/BLOOD_BLD-2022-019210-gr5.jpg)
Gα13 serves as a critical adapter mediating GPIIBIIIA signaling in migration. (A-B) Quantification of spread cells per FOV, cleared area per platelet (μm2) and % of migrating platelets for human platelets treated or not with 100 μM mP6 before (pre) or after (wash) initiation of migration (fibrinogen/albumin, A) or retraction (fibrin, B). One-way ANOVA with post hoc Dunnett’s testing. (C) Representative micrographs of migrating (upper, albumin/fibrinogen) and retracting (lower panels, crosslinked fibrin) human platelets (pre-)treated or not with 100 μM mP6. Scale bar, 5 μm. (D) Representative cell shapes and PH images of migrating or retracting human platelets treated or not with 100 μM mP6. Scale bar, 5 μm. (E) Quantification of cell size (μm2) and circularity [a.u.] of human platelets spreading on fibrin(ogen) matrices for the indicated treatments. One-way ANOVA with post hoc Dunnett's testing, comparing the respective shape changes with sham-treated platelets on the same substrate. (F) Representative images of 3D clot retraction experiments of human PRP treated or not with mP6. (G) Micrographs from live imaging experiments using platelets from LifeAct-eGFP mice before (0, 12 seconds) and after (96-246 seconds) treatment with 100 μM mP6. Pink arrows mark actin nucleation at the leading edge, while asterisks indicate marked filopodia formation. Scale bar, 5 μm. Right panel: quantification of # of actin waves (per minute) moving along the leading edge. Student t test, 2-tailed, unpaired. Unless indicated with asterisks, post hoc testing revealed nonsignificant results (P ≥ .05). P values corresponding to asterisks: ∗P < .05, ∗∗P < .01, ∗∗∗P < .01, ∗∗∗∗P < .001.
Gα13 serves as a critical adapter mediating GPIIBIIIA signaling in migration. (A-B) Quantification of spread cells per FOV, cleared area per platelet (μm2) and % of migrating platelets for human platelets treated or not with 100 μM mP6 before (pre) or after (wash) initiation of migration (fibrinogen/albumin, A) or retraction (fibrin, B). One-way ANOVA with post hoc Dunnett’s testing. (C) Representative micrographs of migrating (upper, albumin/fibrinogen) and retracting (lower panels, crosslinked fibrin) human platelets (pre-)treated or not with 100 μM mP6. Scale bar, 5 μm. (D) Representative cell shapes and PH images of migrating or retracting human platelets treated or not with 100 μM mP6. Scale bar, 5 μm. (E) Quantification of cell size (μm2) and circularity [a.u.] of human platelets spreading on fibrin(ogen) matrices for the indicated treatments. One-way ANOVA with post hoc Dunnett's testing, comparing the respective shape changes with sham-treated platelets on the same substrate. (F) Representative images of 3D clot retraction experiments of human PRP treated or not with mP6. (G) Micrographs from live imaging experiments using platelets from LifeAct-eGFP mice before (0, 12 seconds) and after (96-246 seconds) treatment with 100 μM mP6. Pink arrows mark actin nucleation at the leading edge, while asterisks indicate marked filopodia formation. Scale bar, 5 μm. Right panel: quantification of # of actin waves (per minute) moving along the leading edge. Student t test, 2-tailed, unpaired. Unless indicated with asterisks, post hoc testing revealed nonsignificant results (P ≥ .05). P values corresponding to asterisks: ∗P < .05, ∗∗P < .01, ∗∗∗P < .01, ∗∗∗∗P < .001.
The c-Src inhibitor dasatanib inhibits migration at nanomolar doses
SFKs are crucial therapeutic targets in clinical hematology. The clinically approved oral TKI dasatanib, which potently inhibits ABL and c-Src, is recommended as one of the several first-line treatment options in patients with CML or Ph+ acute lymphoblastic leukemia based on current guidelines.21,27,29,45,46 Dasatinib treatment has previously been associated with platelet dysfunction and a striking bleeding rate in up to 40% of patients through incompletely understood mechanisms.23,47 On the basis of our findings regarding the role of c-Src in migratory capacity, we assessed the effect of dasatinib treatment on platelet migration. In line with the literature,19,22,24-26 high doses of dasatinib (5 μM) affected multiple platelet effector functions (supplemental Figure 5A-E). Interestingly, however, a dose-response curve for migration, 2D single-cell retraction, procoagulant function and in vitro thrombus formation revealed that migration was particularly sensitive to dasatinib, whereas thrombus formation, retraction of crosslinked fibrin, degranulation, and procoagulant activation were only inhibited at doses down to 1000 to 100 nM. Platelet migration was impaired even when using dasatinib at low nanomolar doses (Figure 6A; supplemental Figure 5F-J). Consequently, at 10 nM, dasatinib potently reduced migratory capacity and led to a loss of polarization and lamellipodia formation associated with distinct morphological changes, including reduced circularity and increased filopodia formation, however this concentration had no significant effect on platelet spreading, thrombus formation, retraction of crosslinked fibrin as well as degranulation, and procoagulant activation (Figure 6B-D; supplemental Figure 5I-J). We detected decreased phosphorylation levels of c-Src at this dose, confirming on-target activity (Figure 6E-F; supplemental Figure 5L),48,49 Murine platelets exhibited the same phenotype with no differences in surface activation markers upon stimulation in suspension but a marked migration defect when exposed to dasatinib at low nanomolar concentrations (supplemental Figure 5M-N). These data suggest a higher sensitivity of platelet migration toward c-Src inhibition compared with other platelet functions.

The c-Src inhibitor dasatanib exhibits a low threshold for migration inhibition. (A) Dose-response curves of human platelets in migration, retraction, activation and in vitro thrombus formation assays to doses of dasatinib (10 μM – 1 nM), normalized to sham-treated samples (%). Supplemental Figure 4F-G for corresponding micrographs and statistical analyses. (B) Representative micrographs of confocal images of human platelets migrating on fibrinogen-albumin matrices treated with 10 nM dasatinib or PBS. Quantification of migrating platelets and the cleared area per cell (μm2) (n = 4 biological replicates). Student t test, 2-tailed, unpaired. Scale bar, 50 μm. (C) Cell-based shape analysis of cell size, circularity, aspect ratio and the number (#) of filopodia of migrating platelets exposed to sham treatment or dasatinib 10 nM. Student t test, 2-tailed, unpaired. (D) Representative micrographs of confocal images of whole blood pretreated with sham treatment or dasatinib 10 nM superfused over a collagen matrix (shear rate 1000/s). Scale bar, 50 μm. Quantification of CD41- and P-selectin (PSEL)/CD62P-positive areas. n = 4. Student t test, 2-tailed, unpaired. (E) Representative confocal images of migrating human platelets treated with dasatinib 10 nM or vehicle and stained for CD41 (white), total c-Src (blue) and phospho-c-Src (Tyr418, red). Scale bar, 10 μm. White arrowheads indicate punctual phospho-c-Src localization at the leading edge, white lines represent migration tracks. Right panels: cell-based quantification of MFI of > 50 platelets from n = 2 biological replicates. (F) Representative western blots of human platelet lysates (n = 3) treated with vehicle or dasatinib before activation with ADP and thromboxane. Lower panels: quantification of densitometry. One-way ANOVA with post hoc Dunnett’s testing. Unless indicated with asterisks, post hoc testing revealed nonsignificant results (P ≥ .05). P values corresponding to asterisks: ∗P < .05, ∗∗P < .01, ∗∗∗P < .01, ∗∗∗∗P < .001.
The c-Src inhibitor dasatanib exhibits a low threshold for migration inhibition. (A) Dose-response curves of human platelets in migration, retraction, activation and in vitro thrombus formation assays to doses of dasatinib (10 μM – 1 nM), normalized to sham-treated samples (%). Supplemental Figure 4F-G for corresponding micrographs and statistical analyses. (B) Representative micrographs of confocal images of human platelets migrating on fibrinogen-albumin matrices treated with 10 nM dasatinib or PBS. Quantification of migrating platelets and the cleared area per cell (μm2) (n = 4 biological replicates). Student t test, 2-tailed, unpaired. Scale bar, 50 μm. (C) Cell-based shape analysis of cell size, circularity, aspect ratio and the number (#) of filopodia of migrating platelets exposed to sham treatment or dasatinib 10 nM. Student t test, 2-tailed, unpaired. (D) Representative micrographs of confocal images of whole blood pretreated with sham treatment or dasatinib 10 nM superfused over a collagen matrix (shear rate 1000/s). Scale bar, 50 μm. Quantification of CD41- and P-selectin (PSEL)/CD62P-positive areas. n = 4. Student t test, 2-tailed, unpaired. (E) Representative confocal images of migrating human platelets treated with dasatinib 10 nM or vehicle and stained for CD41 (white), total c-Src (blue) and phospho-c-Src (Tyr418, red). Scale bar, 10 μm. White arrowheads indicate punctual phospho-c-Src localization at the leading edge, white lines represent migration tracks. Right panels: cell-based quantification of MFI of > 50 platelets from n = 2 biological replicates. (F) Representative western blots of human platelet lysates (n = 3) treated with vehicle or dasatinib before activation with ADP and thromboxane. Lower panels: quantification of densitometry. One-way ANOVA with post hoc Dunnett’s testing. Unless indicated with asterisks, post hoc testing revealed nonsignificant results (P ≥ .05). P values corresponding to asterisks: ∗P < .05, ∗∗P < .01, ∗∗∗P < .01, ∗∗∗∗P < .001.
Dasatinib aggravates inflammatory bleeding through inhibition of platelet migration in vivo
Bleeding events previously associated with dasatinib intake have been linked to mucosal inflammation and subsequent hemorrhage.19,27,50 Notably, mucosal and inflammation-associated bleeding share key elements that are distinct from both classical hemostasis and thrombosis, including a critical role for single platelets recruited to the vascular wall.51-56 Previous work has elucidated the importance of platelet function and sufficient platelet counts in inflammatory hemostasis across different tissues and organs.30,52-55 Among these, platelet migration has been shown to be essential for preventing inflammation-associated hemorrhage.16 Given the striking inhibitory effect of dasatinib on migratory capacity observed in vitro, we performed a model of inflammation-associated pulmonary hemorrhage in mice treated with low doses of dasatinib (1 mg/kg BW, Figure 7A). Treatment with dasatinib aggravated inflammatory hemorrhage without affecting neutrophil or platelet recruitment to the lungs or systemic numbers of platelets and neutrophils (Figure 7B-D; supplemental Figure 6A-D). Dasatinib treatment did not affect systemic levels of procoagulant platelets (Figure 7C; supplemental Figure 6E).30
Dasatinib aggravates inflammatory bleeding in vivo through inhibition of platelet migration and blocks platelet migration in CML patients. (A) Experimental scheme of subacute LPS-induced lung injury model, comparing Bl6 mice treated with dasatinib 1 mg/kg BW or vehicle (black arrows) 12 hours before, immediately before and 12 hours after intranasal LPS treatment (red arrow). (B) Representative macroscopic image of BALF derived from experimental groups, collected in 2 mL Eppendorf tubes, and flow-cytometric assessment of BALF red blood cell (RBC) count, neutrophil (PMN) and platelet counts. n = 4 animals per experimental group. Student t test, two-tailed, unpaired. (C) Quantification of blood neutrophil, platelet and procoagulant (proc.) platelet count as assessed by flow cytometry. Student t test, 2-tailed, unpaired. (D) Representative micrographs from immunofluorescence stainings of LPS-treated lung slices from Bl6 mice treated with vehicle or 1 mg/kg dasatinib. Scale bar, 100 μm. Quantification of TER119+ (RBC+) area in % of total area as well as neutrophil and platelet counts per FOV. n = 4. Student t test, 2-tailed, unpaired. (E) Experimental scheme of LPS-induced sepsis model followed by confocal 4D live imaging of mesenterial venules, comparing Bl6 mice treated with dasatinib 1 mg/kg BW or vehicle (black arrows) 12 hours before and immediately before intraperitoneal injection of 1 mg/kg BW LPS (red arrow). (F) Vessel-based quantification of adhering, migrating, procoagulant and neutrophil-bound platelets in vessels of vehicle- or dasatinib-treated animals from 4D confocal live imaging. Student t test, 2-tailed, unpaired. n = 2 to 3 animals per group. (G) Confocal images of migrating platelet (white shape) and its migration path (red line) against the direction of flow (red arrow) in a vehicle-treated mouse. Scale bar, 15 μm. (H) Longitudinal confocal images of adherent platelets (white arrow heads) and neutrophils in a mesenterial venule of a dasatinib-treated mouse. Scale bar, 20 μm. Supplemental Video 8 for live imaging videos corresponding to panels G-H. (I) Scheme of experimental patient (n = 3 dasatinib, n = 1 bosutinib, n = 3 imatinib) and control groups (n = 5) included in the translational platelet phenotyping study. (J) Representative confocal images of migration assays on albumin/fibrinogen matrices using human platelets derived from healthy controls or patients on imatinib vs dasatinib or bosutinib (both c-Src inhibitors). Scale bar, 25 μm. (K) Relative quantification (normalized to controls) of % migrating platelets and cleared area per platelet. One-way ANOVA with post hoc Dunnett’s testing. Unless indicated with asterisks, post hoc testing revealed nonsignificant results (P ≥ .05). P values corresponding to asterisks: ∗P < .05, ∗∗P < .01, ∗∗∗P < .01, ∗∗∗∗P < .001.

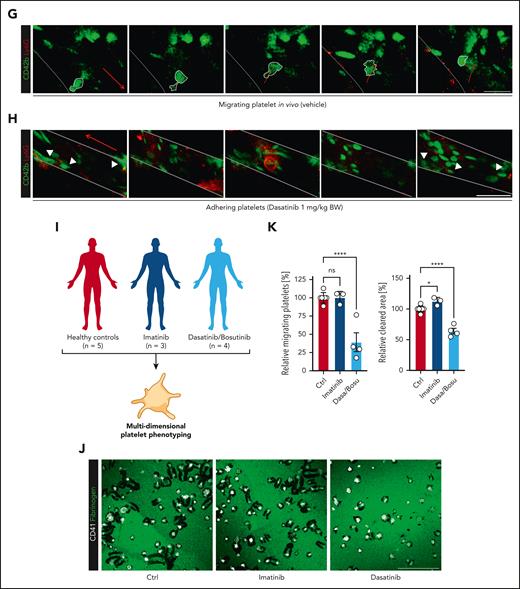
To investigate the effect of systemic dasatinib application on platelet motility in vivo, we performed 4D confocal intravital microscopy of mesenterial vessels in mice treated with the same dose of dasatinib (1 mg/kg BW, Figure 7E) or sham treatment. Although we found no evidence of altered platelet or platelet-neutrophil interactions, dasatinib-treated mice showed a reduction in the number of migrating platelets and a corresponding increase in the number of adherent platelets (Figure 7F-H; supplemental Video 8). Importantly, classical hemostatic or thrombotic platelet phenotypes including hemostasis in a model of tail vein bleeding as well as ferric chloride–induced carotid artery thrombus formation were unaffected at doses of 1 mg/kg BW dasatinib (supplemental Figure 6F-H), confirming previous results.20 Together, these data point toward a platelet migration defect that is present even at low doses of dasatinib and may provide a link between the molecular inhibitory function on c-Src and the observed clinical bleeding phenotype of patients treated with dasatinib.
Platelets from patients treated with dasatinib exhibit prominent migration defects
Finally, to address the translational relevance of our findings in humans, we recruited patients with CML treated with either dasatinib or bosutinib,57 both of which target c-Src, or the multi-TKI imatinib, which does not affect c-Src–mediated signaling (Figure 7J-K; supplemental Figure 6J-O; Table 1). None of the patients included in this pilot study had experienced any clinically relevant bleeding events, and all patients were in stable molecular remission at the time of blood sampling. A total of n = 12 individuals were included in this translational pilot study (n = 3 patients with CML on dasatinib, n = 1 patients with CML on bosutinib, n = 3 patients with CML on imatinib, and n = 5 healthy controls), and both whole blood and isolated platelets were used for in-depth functional phenotyping. Peripheral platelet counts and systemic fibrinogen levels as well as other hemostatic parameters including partial thromboplastin time and international normalized ratio did not differ significantly between groups and were mostly within normal range limits (supplemental Figure 6J). Isolated platelets of all patients treated with dasatinib and bosutinib exhibited a striking reduction in migratory capacity compared with both patients with CML on imatinib and healthy control without affecting the ability to spread on fibrinogen matrices (Figure 7J-K; supplemental Figure 6K-L). Notably, other assessed platelet functions, including clot retraction, integrin activation, degranulation, in vitro thrombus formation, surface expression of key platelet receptors, and formation of platelet-leukocyte and platelet-neutrophil–aggregates were not significantly affected in patients included in this study (supplemental Figure 6J-O).
Clinical characteristics of leukemia patients and healthy subjects included in the human pilot study
| Age | Sex | Drug | Dose | Relevant comedication | |
|---|---|---|---|---|---|
| Control | |||||
| 1 | 62 | F | — | — | — |
| 2 | 31 | F | — | — | — |
| 3 | 52 | M | — | — | — |
| 4 | 28 | M | — | — | — |
| 5 | 48 | F | — | — | — |
| Mean | 44.2 | ||||
| Imatinib | |||||
| 1 | 60 | F | Imatinib | 400 mg | — |
| 2 | 72 | M | Imatinib | 400 mg | — |
| 3 | 49 | M | Imatinib | 400 mg | — |
| Mean | 60.3 | ||||
| Dasatinib/bosutinib | |||||
| 1 | 31 | F | Dasatinib | 140 mg | — |
| 2 | 59 | F | Dasatinib | 140 mg | anagrelide (1 mg OD) |
| 3 | 61 | M | Bosutinib | 500 mg | ASA (100 mg OD) |
| 4 | 66 | F | Dasatinib | 140 mg | — |
| Mean | 54.3 |
| Age | Sex | Drug | Dose | Relevant comedication | |
|---|---|---|---|---|---|
| Control | |||||
| 1 | 62 | F | — | — | — |
| 2 | 31 | F | — | — | — |
| 3 | 52 | M | — | — | — |
| 4 | 28 | M | — | — | — |
| 5 | 48 | F | — | — | — |
| Mean | 44.2 | ||||
| Imatinib | |||||
| 1 | 60 | F | Imatinib | 400 mg | — |
| 2 | 72 | M | Imatinib | 400 mg | — |
| 3 | 49 | M | Imatinib | 400 mg | — |
| Mean | 60.3 | ||||
| Dasatinib/bosutinib | |||||
| 1 | 31 | F | Dasatinib | 140 mg | — |
| 2 | 59 | F | Dasatinib | 140 mg | anagrelide (1 mg OD) |
| 3 | 61 | M | Bosutinib | 500 mg | ASA (100 mg OD) |
| 4 | 66 | F | Dasatinib | 140 mg | — |
| Mean | 54.3 |
OD, once daily.
Discussion
Recent data underscore that platelets act far beyond clot formation, being involved in processes ranging from angiogenesis and maintenance of vascular homeostasis to host defense.16,17,30,58,59 This is reflected in the remarkable diversity of their receptors and associated signaling pathways.2
Through the polarization process dissection that initiates migratory behavior, we show that lamellipodium formation precedes myosin IIa–dependent retraction and that genetic or pharmacological abrogation of Arp2/3 leads to a loss in the polarized phenotype. Surprisingly, soluble agonists appear to be dispensable for platelet migration once initiated, and clinically used antiplatelet therapies, including P2Y12 and thromboxane antagonists, have no inhibitory effect with possible implications for these inhibitors in settings of inflammation. This indicates that (1) immobilized adhesive ligands of the microenvironment are likely to instruct platelet migration and (2) intracellular signaling cascades triggered by mechanosensing are responsible for the initiation and maintenance of migratory behavior. Importantly, fragility and thus pliability of immobilized ligands is paramount for the initiation of platelet migration, as shown for both albumin/fibrinogen matrices on silanized surfaces and biotin-avidin bonds that are ruptured by tractile platelet force.16,32 Consequently, we found that migratory capacity, but not platelet spreading, was abrogated on unpliable surfaces, including plain glass and RGD-functionalized PLL-g-PEG coatings.
Platelet signaling cascades are frequently redundant and show overlapping upstream and downstream regulators, complicating mechanistic studies.2 We circumvented these challenges with a morphodynamics-based screening approach on single platelets to investigate upstream regulators of Arp2/3-dependent lamellipodium formation. We identified GPIIBIIIA outside-in signaling via Gα13 to be of critical importance. This complex then signals via c-Src/14-3-3ζ, which in turn orchestrates Arp2/3 activation, most likely via small GTPases, Rac-1 and Cdc42. Components of this signaling cascade have also been implicated in platelet aggregation and retractile function.60 Interestingly, we found that c-Src/14-3-3ζ inhibition thresholds were significantly lower for polarization and migration than for other assessed platelet functions, such as platelet activation, adhesion, and retraction.6,7 This observation suggests that pathways mediating platelet migration are not redundant and rely on a fully functional GPIIBIIIA/Gα13/c-Src/14-3-3ζ signaling axis.
This axis has also been implicated in platelet spreading, which leads to the question if and how these processes are distinct.6,41,61,62 Our data show that leading edge formation is more sensitive to c-Src inhibition than to stationary spreading. This might be because of the fact that “continuous and dynamic spreading” at the protrusive front during migration most likely requires constant actin polymerization to be maintained. Potentially, this encompasses local integrin and c-Src signaling nodes at the leading edge required for actin nucleation, which could be more susceptible to pharmacological interference and less accessible to compensation by other signaling cascades.63,64 Also, using Cyfip1-deficient mice we have previously highlighted that spreading per se is not necessary for migration if sufficient Arp2/3-driven actin nucleation is maintained.16,65 In animal models, recent data also suggests that platelet spreading is restricted to inflammatory settings in vivo and might occur only in the context of platelet haptotaxis. Intravital microscopy failed to detect platelet lamellipodia in thrombus formation, and Cyfip1-deficient mice that showed prominent spreading defects did not exhibit defects in hemostasis, thrombosis, or inflammatory bleeding.16,65,66
The low inhibition threshold was particularly evident for clinically used c-Src-targeting TKI, dasatinib, which reduced c-Src phosphorylation and efficiently blocked migration at nanomolar doses. In line with the literature, we observed reduced c-Src phosphorylation at residue Y418 with 10 nM dasatinib.48,49 Previous work has convincingly shown that dasatinib, at higher concentrations, potently inhibits core platelet functions, including procoagulant activation, its aggregation, and thrombus formation; we could consistently reproduce these findings (supplemental Figure 5).20,22,24-26 Furthermore, in a pivotal study, Mazharian et al provided strong evidence that dasatinib treatment at higher doses (5 mg/kg BW) reversibly affected megakaryocyte differentiation and proplatelet formation, leading to transient thrombocytopenia and prolonged tail bleeding times.67
Together, these data suggest that even small reductions in c-Src phosphorylation have an impact on platelet migration, whereas platelet production and function beyond migration are affected only at higher levels of c-Src inhibition. In line with this, we found that patients treated with dasatinib showed a prominent defect in platelet migration, which may cumulate in the observed bleeding diathesis, however effects on other assessed platelet functions were less pronounced and inhomogeneous. Even if the small number of patients included in our pilot study may limit the generalizability of our findings, abrogation of platelet migration was the only consistently observed phenotype in all patients who were on c-Src inhibitors, dasatinib or bosutinib. Accordingly, mice treated with low-dose dasatinib (1 mg/kg BW) also exhibited a decrease in platelet migration in vivo and exacerbated inflammatory hemorrhage. These findings align well with the frequently observed mucosal bleeding phenotype of individuals treated with dasatinib, which is mechanistically closely related to inflammatory bleeding.52 Although we cannot exclude that potential off-target effects of dasatinib, including inhibition of bruton kinase or platelet-derived growth factor receptor beta as well as potential effects on endothelial cell integrity68,69 that may contribute to the observed phenotype, both the consistent effects of different c-Src inhibitors (dasatinib, bosutinib, and KB SRC4) as well as reduced Y418 phosphorylation argue for the specificity of c-Src in crucially contributing to migration.45,70,71
Finally, our findings may hold therapeutic potential. We have previously shown that platelet migration can lead to excessive neutrophil activation and increased mortality in a mouse model of Staphylococcus aureus–induced sepsis.17 Although any therapeutic intervention needs to be balanced with protective features of migration at barrier sites, low-dose application of SFK inhibitors, such as dasatinib could represent a novel approach to target platelets in immunopathology and thromboinflammation.
In summary, to the best of our knowledge, we provide novel insights into the regulation of an essential platelet effector function, ie, migration and systematically describe both morphological and behavioral changes along with signaling cascades essential to both. These findings may serve as a mechanistic explanation to the clinically relevant bleeding disorders in patients with normal platelet counts receiving c-Src inhibitors.
Acknowledgments
The authors thank all patients included for participation in this study. The authors also thank all laboratory members for technical support, specifically Coung Tien Kieu for expert help with western blotting. The authors are grateful to Johanna Erber for editorial support.
This study was supported by the Deutsche Herzstiftung e.V., Frankfurt a. M. (individual grants to R.K. and L.N.), Deutsche Forschungsgemeinschaft (DFG), SFB 914 (S.M. [B02 and Z01]), the DFG Project-ID 210592381 – SFB 1054 (T.B., J.K. [B03 and Z01] and J.K. [Z02]), the DFG SFB 1123 (L.N., S.M. [B06]), the DFG FOR 2033 (S.M.), the German Center for Cardiovascular Research (DZHK) (Clinician Scientist Program [L.N.], Start-Up Grant [L.N.], 1.4VD [S.M.]), the DFG Clinician Scientist Program PRIME (413635475, R.K., K.P.) and the FP7 program (project 260309, PRESTIGE [S.M.]). F.G. received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 747687. This work was also supported by the LMUexcellent program (R.K.), the Friedrich-Baur-Stiftung (R.K.) and the European Research Council (ERC-2018-ADG “IMMUNOTHROMBOSIS” [S.M.]).
Authorship
Contribution: F.G. and L.N. initiated the study; R.K. and L.N. conceptualized the study; F.G., R.K., and L.N. created the methodology; R.K., A. Anjum, L.K., Q.L., D.R., A. Akhalkatsi, A.D.z.S., R.E., B.R., C.G., K.P., F.G., and L.N. conducted the investigation; R.K., F.G, T.B., J.K., J.W.H., K.S., S.M., and L.N. collected the resources; R.K., F.G., and L.N. conducted formal analysis; R.K. and L.N. wrote the original draft; all authors edited the draft; R.K. and L.N. handled data curation and software; R.K., F.G., and L.N. visualized the study; R.K., F.G., S.M., and L.N. provided supervision and project administration; and R.K., S.M., and L.N. administered the funding.
Conflict-of-interest disclosure: T.B. and J.K. have an exclusive licensing agreement with BioLegend, Inc for the commercialization of mC1-multimer. The remaining authors declare no competing financial interests.
Correspondence: Rainer Kaiser, Medizinische Klinik und Poliklinik I, University Hospital Ludwig-Maximilian-University Munich, Marchioninistr 15, 81377, Munich, Germany; e-mail: rainer.kaiser@med.uni-muenchen.de; and Leo Nicolai, Medizinische Klinik und Poliklinik I, University Hospital Ludwig-Maximilian-University Munich, Marchioninistr 15, 81377, Munich, Germany; e-mail: leo.nicolai@med.uni-muenchen.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal