Key Points
PML/RARA-mediated downregulation of Irf8 accelerates APL initiation.
Irf8−/− mutants show expansion of promyelocyte compartment and PML/RARA Irf8−/− mutants show reduced leukemia-free survival.

Abstract
Although the role of promyelocytic leukemia/retinoic acid receptor α (PML/RARA) fusion protein is well recognized in acute promyelocytic leukemia (APL), its contribution to initiation and maintenance of leukemogenesis is not completely understood. Transcriptome analysis in the murine MRP8-PML/RARA APL model has demonstrated modest alterations in gene expression accompanied by expansion of the promyelocyte compartment. Of particular interest, mice expressing PML/RARA showed downregulation of the transcription factor Irf8 mRNA. Interferon regulatory factor 8 (IRF8) is a known regulator of hematopoiesis. Previous research had implicated IRF8 as a tumor suppressor for myeloid neoplasia, and mice lacking IRF8 develop a well-differentiated myeloproliferative neoplasm characterized by expansion of neutrophilic lineage cells. We hypothesized that PML/RARA-mediated downregulation of Irf8 transcript levels contributes to the initiation of APL. We observed significant downregulation of IRF8 protein levels in highly purified promyelocyte populations of PML/RARA transgenic mice. We also found that loss of IRF8 results in expansion of promyelocytes in vivo, partially phenocopying the impact of PML/RARA expression. Moreover, survival experiments showed that complete loss of IRF8 leads to acceleration of APL onset in our PML/RARA mice. Collectively, these data identify IRF8 downregulation as an important factor in APL initiation and highlight a tumor-suppressor role for IRF8 in this acute leukemia.
Introduction
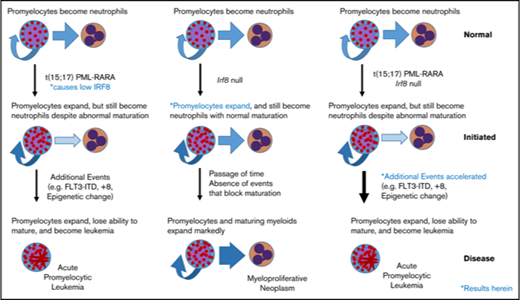
Acute promyelocytic leukemia (APL) is marked by the accumulation of promyelocytes due to a blockade of differentiation coupled with unlimited expansion. Approximately 97% of patients with APL express the promyelocytic leukemia/retinoic acid receptor α (PML/RARA) fusion protein as a result of t(15;17) (q22;q11.2), and although the paradigm has long suggested that PML/RARA is a potent repressor of key myeloid maturation genes, we recently challenged this model,1 reopening fundamental questions as to the precise contribution of the fusion protein to leukemic transformation.
IRF8 is a known regulator of hematopoiesis. IRF8 plays an important role in orchestrating specification and differentiation of B cells, dendritic cells, and monocytes.2 Particularly in bipotential granulocyte/monocyte precursors, IRF8 expression is necessary to direct cells down the monocytic lineage.3,4 IRF8 is also a key mediator of innate immunity and will drive targeted transcriptional programs in response to interferon signaling, following binding to specific DNA elements and posttranslational modifications allowing association with partner proteins.5,6
We recently identified that Irf8 is downregulated in murine PML/RARA preleukemic promyelocytes compared with their wild type (WT) counterparts.1 Given the robustness of the Irf8 downregulation that we observed in the absence of major alterations of other common myeloid transcription factors, we hypothesized that lower Irf8 levels mediated by PML/RARA play a key role in the leukemogenic process. Further, we observed that Irf8 is additionally downregulated in PML/RARA leukemic promyelocytes compared with their preleukemic counterparts,1 suggesting that downregulation of Irf8 levels could be important to maintain the leukemogenic program. Intrigued by these results, we investigated how IRF8 downregulation impacts the promyelocyte compartment in preleukemic mice as well as the kinetics of APL initiation in our MRP8-PML/RARA model. We show that young Irf8-null animals show an expansion of promyelocytes that phenocopies the expansion seen in PML/RARA mice. We also show that acute leukemia onset is strongly accelerated in PML/RARA Irf8−/− recipients compared with PML/RARA mice. These data support the hypothesis that downregulation of IRF8 by PML/RARA is a key mechanism in the initiation of APL.
Methods
Murine model, BM harvest, and transplantation experiments
The MRP8-PML/RARA7 transgenic and Irf8−/−8 genetically modified models were previously described. Transplantation experiments (Figure 1D-F) were performed as described in Jones et al,9 and each cohort included recipients of 6 individual bone marrow (BM) donors.
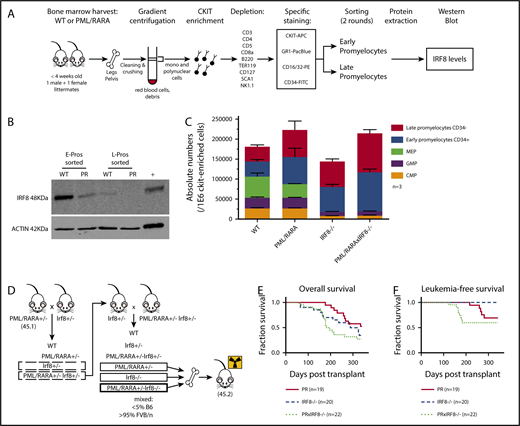
IRF8 is downregulated by PML/RARA in preleukemic promyelocytes and acts as a tumor suppressor in PML/RARA-driven APL. (A) Details of the experimental strategy used to investigate IRF8 protein levels in highly purified populations of sorted promyelocytes, in the presence or absence of PML/RARA. (B) Western blot analysis of IRF8 in sorted early and late promyelocytes of PML/RARA mice. Total spleen from WT animals was loaded as a positive control. IRF8 is detected at a size band of 48 kDa. Actin loading control is detected at a size band of 42 kDa. Images were obtained on LI-COR scanner. (C) Enumeration of c-Kit+ progenitor populations in the BM of PML/RARA, Irf8−/−, and PML/RARA Irf8−/− mice (n = 3 for each group). Error bar represents mean ± standard error of the mean. (D) Mating strategy used to investigate kinetics of APL initiation in PML/RARA, Irf8−/−, and PML/RARA Irf8−/− backgrounds. (E-F) Overall (E) and acute (F) leukemia-free survival of lethally irradiated recipients of BM from young PML/RARA, Irf8−/−, and PML/RARA Irf8−/− donor mice. Leukemia-free survival in the PML/RARA Irf8−/− cohort was shorter than that of the PML/RARA cohort (P = .03, Gehan-Breslow-Wilcoxon test). APC, allophycocyanin; CMP, common myeloid progenitor; GMP, granulocyte/macrophage progenitor; FITC, fluorescein isothiocyanate; MEP, megakaryocyte-erythrocyte progenitor; PE, phycoerythrin; PR, PML/RARA.
IRF8 is downregulated by PML/RARA in preleukemic promyelocytes and acts as a tumor suppressor in PML/RARA-driven APL. (A) Details of the experimental strategy used to investigate IRF8 protein levels in highly purified populations of sorted promyelocytes, in the presence or absence of PML/RARA. (B) Western blot analysis of IRF8 in sorted early and late promyelocytes of PML/RARA mice. Total spleen from WT animals was loaded as a positive control. IRF8 is detected at a size band of 48 kDa. Actin loading control is detected at a size band of 42 kDa. Images were obtained on LI-COR scanner. (C) Enumeration of c-Kit+ progenitor populations in the BM of PML/RARA, Irf8−/−, and PML/RARA Irf8−/− mice (n = 3 for each group). Error bar represents mean ± standard error of the mean. (D) Mating strategy used to investigate kinetics of APL initiation in PML/RARA, Irf8−/−, and PML/RARA Irf8−/− backgrounds. (E-F) Overall (E) and acute (F) leukemia-free survival of lethally irradiated recipients of BM from young PML/RARA, Irf8−/−, and PML/RARA Irf8−/− donor mice. Leukemia-free survival in the PML/RARA Irf8−/− cohort was shorter than that of the PML/RARA cohort (P = .03, Gehan-Breslow-Wilcoxon test). APC, allophycocyanin; CMP, common myeloid progenitor; GMP, granulocyte/macrophage progenitor; FITC, fluorescein isothiocyanate; MEP, megakaryocyte-erythrocyte progenitor; PE, phycoerythrin; PR, PML/RARA.
Staining of promyelocytes and flow cytometry
As previously described we identified early promyelocytes (c-Kit+GR1intCD16/32+CD34+; “E-Pro”) and late promyelocytes (c-Kit+GR1intCD16/32+CD34−; “L-Pro”) as well as granulocyte/macrophage progenitors (c-Kit+GR1−CD16/32+CD34+; GMPs), common myeloid progenitors (c-Kit+GR1−CD16/32−CD34+), and megakaryocyte-erythrocyte progenitors (c-Kit+GR1−CD16/32−CD34−).1
Detection of IRF8 (interferon consensus sequence–binding protein) by western blot
Following 2 rounds of sorting, protein lysates of highly purified cells (65 000), were electrophoresed, transferred to nitrocellulose, and examined with anti-IRF8 (39-8800, Invitrogen) and goat anti-mouse IRDye (926-32210, LI-COR Biosciences). Additional details are provided in supplemental Methods.
Results and discussion
The role of IRF8 as a possible myeloid leukemia tumor suppressor has been documented by several groups in various hematologic contexts (Table 1 10-18 ). A few groups, including ours, have suggested that alteration of Irf8 occurs in the context of PML/RARA (Table 1, bottom), and human APLs have lower levels of IRF8 expression in comparison with a number of other subtypes of acute myeloid leukemia (supplemental Figure 1). Our gene expression studies showed that PML/RARA expression leads to lower Irf8 transcript levels in preleukemic promyelocytes (∼4.8-fold compared with WT), with further loss in fully leukemic cells (∼90-fold compared with preleukemic),1 suggesting that downregulation of this transcription factor mediated by and in association with PML/RARA could participate in both the emergence and maintenance of a leukemic clone. To interrogate if lower transcript abundance in our mice would be reflected as lower protein levels, we used the experimental strategy described in Figure 1A to investigate IRF8 protein levels in our highly purified promyelocyte populations from MRP8-PML/RARA transgenic animals or their WT littermates.
We confirmed a strong downregulation of IRF8 in our PML/RARA early promyelocyte population, with a similar trend in late promyelocytes (Figure 1B). IRF8 is normally downregulated with neutrophilic maturation, and the presence of PML/RARA results in precocious loss during granulocyte development. To delineate the impact of this early loss of IRF8, we assessed promyelocyte numbers in young healthy Irf8−/− and PML/RARA Irf8−/− mice in comparison with WT Irf8+/+ mice and Irf8+/+ mice expressing the PML/RARA transgene (Figure 1C). As previously observed, young PML/RARA Irf8+/+ mice had a substantially increased number of marrow promyelocytes in comparison with WT mice.1 Fascinatingly, loss of Irf8 alone resulted in an essentially identical expansion of promyelocytes (as well as a loss of earlier myeloid progenitors in the BM, not seen in PML/RARA Irf8+/+ mice), and a combination of PML/RARA expression and IRF8 loss did not result in a statistically significant further expansion of promyelocytes. These results suggest an epistatic relationship between PML/RARA and IRF8, compatible with downregulation of IRF8 by PML/RARA as being a key mechanism by which t(15;17) expands promyelocytes in the initiation of APL. The prior observation that PML/RARA binds near the IRF8 gene in the human NB4 cell line and primary human APL cells16 suggests that PML/RARA is able to downregulate IRF8 through direct transcriptional repression.
Given the concordance of our experimental results with prior studies of IRF8 in myeloid neoplasia, we hypothesized that downregulated Irf8 levels mediated by PML/RARA could play a critical role in disease initiation in our mouse model. If so, then further depletion of Irf8 should cooperate in APL development. To test this hypothesis, we compared the development of leukemia in mice expressing PML/RARA and lacking IRF8 in their BM with that seen in mice having only the single genetic changes. The experimental design is depicted in Figure 1D, and representative pathology of ill animals in these cohorts is presented in supplemental Figure 2. As discussed above, Irf8−/− recipients develop a myeloproliferative neoplasm, requiring euthanasia of most animals within one year. These non-acute leukemia deaths are reflected as similar overall survival curves observed in the Irf8−/− and PML/RARA Irf8−/− cohorts (Figure 1E). When looking specifically at deaths due to acute leukemias, loss of Irf8 strongly accelerated PML/RARA-mediated leukemia initiation (Figure 1F). In the PML/RARA-only cohort, the leukemias that developed arose at 215 to 279 days. In the PML/RARA Irf8−/− cohort, the leukemias that developed arose at 122 to 182 days. It was notable that all the acute leukemias in the PML/RARA Irf8−/− cohort occurred prior to the first appearance of acute leukemia in the PML/RARA cohorts. These data demonstrate that further amplifying the aberrant PML/RARA-mediated downregulation of IRF8 cooperates in leukemic transformation. Nevertheless, even in the absence of IRF8, there is still substantial disease latency. One limitation of the present study is a lack of in vivo data herein on the effects of IRF8 overexpression in APL cells. To bridge this gap, a recently published study demonstrated that overexpression of IRF8 in leukemic cells from our MRP8-PML/RARA mouse model7 inhibits transplantation of established leukemia.19
Our work complements existing data to indicate an important role for IRF8 suppression in APL pathogenesis. Altogether, our data support a model of APL leukemogenesis in which the translocation of chromosomes 15 and 17 initiates leukemia development, in part by downregulating IRF8, and in which the resulting expansion of the promyelocyte compartment contributes to acquisition of additional cooperating events (eg, trisomy of chromosome 8,20 mutation of FLT321 ) that complete leukemic transformation.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the UCSF Comprehensive Cancer Center Shared Resources in Mouse Pathology and Cell Analysis, the UCSF Flow Cytometry Core, Keiko Ozato for kindly providing authorization to use the Irf4/Irf8 knockout model, and Damien Reynaud for helping to acquire this line.
This work was funded by the National Cancer Institute, National Institutes of Health (R01-CA95274).
Authorship
Contribution: C.G. cowrote the manuscript, designed and executed experiments, analyzed data, and prepared figures; S.S. designed and executed experiments, cowrote the manuscript, analyzed data, and prepared figures; T.B. and M.R.W. executed experiments and analyzed data; B.F and H.G. analyzed data; E.P. and H.d.T. provided project direction; and S.C.K. cowrote the manuscript, provided project direction, designed experiments, and analyzed data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for C.G. is Genentech, Inc., South San Francisco, CA.
The current affiliation for T.B. is Allogene Therapeutics, South San Francisco, CA.
The current affiliation for M.R.W. is Gilead Sciences, Inc., Foster City, CA.
The current affiliation for E.P. is Columbia Stem Cell Initiative, Columbia University Medical Center, New York, NY.
Correspondence: Scott C. Kogan, Department of Laboratory Medicine and Helen Diller Family Comprehensive Cancer Center, 513 Parnassus Ave, Room S-561, Box 0451, University of California, San Francisco, San Francisco, CA 94143-0451; e-mail: scott.kogan@ucsf.edu.