

Genetic aberrations associated with the development of extranodal high-grade large B-cell lymphoma originating in the stomach have not been fully identified yet. We analyzed 31 such lymphomas using 73 microsatellite markers for allelic imbalance and microsatellite instability. The highest frequency (42%) of loss of heterozygosity (LOH) was found on the long arm of chromosome 6. We identified 2 LOH hot spots on 6q21-22.1 and 6q23.3-25, flanked by markers D6S246-D6S261 and D6S310-D6S441, respectively, containing putative tumor suppressor genes (TSGs). These 6q aberrations were found to be the sole allelic imbalance in 1 patient only; they were mostly accompanied by additional abnormalities. Several known TSGs, namely, the APC, p15/p16, p53, and DCC genes, were found to suffer frequent LOH during lymphomagenesis. LOH was also detected in regions containing putative TSGs on 7q and 13q14. Frequent amplification of genomic material was found in the 2p, 3q27 at the BCL-6 gene locus, 6p, 7q, 11q23-24 at the MLL gene locus, and 18q regions. Analysis of the pattern of occurrence of these aberrations revealed an association of the amplification of the MLL gene region with LOH at the p53 locus (P = .02). Only low frequency of microsatellite instability (MSI) was detected in these lymphomas and MSI incidence increased with age (P = .01). Karyotypic instability thus plays the main role in the development of gastric high-grade large B-cell lymphoma. Common genetic aberrations responsible for lymphomagenesis are deletions of 6q, loss of p53, and amplification of the 3q27 and the MLL gene regions.
Twenty-five percent to 40% of malignant non-Hodgkin's lymphomas (NHL), the so-called primary extranodal lymphomas, arise outside the lymph nodes, most frequently in the gastrointestinal tract.1 Studies of gastric lymphomas, which constitute the most common extranodal lymphoma, have suggested that their clinicopathologic features are more closely related to the structure and function of mucosa-associated lymphoid tissue (MALT) than of peripheral lymph nodes.2 Recently, marginal zone B-cell lymphoma of MALT-type was established as a distinct clinicopathologic entity in the group of extranodal lymphomas.3 It presents either as a low-grade indolent disease showing characteristic aberrations like translocation t(11;18)(q21;q21)4 or a more aggressive high-grade lymphoma.5 In contrast to their low-grade counterparts, only few data are available regarding cytogenetic and molecular aberrations in high-grade large B-cell lymphomas originating in the stomach. These tumors show mostly complex aberration patterns with several recurrent features such as frequent deletions of chromosome 6q, or partial or whole gains of chromosomes 1, 3, 7, 11, 12, 17, 18, and 21 detected in cytogenetic and comparative genomic hybridization (CGH) studies (unpublished data).4,6Several frequently occurring molecular abnormalities have been studied and well documented. Du et al7 found p53 deletions and mutations in low- and high-grade extranodal, primarily GI tract lymphomas, and suggested that partial inactivation of the p53 gene might play an important role in the development of the low-grade lymphomas, whereas complete inactivation might be associated with high-grade transformation. It has been shown that some gastric high-grade large B-cell lymphomas overexpress the BCL-6 protein8 or demonstrate rearrangements of the BCL-6 locus on chromosome 3q27.9 Homozygous deletion of the p16 gene was found in 14% of patients with gastric high-grade large B-cell lymphoma.10 On the other side, some of the genetic abnormalities occurring in nodal B-cell NHL, namely, rearrangements of BCL-1, BCL-2, and c-myc, are absent in extranodal lymphomas.11-14 However, all these molecular studies have usually narrowly focused on a role of a particular individual gene such as p53 in the pathogenesis of the disease and did not investigate the sequence and relationship between the various gene abnormalities detected.
Genomic instability is a basic property of tumor cells. It generates the diversity necessary for a cancer cell to escape from inherent restraints on growth. One form of genomic instability is the result of inactivation of TSGs, which is the hallmark of tumor suppressor pathway of oncogenesis. The other form results from the malfunction of the DNA mismatch repair system and leads to replication error phenotype (RER+) characteristic of mutator pathway of oncogenesis.15,16 The RER+ phenotype is infrequently detected in nodal B-cell NHL,17 but recent findings suggested that the mismatch repair system defects might play a significant role in the pathogenesis of extranodal lymphoma.18 Both aspects of genomic instability can be evaluated using 1 method, microsatellite analysis.
To assess the contribution of both the tumor suppressor and mutator pathways to the extranodal gastric high-grade large B-cell lymphomagenesis, we analyzed 31 such patients with 73 microsatellite markers. We characterize the genetic aberrations common in the tested subjects and show that abnormalities typical for the tumor suppressor pathway play a major role in the pathogenesis of this disease.
Patients and methods
Patients and samples
Thirty-one consecutive patients with extranodal gastric high-grade large B-cell lymphoma from the lymph node registry at the Institute of Pathology in Wuerzburg on whom fresh frozen tissue from gastrectomy specimens was available were selected for the study. The diagnosis was established according to the criteria of the Revised European-American Lymphoma Classification19 and Chan et al20 by morphologic and immunophenotypic analyses of paraffin-embedded and fresh frozen tissue sections, using standard staining methods as described recently.5 All the cases were diffuse high-grade large B-cell lymphomas originating in the stomach; however, in 6 of them (patients 1 through 6), a concomitant low-grade MALT component was present and these subjects were therefore diagnosed with extranodal high-grade marginal zone B-cell lymphoma of MALT-type. Collectivelly, these 31 lymphomas are best referred to as “gastric high-grade large B-cell lymphomas.” Patients presented in various stages of disease: 4 subjects were in stage EI1; 13 in stage EI2; 9 were stage EII1; 4 were stage EII2; and 1 was stage EIII.21 The tumors were localized at presentation with no clinical evidence of generalized disease. They did not show spread beyond the regional gastric lymph nodes, except for the patient in stage EIII, who is nevertheless still relapse-free 3 years after diagnosis. The patient population showed an age distribution from 31 to 79 years, with a mean of 58. The overall male-to-female ratio was 2.44; there were 22 men and 9 women included in the study.
Microscopic dissection and DNA extraction
In each case, 16 serial 10 μm fresh frozen tissue sections were cut. The first and last cuts were stained with hematoxylin and eosin to assure high tumor content and as a guidance for the following dissection. Under polarized light, an area showing high-grade large B-cell lymphoma was scraped to be used for microsatellite analysis. In a similar way, control genomic DNA was derived from separate tissue blocks not involved by the tumor. DNA extraction was performed with proteinase K and phenol-chloroform, according to routine molecular biology protocols.22
Microsatellite analysis
Primer sequences for the amplification of microsatellite repeats listed in Table 1 were retrieved from Genome Database (GDB). PCR primers were synthesized at MWG Biotech (Munich, Germany) and 1 oligonucleotide of each primer pair labeled with fluorescent dye phosphoramidites FAM, TAMRA, or HEX. Paired normal and tumor DNA samples from each patient were amplified with PE AmpliTaq Gold enzyme (Perkin-Elmer, Foster-City, CA) in multiplex PCR reactions using 50 ng of genomic DNA as a template, under conditions specified by GDB. Thirty cycles were carried out in a PE-2400 thermal cycler (Perkin-Elmer) in a total volume of 20 μL. Aliquots of the PCR reactions were then mixed with size standard and formamide, denatured, and subjected to electrophoresis on a 373 DNA Sequencer (ABI, Foster City, CA). The automatically collected data were analyzed with GENESCAN software as described in the manufacturer's manual. Only patients heterozygous for a given locus were regarded to be informative; homozygosity and microsatellite instability rendered the particular locus unevaluable for LOH or amplification. In heterozygous cases, ratios of both alleles in normal and tumor tissues were calculated. If these ratios showed a difference of more than 20%, the locus was further evaluated for possible allelic imbalance. For determination of LOH or amplification in a locus, first the unchanged allele was identified (by comparison to other markers showing no change in the same multiplex PCR), then the ratios of the allele showing decreased or increased signal to the unchanged allele were calculated, first for control DNA, then for the tumor. Increase of the ratio by 40% in the tumor compared with the control was called amplification, decrease by 40% LOH. All aberrations were confirmed 2 times.
Seventy-three microsatellite markers used in the analysis of gastric high-grade large B-cell lymphoma
| Marker | Location |
|---|---|
| BAT-40 | 1p13.1 |
| D1S237, D1S2827 | 1q32.1 |
| BAT-26 | 2p21-22 |
| D2S391, D2S123 | 2p16-22 |
| D2S2112 | 2p13 |
| D3S1609, TGFbR2-(A)10, D3S1612, D3S1298, D3S1611 | 3p22-24.2 |
| D3S4103, D3S1300 | 3p14.2 |
| D3S1261 | 3p14.1 |
| D3S1206 | 3q21.3-25.2 |
| D3S1212, D3S1229 | 3q26.2-27 |
| D3S1530, D3S1580, D3S1314, D3S1311 | 3q27-qter |
| D4S43 | 4p16.3 |
| D5S82, D5S346 | 5q21 |
| D6S1721 | 6p23 |
| D6S246, D6S268, D6S1592, D6S447, D6S1698, D6S302, D6S261 | 6q21-q22.1 |
| D6S287 | 6q21-q23 |
| D6S292 | 6q22.3-q23 |
| D6S310 | 6q23.3-q25 |
| D6S441 | 6q24-q25.3 |
| IGF2R-3′UTR, IGF2R-(G)8 | 6q26 |
| D6S297, D6S281 | 6q27 |
| D7S501, D7S523 | 7q31 |
| D7S486, D7S522 | 7q31.1 |
| D7S649 | 7q31.1-31.2 |
| D7S483 | 7q36.1 |
| C-MYC | 8q24.12-24.13 |
| D9S2136, D9S1748, D9S1752 | 9p21 |
| MXI1-3′UTR | 10q25 |
| D11S987 | 11q13.3 |
| D11S2179, D11S1356, D11S925, D11S1345, D11S933 | 11q23-24 |
| D12S89, D12S98, D12S70, D12S269 | 12p12-13 |
| D13S153, D13S319, D13S272, AFMa301wb5 | 13q14 |
| E2F-4-(AGC)n | 16q22.1 |
| D17S250 | 17p12 |
| TP53CA | 17p13.1 |
| D18S474, DCC.PCR1 | 18q21 |
| BAX-(G)8 | 19q13 |
| D20S110 | 20q22.1 |
| Marker | Location |
|---|---|
| BAT-40 | 1p13.1 |
| D1S237, D1S2827 | 1q32.1 |
| BAT-26 | 2p21-22 |
| D2S391, D2S123 | 2p16-22 |
| D2S2112 | 2p13 |
| D3S1609, TGFbR2-(A)10, D3S1612, D3S1298, D3S1611 | 3p22-24.2 |
| D3S4103, D3S1300 | 3p14.2 |
| D3S1261 | 3p14.1 |
| D3S1206 | 3q21.3-25.2 |
| D3S1212, D3S1229 | 3q26.2-27 |
| D3S1530, D3S1580, D3S1314, D3S1311 | 3q27-qter |
| D4S43 | 4p16.3 |
| D5S82, D5S346 | 5q21 |
| D6S1721 | 6p23 |
| D6S246, D6S268, D6S1592, D6S447, D6S1698, D6S302, D6S261 | 6q21-q22.1 |
| D6S287 | 6q21-q23 |
| D6S292 | 6q22.3-q23 |
| D6S310 | 6q23.3-q25 |
| D6S441 | 6q24-q25.3 |
| IGF2R-3′UTR, IGF2R-(G)8 | 6q26 |
| D6S297, D6S281 | 6q27 |
| D7S501, D7S523 | 7q31 |
| D7S486, D7S522 | 7q31.1 |
| D7S649 | 7q31.1-31.2 |
| D7S483 | 7q36.1 |
| C-MYC | 8q24.12-24.13 |
| D9S2136, D9S1748, D9S1752 | 9p21 |
| MXI1-3′UTR | 10q25 |
| D11S987 | 11q13.3 |
| D11S2179, D11S1356, D11S925, D11S1345, D11S933 | 11q23-24 |
| D12S89, D12S98, D12S70, D12S269 | 12p12-13 |
| D13S153, D13S319, D13S272, AFMa301wb5 | 13q14 |
| E2F-4-(AGC)n | 16q22.1 |
| D17S250 | 17p12 |
| TP53CA | 17p13.1 |
| D18S474, DCC.PCR1 | 18q21 |
| BAX-(G)8 | 19q13 |
| D20S110 | 20q22.1 |
Comparative genomic hybridization (CGH)
CGH was performed according to a standard protocol.23Tumor DNA was labeled with biotin-16-dUTP, and normal DNA extracted from peripheral blood lymphocytes of a healthy donor was labeled with digoxigenin-11-dUTP (Boehringer, Mannheim, Germany). Equal amounts of test and reference DNA (1 μg each) were cohybridized on commercially available metaphase slides (Vysis, Downers Grove, IL). Detection of biotin- and digoxigenin-labeled probes was accomplished with fluorescein-isothiocyanate (FITC) antiavidin (Vector Laboratories, Burlingame, CA) or Cy3-conjugated antidigoxigenin (Dianova, Hamburg, Germany), respectively. The 4,6-diamidino-2-phenylindole counterstain was used for chromosome identification after antibody detection. Signals were visualized under a Zeiss Axiophot fluorescence microscope (ZEISS, Jena, Germany) and analyzed with the ISIS digital image analysis system (MetaSystems, Altlussheim, Germany). At least 15 metaphases per case were analyzed. For identification of chromosomal imbalances, ratios of 1.25 and 0.8 were used as upper and lower threshold, respectively. A high-level amplification was defined as an overrepresentation of genetic material with fluorescence ratio exceeding 2.0, or on the basis of observation of strong focal signals in the FITC fluorescence with corresponding ratio profile being diagnostic for overrepresentation.6 The final evaluation of CGH profiles was performed by visual control of the digital images to rule out artifacts.
Fluorescence in situ hybridization (FISH)
The interphase FISH MLL assay was established by selecting YAC DNA clone 785c6 localized to the MLL gene locus and as a control YAC 878c12 assigned to 11q21. After amplification of human sequences by Alu-PCR,24 probes were generated by nick translation with biotin-16-dUTP or digoxigenin-11-UTP (Roche Diagnostics, Mannheim, Germany). FISH was performed on cytogenetic preparations or tumor cells isolated from frozen tumor tissue according to standard protocols.25 To determine the cut-off level in normal interphase nuclei, cytogenetic preparations of 5 reactive lymph node specimens served as a negative control. At least 100, in the majority of cases 200, intact nuclei per slide were evaluated by a Zeiss Axiophot fluorescence microscope. Illustrations were generated by using the ISIS imaging system.
Immunostaining for MSH2 and MLH1
Immunohistochemical staining for MSH2 (hybridoma clone FE11, Calbiochem, Germany) and MLH1 (hybridoma clone G168-15, PharMingen, Germany) was performed on pressure cooked, pretreated, formalin-fixed, paraffin-embedded tissue sections and visualized using standard immunoperoxidase technique. Normal tissue of the same patient served as a control. A case was considered positive for each antigen, when more than 80% of the tumor cells showed strong nuclear staining compared with normal cells on the same slide.
Results
LOH on chromosome 6q is the most frequent allelic imbalance detected in gastric high-grade large B-cell lymphoma
Seventy-three microsatellite markers were evaluated for loss or gain of chromosomal material (Table 1). The markers were chosen to cover loci of known and putative TSGs, or chromosomal regions shown to harbor gross chromosomal aberrations in a previous cytogenetic study, using a part of the same patient material.4 To reliably distinguish LOH from genomic amplification, multiplex PCR reactions with the marker in question and minimally 1 other marker used as an internal control were performed. The results were further confirmed by CGH in the great majority of patients (Figure 1), (unpublished data). Several loci on various chromosomes showed allelic imbalance in minimally 2 cases and are depicted in Table2. The most frequent allelic imbalance detected by microsatellite analysis in this study was localized to chromosome 6. Altogether, 13 (42%) patients showed a deletion of a part or the whole long arm of chromosome 6. To determine more closely the extent of the deletions, we analyzed this chromosomal arm with 15 markers. Two LOH hot spots were identified (Table3). One located in the 6q21-22.1 region (7 patients, 23% of informative cases), defined by markers D6S246 and D6S261, and the second one in the 6q23.3-25 region (10 patients, 32% of informative cases), between markers D6S310 and D6S441. Frequent allelic imbalance was also displayed on the short arm of chromosome 6 (7 patients, 4 amplifications, 2 deletions). However, 2 of the patients showing amplification of 6p had concomitant isochromosome 6p(unpublished data) and LOH with all 6q markers pointing to a loss of the whole long arm of chromosome 6. Therefore, the 6p amplification in these 2 subjects was caused by presence of isochromosome 6p and, thus, was a result of the 6q deletion, not expression of an isolated amplification of 6p.

Amplification of the 3q27 region as detected with the D3S1311 microsatellite marker and confirmatory CGH for chromosome 3 in patient 22.
Control and tumor DNA samples were amplified with markers D3S1311 located in the 3q27 region and D7S496 used as an internal control in a multiplex PCR reaction, and the ratios of the alleles determined. CGH profile of chromosome 3 of the same patient is shown on the right side. The cut-off values for losses (0.80) and gains (1.25) are depicted as blue and magenta lines, respectively.
Amplification of the 3q27 region as detected with the D3S1311 microsatellite marker and confirmatory CGH for chromosome 3 in patient 22.
Control and tumor DNA samples were amplified with markers D3S1311 located in the 3q27 region and D7S496 used as an internal control in a multiplex PCR reaction, and the ratios of the alleles determined. CGH profile of chromosome 3 of the same patient is shown on the right side. The cut-off values for losses (0.80) and gains (1.25) are depicted as blue and magenta lines, respectively.
Chromosomal regions showing frequent allelic imbalance
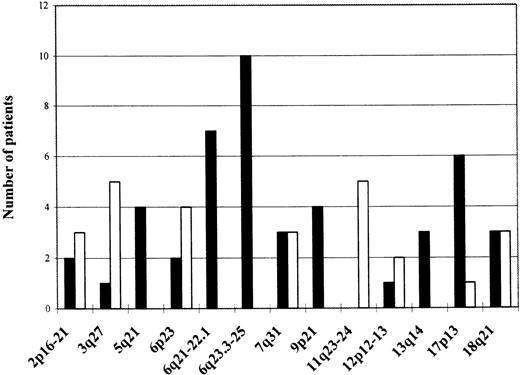
Amplified regions are depicted as empty bars; regions showing LOH as black bars; and the absolute frequency of the aberrations expressed as number of patients showing an aberration in a specific region is shown on the left side.
LOH hot spots on chromosome 6q in gastric high-grade large B-cell lymphoma

Detailed LOH map of chromosome 6q was assembled using 15 microsatellite markers. Thirteen cases that showed LOH at 1 or more loci are presented. Status of each locus is indicated: (horizontal stripes) retention of heterozygosity; (falling stripes) not informative; (vertical stripes) microsatellite instability; and (black) LOH. Numbers of patients are listed at the top of each column. LOH hot spots at 6q21-22.1 (♦) and 6q23.3-25 (♠) are indicated.
Other regions showing consistent LOH
Locus of the p53 gene on chromosome 17p13.1 assayed for with the TP53CA marker showed the second most frequent LOH (Table 2), evident in 6 patients (22% of informative cases). This frequency is similar to that reported in previous MALT lymphoma studies.7 Other tumor suppressor loci displaying consistent LOH were the APC gene locus on 5q21, showing LOH in 4 patients (14%), and the p15/p16 locus on chromosome 9p21 that displayed LOH also in 4 patients (14%). The 13q14 chromosomal band containing the loci of the retinoblastoma gene and a putative TSG called Leu5 deleted in chronic lymphocytic leukemia26 was investigated with several markers. Three patients (10%) displayed LOH, but all 3 deletions detected were rather large and comprised both the retinoblastoma and Leu5 gene loci.
High-level amplification of the MLL gene region
Several microsatellites on the long arm of chromosome 11 (Table 1) were used in the analysis of the 11q chromosomal arm, which is the breakpoint of the reciprocal translocation t(11;18)(q21;q21) found in low-grade MALT lymphoma. Markers positioned in the 11q23-24 region, but not a marker for the cyclin D1 gene locus on 11q13.3 showed amplification of genomic DNA in 5 cases (16%). The 11q23 region contains the MLL gene involved in recurrent translocations in leukemias and lymphomas. Because a concomitant CGH study revealed a high-level 11q23 amplification in 1 of these patients, we further performed FISH analysis with YAC 785c6 contained in the MLL gene locus on 3 patients whose material was available. One of these patients showed high-level amplification detected by FISH, CGH, and microsatellite analyses; the other 2 demonstrated 3 copies of the MLL gene (Figure 2 and raw data not shown).

High-level amplification of the 11q23-24 region containing the MLL gene locus in patient 15.
(A) Results of a multiplex PCR for repeats D4S43 and MXI1 used as an internal control and microsatellite D11S1356 located in the MLL gene region. The individual amplificates show decreasing signal with increasing product size in reaction 15 M (tumor) as compared with 15 N (control); however, the 208 bp allele of the D11S1356 does not follow this pattern and its signal is actually increased. Comparison of the D11S1356 208/198 bp allele ratios in the normal and tumor tissues, 0.7 and 3.09, respectively, shows an increase of the 208 bp allele signal by 341% in the tumor. (B) CGH results for chromosome 11 performed with the same tumor DNA showing high-level amplification of the 11q23-24 region. The cut-off values for losses (0.80) and gains (1.25) are depicted as red and green lines, respectively. (C) Interphase FISH assay performed with the YAC clone 785c6 (green) localized to the MLL gene locus and the control YAC 878c12 (red) assigned to 11q21, showing multiple copies of the MLL gene region. The patient was tetraploid, the fourth signal associated with the control YAC is out of focus.
High-level amplification of the 11q23-24 region containing the MLL gene locus in patient 15.
(A) Results of a multiplex PCR for repeats D4S43 and MXI1 used as an internal control and microsatellite D11S1356 located in the MLL gene region. The individual amplificates show decreasing signal with increasing product size in reaction 15 M (tumor) as compared with 15 N (control); however, the 208 bp allele of the D11S1356 does not follow this pattern and its signal is actually increased. Comparison of the D11S1356 208/198 bp allele ratios in the normal and tumor tissues, 0.7 and 3.09, respectively, shows an increase of the 208 bp allele signal by 341% in the tumor. (B) CGH results for chromosome 11 performed with the same tumor DNA showing high-level amplification of the 11q23-24 region. The cut-off values for losses (0.80) and gains (1.25) are depicted as red and green lines, respectively. (C) Interphase FISH assay performed with the YAC clone 785c6 (green) localized to the MLL gene locus and the control YAC 878c12 (red) assigned to 11q21, showing multiple copies of the MLL gene region. The patient was tetraploid, the fourth signal associated with the control YAC is out of focus.
Other consistent allelic imbalances
A distinct patient group was formed by cases showing partial or complete amplification of chromosome 3. Five subjects (16%) displayed amplification of the 3q27 chromosomal region containing the locus of the BCL-6 gene (Figure 1), but 2 of the patients had actual amplification with all markers located on chromosome 3 and had trisomy 3 confirmed by cytogenetic and FISH analyses.27 Six (19%) patients showed allelic imbalance in the 18q chromosomal region. Three (10%) of them had LOH at the DCC microsatellite located in the DCC gene locus, but another 3 subjects showed amplification with the marker D18S474 located more centromerically. Region 12p12-13 containing the TEL gene was assayed for, with several markers showing amplification of this region in 2 cases (6% of informative cases). Five patients (16%) revealed allelic imbalance in the 2p16-21 region.
Grouping of LOHs and amplifications
Besides the frequency of genomic deletions and amplifications, the pattern of occurrence of these changes and their associations were studied. When the results of the analysis were plotted in a diagram (Table 4), it became obvious that the 6q LOH is an unifying characteristic feature present in 52% of patients showing any allelic imbalance. Thus, it seems that this deletion is one of the most important abnormalities characteristic of gastric high-grade large B-cell lymphoma. A much smaller group was formed by 5 subjects showing the 3q27 amplification. Only 1 of these subjects displayed both the 3q27 amplification and LOH on 6q, pointing at the possibility that these aberrations might belong to different pathogenetic pathways. However, independence of these 2 aberrations could not be reliably statistically investigated because of the small sample size. Another characteristic group was formed by 5 patients showing amplification of the 11q23-24 MLL gene region. Four of the patients had simultaneous LOH at the p53 locus, and this association between the 11q23-24 and p53 aberrations proved to be statistically highly significant (P = .02, χ2 test, exact). 9p21 LOH occurred together with the 6q LOH and also as an isolated abnormality in 2 patients, but not in the 3q27 group. LOH at the p53 gene locus was evident in both the 3q27 amplification and 6q LOH groups, as was the case with other allelic imbalances detected in these patients. Another interesting association was revealed by age analysis of patients showing specific allelic imbalances, namely, amplifications of 12p12-13 and LOH in the 13q14 region were exclusively associated with older age (> 66 years). When the patients were stratified according to the stage of disease at presentation, only those displaying stage EII and beyond demonstrated the 13q14 LOH. None of the subjects showing 4 or more microsatellite instability (MSI) positive markers had a p53 LOH or 6q LOH. On the other side, 3 of 4 patients who had no MSI positive repeats showed 6q LOH.
Pattern of LOHs and genomic amplifications in gastric high-grade large B-cell lymphoma
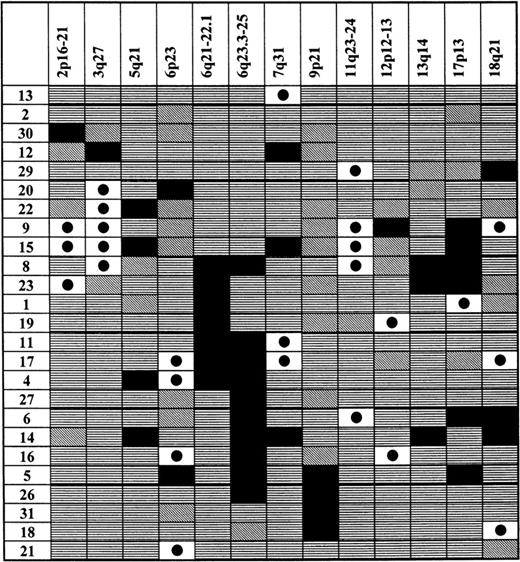
Chromosomal regions showing allelic imbalances are shown at the top of each column. Patients from the analyzed population, who showed an allelic imbalance, are listed in the first left-hand side column. Status of each locus is indicated: (horizontal stripes) retention of heterozygosity; (falling stripes) not informative; (black dot on white background) genomic amplification; and (black) LOH.
Only a minor group of gastric high-grade large B-cell lymphomas shows significantly increased MSI
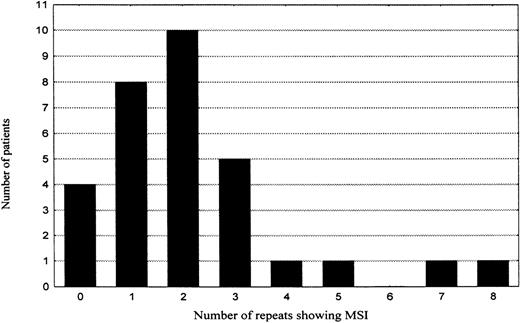
To assess the role of MSI in lymphomagenesis, all microsatellites were also evaluated for MSI. However, only 67 (3%) of 2263 genotypes revealed features of MSI. The majority (74%) of the novel alleles showed only 1 repeat difference to the original allele, 78% were additions, 22% deletions of 1 repeat from the original allele. All the MSI events were type II mutations (only 1 novel allele occurred per marker). When the frequency of MSI per individual patient was plotted in a diagram (Table 5), 2 groups of patients emerged. The first group was composed of patients who either showed no MSI (4 patients, 13%) or displayed a low level of MSI (25 patients, 81%). In this group, the MSI frequency showed a tendency to fit a binomial distribution with mean of about 2 MSI positive repeats and SD of 1.7. The second group consisted of only 2 patients (6%), showing 7 (10%) and 8 (11%) MSI positive markers, respectively. They were located more than 2 SDs apart from the mean of the first group and thus not included in the 95% confidence interval. However, for the increased frequency of MSI in these 2 cases to manifest a biologic process different from that one characteristic of the majority of gastric high-grade large B-cell lymphomas, the difference in MSI frequency is too small and statistically insignificant. Moreover, search for mutations at the polydeoxyadenine tract of the transforming growth factor beta type 2 receptor gene (TGF-βRII), polydeoxyguanine tracts of insulin-like growth factor II receptor (IGF2R) and BAxgenes, or the AGC repeat in the coding region of the E2F-4 gene, mutations that are characteristically associated with MSI-H phenotype in colorectal carcinoma, did not reveal any MSI at these repeats in any of the subjects studied. Immunostains for protein products of the mismatch repair genes MLH1 and MSH2 showed a strong nuclear staining comparable with surrounding normal tissue in more than 90% of the tumor cells in every patient (raw data not shown).
Increasing MSI incidence with age.
The patients were divided according to their age into 3 groups (< 60 years, 60-69 years, and 70 years and older) and comparison of MSI incidence among the individual groups (means and SDs were 1.3 ± 0.8, 1.9 ± 1.3, and 3.8 ± 2.7 MSI event, respectively) was then performed (Table 6). A significant difference in the MSI incidence was found when the groups of patients younger than 60 years of age and older than 70 years were compared (Mann-Whitney U test, 1-tailed). With increasing age, the MSI incidence showed a trend to rise (P = .012, Jonckheere-Terpstra test), and the variability of the MSI incidence increased (P = .02, F test).
Increasing MSI incidence with age
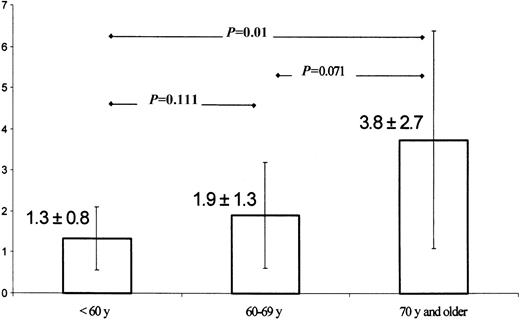
Mean MSI incidence and SD of 3 groups of patients stratified according to age were compared using Mann-Whitney U test (exact, 1-tailed). MSI shows a trend to increase with older age (P = .012, Jonckheere-Terpstra test), and there is also significant increase in MSI variability (P = .02, F test).
Discussion
Compared with nodal high-grade non-Hodgkin's lymphomas, few data are available concerning the characteristic chromosomal abnormalities, genetic, and molecular features of extranodal gastric high-grade large B-cell lymphomas. Those studies performed up to date failed to identify changes that would be specific for these neoplasms. They are in the majority aneuploid tumors showing a high degree of karyotypic instability. Those few nonrandom chromosomal deletions or amplifications identifiable in these tumors reflect loss or gain of genetic material and suggest the presence of TSGs or oncogenes whose function is altered during the process of malignant transformation. We performed a genomic search aiming to identify gene loci involved in the pathogenesis of high-grade large B-cell lymphoma originating in the stomach, and grouped these tumors according to their allelotype with 73 highly informative microsatellite markers located in the vicinity of either known or putative TSGs and oncogenes.
The most frequent LOH in our study occurred on chromosome 6q, as 42% of subjects tested showed a deletion of a part or the whole long arm of chromosome 6. This deletion is thus a more frequent phenomenon than has been reported for extranodal high-grade large B-cell lymphoma so far.4,9 A 6q deletion has been found in a variety of other human malignancies as well, including breast carcinoma,28malignant melanoma,29 renal cell carcinoma,30salivary gland adenocarcinoma,31 ovarian carcinoma,32 acute lymphoblastic leukemia,33and nodal non-Hodgkin's lymphomas.34 In nodal NHL, 3 regions, 6q21, 6q23, and 6q25-27, were determined to show by cytogenetic analysis frequent deletions.35 However, an exclusive association of a specific 6q deletion with a distinct grade of NHL could not be confirmed.36 In our study, in addition to patients showing loss of the whole long arm of chromosome 6 or a large deletion from 6q21 to 6q25, several cases had interstitial 6q21-22.1 or 6q23.3-25 deletions, flanked by markers D6S246 and D6S261, and D6S310 and D6S441, respectively. We thus found 2 distinct regions showing LOH on 6q, indicative of the presence of a TSG. The D6S246-D6S261 interval is rather large (Table 3) encompassing a recently identified region of minimal deletion in acute lymphoblastic leukemia containing a putative TSG.37,38 The second region of minimal deletion on 6q23.3-25 is about 6 cM long and gene rich. It contains, in addition to tens of unidentified ESTs, several already cloned genes. One of these known genes is the ZAC TSG that has been shown to inhibit tumor cell proliferation in vitro and in vivo in nude mice39 and whose expression was found to be lost or reduced in breast cancer cell lines and primary breast tumors.40 On the basis of these results, we suggest, albeit with a small patient sample size, that the 6q deletions play a pivotal role in the pathogenesis of gastric diffuse high-grade large B-cell lymphoma. Because cytogenetic studies of low-grade gastric lymphoma failed to show any of these deletions,4 the 6q LOH appears to characterize the high-grade stage of the disease and could play a role in the transition from low-grade to high-grade gastric lymphoma.
There could be several reasons why we do not see the 6q deletions in a higher percentage of patients. We analyzed the 6q21-22.1 region with 7 markers and the 6q23.3-25 1 with 2 markers. As the latter deletion is relatively small, it is possible that a more detailed analysis that is currently under way could reveal more 6q23.3-25 LOH–positive cases. Another reason not more of the subjects show the 6q deletion might be the genetic heterogeneity of the high-grade lymphomas originating in the stomach. There is no way at present of making the distinction between a fully transformed high-grade MALT-type lymphoma without a concomitant low-grade MALT component and a primary extranodal high-grade large B-cell lymphoma, and it remains uncertain whether such a distinction is of biologic or clinical significance.2 The only difference in the allelotype between the 6 patients with secondary high-grade MALT lymphoma (patients 1 through 6) and the rest of our study population without the low-grade MALT component we saw, was a statistically insignificant absence of aberrations on chromosome 7 and absence of the 3q27 amplification in the former group (Table 4, patient 3 had no allelic imbalance). Otherwise, we detected a similar pattern of aberrations in both secondary high-grade MALT lymphomas and other extranodal diffuse high-grade large B-cell lymphomas.
Intriguing association was revealed between the amplification of the 11q23-24 region comprising the MLL gene and LOH at the p53 locus (P = .02, χ2 test, exact). Several chromosomal regions showed amplifications in this study, but only the MLL region was connected with a concomitant LOH at the p53 locus and herewith a loss of the second p53 allele. Disruption or mutation of both p53 alleles, not only one, is correlated with the acquisition of the ability to amplify endogenous genes; however, alternate pathways can modulate gene amplification as well.41 Why specifically loss of the second wild-type p53 allele is associated with MLL gene amplification is not known and needs to be further investigated. The MLL gene appears to be unique in the human genome and has been conserved during evolution. It is leukemogenic when it fuses with itself,42 as well as when it fuses with genes on other chromosomes. Recurring chromosomal translocations involving chromosome 11, band q23, occur in acute lymphoid and myeloid leukemias;43 rare amplifications of the MLL gene were observed in myeloid leukemia.44 We are the first to show amplification of the MLL locus in lymphoma.
Another common chromosomal aberration detected in this study is amplification of the 3q27 region on the long arm of chromosome 3. Whole or partial trisomy 3 has been described in several cytogenetic studies on both low- and high-grade NHL.27,34,45 We assayed this chromosome for allelic imbalance using 12 microsatellite markers and found the 3q27 region to be amplified in 5 patients (16%). However, 2 of these patients showed amplification with all informative markers on chromosome 3 and had actually trisomy 3. Which gene or genes are involved in the amplification has not been determined yet. BCL-6 might be a candidate; it maps to 3q27 and shows high frequency of rearrangement in nodal diffuse large B-cell lymphomas (DLBL).46,47 However, there were no BCL-6 rearrangements detected in low-grade MALT cases showing trisomy 3, and the frequency of BCL-6 rearrangement in gastric high-grade large B-cell lymphomas is much lower than in nodal DLBL.9
In light of the high level of karyotypic instability typical for the investigated gastric high-grade large B-cell lymphomas and the degree of allelic imbalance we detected in this study, it seems unlikely that the mutator pathway of oncogenesis plays a significant role in its lymphomagenesis. A report by Peng et al18 suggested that the incidence of MSI in extranodal lymphomas could be radically different from the incidence in nodal lymphomas, and the mutator pathway could be the major route of pathogenesis in the former. Our analysis showed only a low background MSI with about 3% of the microsatellites assayed for in the majority of patients. The MSI frequency in patients older than 70 years was about twice as high compared with subjects under 60 (P = .01), suggesting that MSI was rather an epiphenomenon associated with aging. Only 2 patients showed somewhat more frequent MSI with 10% and 11% of the microsatellites used, respectively. However, compared with epithelial cancers and especially colorectal cancer, an 11% level of MSI is still too low to consider these 2 cases to be MSI-H.48 Only cases with more than 40% of all markers unstable are diagnosed as MSI-H in sporadic colorectal carcinoma.49,50 A small fraction of many tumor types, in addition to those of the colon, display some level of MSI.51 However, in most of these tumors, the instability is considerably less pronounced than that observed in colon tumors and it is questionable if it is due to mismatch repair gene defects.
In summary, from the multitude of genomic aberrations and the low level of MSI detected in this study, the conclusion can be drawn that the tumor suppressor pathway is the major pathogenetic pathway in gastric high-grade large B-cell lymphomagenesis. Because of the high percentage of LOH on chromosome 6q, we hypothesize that important gatekeeper genes are situated on this chromosome at the 6q21-22.1 and 6q23.3-25 regions. The identification of these regions as deletion hot spots in gastric high-grade large B-cell lymphoma is a novel finding and should contribute to the understanding of its pathogenesis. However, there is a spectrum of less frequent aberrations that occur during the genesis of this lymphoma and its progression as well. Complete loss of p53 function caused by LOH in the p53 locus was the second most frequent LOH detected and appears to be of special significance as evidenced by its association with amplification of the MLL gene locus. Other genes that we show suffer LOH of the second wild-type allele during lymphomagenesis are important checkpoint genes, such as TSGs APC, DCC, p15/p16, and an unknown gene showing LOH on 13q14 (if it is the retinoblastoma or the Leu5 gene still remains to be decided). In addition to the 11q23-24, regions 3q27 and 18q harboring potential oncogenes show frequent amplification of genomic material as well. We thus identified some of the key players in the pathogenesis of this lymphoma, either already known genes or chromosomal regions harboring candidate genes. This is a necessary prerequisite for the establishment of a lymphomagenesis model. To determine in which sequence these aberrations actually occur, which are the early ones, and if they are specific for the high-grade disease, we just started to analyze low-grade MALT lymphomas with the same markers.
Supported by grants from the IZKF (B3) and the Sonderforschungsbereich 172, B13 of the Deutsche Forschungsgemeinschaft.
Reprints:Petr Starostik, Institute of Pathology, Wuerzburg University, Luitpoldkrankenhaus, Josef Schneider Strasse 2, D-97080 Wuerzburg, Germany; e-mail: peter.starostik@mail.uni-wuerzburg.de.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal