Key Points
Tisagenlecleucel expanded in vivo and provided clinical benefit in r/r DLBCL patients in CR after bridging therapy.
Tisagenlecleucel produced durable responses in r/r DLBCL patients without detectable disease before infusion.

Abstract
Tisagenlecleucel demonstrated high rates of durable responses in adult patients with relapsed or refractory diffuse large B-cell lymphoma (r/r DLBCL) in the JULIET trial. Most patients (92%) received bridging therapies to control disease after study entry and before tisagenlecleucel infusion. Here, we examine the efficacy and safety of tisagenlecleucel in the subset of 7 patients who achieved complete response (CR) after bridging therapy and before tisagenlecleucel infusion. Tisagenlecleucel rapidly expanded in all 7 patients, and the transgene levels were measurable for up to 2 years after infusion. After infusion, all 7 patients were still in CR at the month 3 evaluation, and 5 of 7 patients remained progression-free >12 months. Adverse events were similar to the overall JULIET population. Cytokine release syndrome (CRS) was reported in 4 of 7 patients (grade 2 = 2 and grade 3 = 2 using the Penn grading scale), and 1 patient experienced grade 1 neurotoxicity. No patient required tocilizumab or steroids for CRS management. These data provide preliminary evidence of tisagenlecleucel efficacy in patients with r/r DLBCL without detectable disease after bridging or salvage therapies and warrant further investigation of tisagenlecleucel as consolidative therapy in future trials. This trial was registered at www.clinicaltrials.gov as #NCT02445248.
Introduction
Relapsed or refractory diffuse large B-cell lymphoma (r/r DLBCL) has a poor prognosis.1 Results from the SCHOLAR-1 and Collaborative Trial in Relapsed Aggressive Lymphoma (CORAL) studies suggest that response rates to available rituximab-based salvage regimens for r/r DLBCL remain dismal and are unlikely to be durable.1-3 In SCHOLAR-1, the objective response rate was 26% with a 7% complete response (CR) rate and a median overall survival (OS) of 6.3 months.2
Tisagenlecleucel, an autologous anti-CD19 chimeric antigen receptor (CAR) T-cell therapy, results in high rates of durable responses in patients with r/r DLBCL.4,5 Results from JULIET (NCT02445248), a single-arm, open-label, multicenter, global phase 2 trial of tisagenlecleucel in adults with r/r DLBCL, demonstrated an objective response rate of 52%, with CR and partial response (PR) rates of 40% and 12%, respectively.5 Long-term follow-up in JULIET demonstrated durable responses with a consistent safety profile and improved OS compared with historical treatment options.6
Among the 111 patients who received tisagenlecleucel in JULIET, most patients (92%) received various bridging therapies, including conventional combination chemotherapy to control disease during the period between enrollment and start of lymphodepleting chemotherapy.5 However, a subset of 7 patients had no evidence of active disease after bridging therapy and received tisagenlecleucel infusion per protocol. Data from these patients were excluded from the efficacy table in the US package insert7 for the DLBCL indication with the US Food and Drug Administration.
Because tisagenlecleucel targets CD19+ B cells and the study required measurable disease at enrollment, it was unknown whether tisagenlecleucel would expand and provide clinical benefit in patients without measurable disease before infusion. This post hoc exploratory analysis reports on expansion, safety, and outcomes of tisagenlecleucel therapy in the subset of patients with r/r DLBCL who had no measurable disease after bridging therapy and before receiving tisagenlecleucel infusion.
Study design
Study design and patients
Details of JULIET were described previously.5 Briefly, patients were ≥18 years of age at entry and had ≥2 prior lines of therapy including rituximab and an anthracycline. Key eligibility criteria included progressive disease (PD) after, or ineligibility for, autologous stem cell transplantation. Eligible patients underwent leukapheresis and cryopreserved material was shipped to a central manufacturing facility. Patients could receive bridging therapy if deemed necessary by their treating physician. An independent review committee assessed disease status and response using the positron emission tomography (PET) 5-point Deauville scale (Lugano classification).8 A restaging PET–computed tomography scan before lymphodepletion was added as a protocol amendment, but was not completed for all patients. Thus, it is possible that additional patients may have been treated without measurable disease, due to lack of imaging after bridging therapy. Most patients (n = 103) underwent lymphodepleting chemotherapy with fludarabine and cyclophosphamide or bendamustine before tisagenlecleucel infusion; patients with white blood cell counts <1 × 109/L within 1 week of infusion could omit lymphodepleting chemotherapy per protocol. Patients received a single infusion of tisagenlecleucel with a target dose of 5 × 108 viable CAR+ T cells.
Efficacy and safety end points
End points for this analysis included tisagenlecleucel cellular kinetics, response type and duration, survival status, and safety in this patient subset of JULIET. Adverse events were reported using the Medical Dictionary for Regulatory Activities, version 20.1, and Common Terminology Criteria for Adverse Events, version 4.03.5 Cytokine release syndrome (CRS) was graded using the University of Pennsylvania grading scale and was managed by a protocol-specific algorithm.5,9,10 A retrospective analysis of CRS severity using the Lee grading scale11 was also conducted.
Tisagenlecleucel and cellular kinetics
Tisagenlecleucel expansion and persistence were characterized using time course of transgene levels (in copies per microgram of genomic DNA) in peripheral blood, measured as previously described using a TaqMan-based quantitative polymerase chain reaction assay.5,12
Results and discussion
As of 8 December 2017, 111 patients had received tisagenlecleucel in the JULIET trial. Among these patients, 7 had no evidence of active disease after bridging therapies and before tisagenlecleucel infusion.
Demographics and baseline disease characteristics of the 7 patients are shown in Table 1 and were similar to the overall JULIET patient population.5 All 7 patients received both bridging therapy and lymphodepleting chemotherapy. The mean absolute lymphocyte counts before lymphodepleting chemotherapy were similar between the 7 patients and overall population, and the mean absolute lymphocyte counts after lymphodepleting chemotherapy were similar between the 7 patients and the overall JULIET population (data not shown).
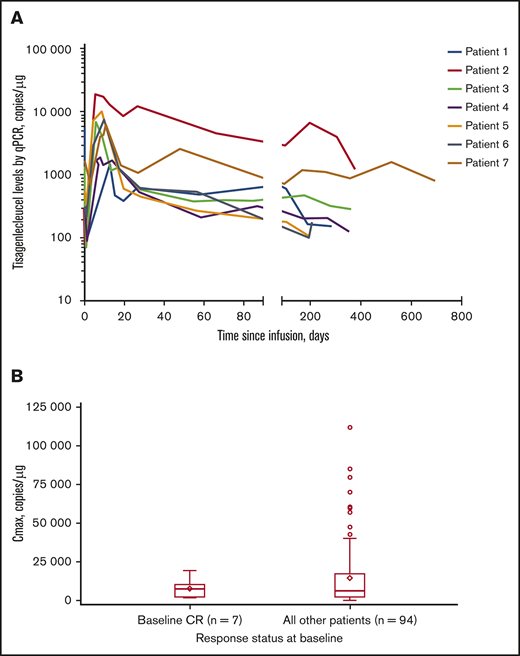
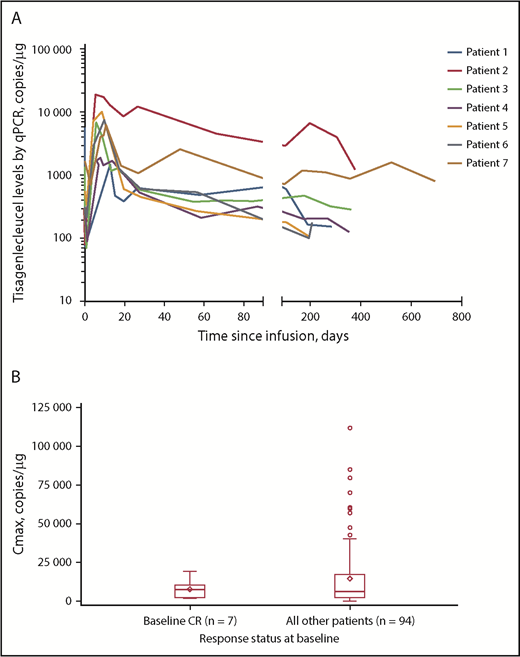
Tisagenlecleucel rapidly expanded during the first 28 days after infusion in all 7 patients, and CAR transgene levels were measurable at a maximum of ∼2 years (Figure 1A). Median time to last quantifiable transgene level was 351 days (range, 190-693 days) and is expected to increase with longer follow-up. Mean transgene levels at peak expansion (maximum concentration [Cmax]) in the 7-patient subset (geometric mean Cmax [percentage of coefficient variation]: 5760 copies per microgram [112%]) were comparable with those of the rest of the patients in the JULIET trial with reliable parameter estimates (n = 94; geometric mean Cmax [percentage of coefficient variation]: 5790 copies per microgram [291%]) (Figure 1B). In the JULIET trial population, the mean expansion was similar between responders and nonresponders.5 Additionally, median time to peak expansion of 9 days from the 7-patient subset was similar to that observed in the rest of the patients in the JULIET population.

Tisagenlecleucel expansion kinetics. (A) Cellular kinetic plots demonstrating in vivo expansion and persistence of tisagenlecleucel in patients with CR at baseline. (B) Box plot of Cmax by response status at baseline (pharmacokinetic analysis set). Geometric mean Cmax (% coefficient variation) in patients with CR at baseline and in the rest of the patients was 5760 copies per microgram (112%) and 5790 copies per microgram (291%), respectively. Diamonds represent the mean and circles represent values outside of 1.5× interquartile range (IQR). Lower and upper whiskers extend to most extreme points within 1.5× IQR or quartile 1 and quartile 3, respectively. qPCR, quantitative polymerase chain reaction.
Tisagenlecleucel expansion kinetics. (A) Cellular kinetic plots demonstrating in vivo expansion and persistence of tisagenlecleucel in patients with CR at baseline. (B) Box plot of Cmax by response status at baseline (pharmacokinetic analysis set). Geometric mean Cmax (% coefficient variation) in patients with CR at baseline and in the rest of the patients was 5760 copies per microgram (112%) and 5790 copies per microgram (291%), respectively. Diamonds represent the mean and circles represent values outside of 1.5× interquartile range (IQR). Lower and upper whiskers extend to most extreme points within 1.5× IQR or quartile 1 and quartile 3, respectively. qPCR, quantitative polymerase chain reaction.
With a median follow-up of 14.5 months, all 7 patients maintained CR at the initial assessment 28 days after infusion and at the month 3 assessment (Table 1); 5 patients (1, 2, 3, 4, and 7) remained progression-free for >12 months at the data cutoff of 8 December 2017. Patient 7 declined all scans beyond the month 3 visit but continues in follow-up with no sign of disease progression >2 years after tisagenlecleucel infusion. Patient 5 experienced PD on day 274, began new anticancer therapy, and died on day 544 of PD. Patient 6 experienced PD on day 196 and withdrew consent to further follow-up (Table 1). Median time from last bridging therapy to the start of lymphodepleting chemotherapy was 44 days in the 7 patients and 24 days for the overall infused population.
Adverse events of special interest in this subset (Table 1) were similar to those in the overall JULIET population. Four of 7 patients experienced CRS (grade 2 = 2 and grade 3 = 2, using the Penn scale9 ) with a median duration of 5 days. None of these 4 patients required tocilizumab or steroids for CRS management. Only 1 patient experienced grade 1 neurologic events.13 No deaths were observed due to tisagenlecleucel, CRS, or cerebral edema/neurotoxicity. Retrospective analysis of CRS severity using the Lee grading scale11 found that all 4 patients had grade 2 CRS.14
In summary, tisagenlecleucel expansion occurred in all 7 patients with no measurable disease after bridging therapy, and mean expansion was similar to the JULIET trial. More than one-half of the patients (5 of 7) remained progression free >12 months after infusion, which is longer than would be expected with bridging therapy alone. One hypothesis to explain these observations is that CAR T cells can expand in response to residual disease undetected by PET imaging. These findings are novel because data from these 7 patients were excluded from the DLBCL efficacy table in the Kymriah (tisagenlecleucel) US package insert,7 and, in the commercial setting for axicabtagene ciloleucel therapy,15 patients who attained CR after bridging therapy did not receive their CAR T-cell infusion.
In conclusion, these results provide preliminary evidence of tisagenlecleucel efficacy in patients with r/r DLBCL without detectable disease after bridging or salvage therapies. Additionally, these 7 patients experienced low rates of CRS and neurotoxicity. These data warrant further exploration in clinical trials of tisagenlecleucel as consolidative therapy for patients with relapsed and high-risk DLBCL in CR.
Acknowledgments
The authors thank the patients enrolled in this study and their families. Medical writing support was provided by Healthcare Consultancy Group.
This work, including medical writing support, was supported by Novartis Pharmaceuticals.
Authorship
Contribution: M.R.B., R.T.M., E.K.W., U.J., J.R.W., J.P.M., I.F., H.H., P.B., and S.J.S. enrolled patients, performed research, and contributed to data collection and interpretation; C.d.C., R.T., Ö.A., R.A., L.P., and V.V.R. performed cellular kinetic and/or statistical analyses and/or contributed to data interpretation; M.R.B. and S.J.S. wrote the first draft; and all authors were involved in revising the manuscript and approved the final version.
Conflict-of-interest disclosure: M.R.B. provided consultant services to, was a member of an entity’s board of directors or advisory committees for, and received research funding from Novartis, Kite Pharma, and Juno Therapeutics; provided consultant services to and received honoraria from Novartis, Kite Pharma, Juno Therapeutics, Agios Pharmaceuticals, and CRISPR Therapeutics; was on a speaker’s bureau for, and received travel and honoraria from, Celgene, Kite Pharma, and Agios Pharmaceuticals; and was employed by, provided consultant services to, and received honoraria from Optum. R.T.M. received honoraria from, was a member of an entity’s board of directors or advisory committees for (Scientific Steering Committee for JULIET), and received research funding from Novartis; provided consultant services to and received honoraria from Incyte Inc, Celgene/Juno Therapeutics, and CRISPR Therapeutics; received honoraria from Kite Therapeutics; has patents for and received royalties from Athersys, Inc; was employed by Oregon Health & Science University (OHSU); and provided consultant services to, and received payment from, Novartis (this potential conflict of interest has been reviewed and managed by OHSU). E.K.W. provided consultant services to, was a member of an entity’s board of directors or advisory committees for, and received research funding from Novartis; received travel expenses from European Hematology Association; received research funding from Pharmacyclics and Celldex; provided consultant services to Kalytera; and provided consultant services to and had equity ownership in Cambium Medical Technologies and Cambium Oncology. U.J. provided consultant services to, received honoraria from, was a member of an entity’s board of directors or advisory committees for, and received research funding from Roche and Gilead; provided consultant services to, received honoraria from, and was a member of an entity’s board of directors or advisory committees for Janssen and Celgene; provided consultant services to and received honoraria from AbbVie; was a member of an entity’s board of directors or advisory committees for and received research funding from Novartis; was a member of an entity’s board of directors or advisory committees for Mundipharma, Takeda-Millennium, Amgen, AOP Orphan, GSK, Infinity, and Bioverativ; and research funding from Merck Sharp & Dohme Corporation. J.R.W. was a member of an entity’s board of directors or advisory committees for Novartis, Apotex, Kite Pharma, and Celgene. J.P.M. received honoraria, travel accommodations, and expenses from, and was a speaker for, Kite Pharma; received honoraria from, was a speaker for, and received research funding from Novartis; and received research funding from Fresenius Biotech, Astellas Pharma, Bellicum Pharmaceuticals, Gamida Cell, and Pluristem Ltd. I.F. provided consultant services to AbbVie, Novartis, Merck, Janssen, Seattle Genetics, Gilead, Lundbeck, F. Hoffmann–La Roche Ltd, and Celgene. H.H. was a member of an entity’s board of directors or advisory committees for Novartis, Takeda, Roche, and Celgene, and received research funding from Roche. P.B. provided consultant services to and received honoraria from Novartis. C.d.C., L.P., and V.V.R. were employed by Novartis. R.T. was employed by Novartis Healthcare Private Limited. Ö.A. was employed by Novartis Pharma AG. R.A. was employed by Novartis Institutes for BioMedical Research, and holds equity ownership in Novartis, Cara Therapeutics, Aeterna Zentaris, Exelixis, and Celgene. S.J.S. provided consultant services to, received honoraria from, was a member of an entity’s board of directors or advisory committees for, and received research funding from Celgene; provided consultant services to and received honoraria from Dava Oncology; received honoraria and research funding from Genentech; was a member of an entity’s board of directors or advisory committees for Gilead and Pfizer; provided consultant services to, received honoraria from, and received research funding from Merck; received honoraria from, was a member of an entity’s board of directors or advisory committees for and received research funding from Novartis; provided consultant services to, received honoraria from, and was a member of an entity’s board of directors or advisory committees for Nordic Nanovector; and received honoraria from OncLive and Physician’s Education Source.
Correspondence: Michael R. Bishop, University of Chicago, 5841 S Maryland Ave, Chicago, IL 60637; e-mail: mbishop@medicine.bsd.uchicago.edu.