Key Points
Dinaciclib enhances NK-cell activity against leukemia cells in preclinical AML models.
Enhanced NK-cell activation by dinaciclib-treated AML is associated with downregulation of inhibitory NK ligand HLA-E on leukemia cells.

Introduction
Although targeting specific oncogenic pathways has improved outcomes for acute myeloid leukemia (AML), in general, survival remains poor. Dinaciclib, a cyclin-dependent kinase (CDK) 1, 2, 5, and 9 inhibitor, has potent anticancer activity against several malignancies and proapoptotic activity against AML.1,2 A major limitation for targeted cancer treatment strategies is treatment resistance, leading many to believe that multimodal combination therapy will be required for the most robust and durable treatment responses. Dinaciclib has been shown to influence the recognition and killing of cancer cells by the immune system, making it an attractive candidate for combined targeted and immune therapy for AML.3
Natural killer (NK) cells can induce a remission in 30% to 50% of patients with chemotherapy-refractory AML.4-6 NK cells respond to target cells through production of proinflammatory cytokines such as interferon-γ (IFNγ) and tumor necrosis factor-α and the polarized release of cytotoxic granules. The balance of activating and inhibitory signals regulates NK-cell killing. Monoclonal antibodies,7 bispecific or trispecific killer engager molecules,8-10 and hypomethylating agents11 have been used to enhance NK-cell killing of cancer targets, but little is known about the therapeutic potential for combined pathway targeted and NK-cell immunotherapy.
Methods
Cell culture, NK-cell functional assays, and flow cytometry
Primary leukemic cells and healthy donor peripheral blood mononuclear cells (PBMCs) were obtained in accordance with the University of Minnesota Institutional Review Board and the Declaration of Helsinki. Human cell lines were obtained from ATCC (Manassas, VA) and validated by short tandem repeat analysis. NK cells were enriched using the EasySep Human NK-cell enrichment kit (StemCell Technologies, Vancouver, BC, Canada). The NK-cell dose for in vivo experiments was calculated based on the preenrichment PBMC NK-cell percentage. Cell culture, fluorochrome-conjugated antibodies, flow cytometry, and analysis were as previously reported.12 Dinaciclib was obtained from Selleck Chemicals (Houston, TX). AML cells were incubated with dinaciclib and then washed prior to coincubation with effector cells. For the NK-cell–killing assay, targets were cocultured with NK cells at an effector-to-target (E:T) ratio of 2:1 for 24 hours (Figure 1A). Viable target cells were enumerated by flow cytometry using Fixable Aqua Dead Cell Dye to exclude dead cells and using AccuCheck Counting Beads (Thermo Fisher, Waltham, MA). For the NK-cell activation assay, targets were cocultured with effectors at an E:T ratio of 2:1 for 4 hours as previously described.12
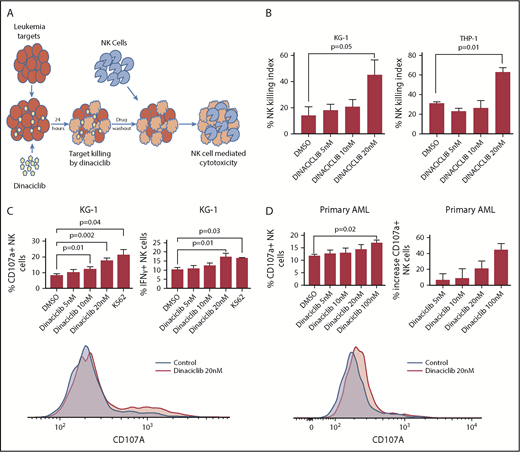
Dinaciclib enhances NK-cell activation and cytotoxicity against human AML targets. (A) Study scheme for NK-cell killing and activation assay. (B) NK-cell–killing assay. KG-1 and THP-1 cell lines were incubated with dinaciclib for 24 hours. After drug washout, NK cells (an E:T ratio of 2:1) or media were incubated with targets for 24 hours. The NK-killing index (percentage) was calculated by comparing the percentage of AML targets killed with drug treatment combined with donor NK cells relative to drug treatment alone. (C-D) Leukemia targets were incubated with dinaciclib overnight. Healthy donors’ PBMCs were added to and incubated with targets for 4 hours with an E:T ratio of 2:1. CD107a and/or IFNγ of NK cells were measured by flow cytometry. (C) NK-cell activation against KG-1. K562 serves as a positive control. (D) NK-cell activation against primary AML samples. The data represent replicates using NK cells from 4 healthy donors and representative histograms are shown at the bottom. Error bars represent mean ± standard error of the mean. DMSO, dimethyl sulfoxide.
Dinaciclib enhances NK-cell activation and cytotoxicity against human AML targets. (A) Study scheme for NK-cell killing and activation assay. (B) NK-cell–killing assay. KG-1 and THP-1 cell lines were incubated with dinaciclib for 24 hours. After drug washout, NK cells (an E:T ratio of 2:1) or media were incubated with targets for 24 hours. The NK-killing index (percentage) was calculated by comparing the percentage of AML targets killed with drug treatment combined with donor NK cells relative to drug treatment alone. (C-D) Leukemia targets were incubated with dinaciclib overnight. Healthy donors’ PBMCs were added to and incubated with targets for 4 hours with an E:T ratio of 2:1. CD107a and/or IFNγ of NK cells were measured by flow cytometry. (C) NK-cell activation against KG-1. K562 serves as a positive control. (D) NK-cell activation against primary AML samples. The data represent replicates using NK cells from 4 healthy donors and representative histograms are shown at the bottom. Error bars represent mean ± standard error of the mean. DMSO, dimethyl sulfoxide.
Human AML cell line xenograft model
Mouse studies were approved by the University of Minnesota Institutional Animal Care and Use Committee. Luciferase-transduced human HL60 mouse xenografts were established in 6- to 10-week-old NSG mice (The Jackson Laboratory, Bar Harbor, ME) as outlined in Figure 2. Roughly equal numbers of male and female mice were used for all experiments and evenly allocated to experimental groups. Dinaciclib was diluted in 20% hydroxypropyl-β-cyclodextrin and administered intraperitoneally. Recombinant human interleukin-15 (IL15; R&D Systems, Minneapolis, MN) was administered subcutaneously. HL60 leukemic burden was determined using an IVIS 100 Imaging System (Perkin-Elmer, Waltham, MA). Human blood NK-cell numbers in mouse peripheral blood were determined by collecting 100 µL of peripheral blood by cheek bleed and enumerating human CD45+CD3−CD56+ NK cells during a 60-second collection using flow cytometry.
Statistical analysis
Prism software (GraphPad, San Diego, CA) was used for statistical analysis.
Results and discussion
We first tested the effects of dinaciclib on NK-cell–mediated cytotoxicity against human AML cells. AML cell lines were cultured in RPMI-1640 with 10% fetal bovine serum containing 1% penicillin/streptomycin and treated with dinaciclib or control vehicle for 24 hours. Cells were then washed to eliminate the drug and then cocultured with primary NK cells at an NK E:T ratio of 2:1 for 24 hours. Viable AML target cells were enumerated by flow cytometry. Compared with the vehicle control group without NK cells, treatment with 20 nM dinaciclib alone resulted in 74.9% and 76.1% target killing whereas NK cells combined with 20 nM dinaciclib killed 86.3% and 91.3% of the targets for KG-1 and THP-1 cells, respectively. The NK-killing index (percentage) was determined by comparing the viable leukemic cell number after treatment with drug alone to drug plus NK cells. Treatment with 20 nM dinaciclib increased the NK-killing index compared with vehicle control (KG-1, 45.6% vs 14.5% [P = .05]; THP-1, 63.7% vs 31.7% [P = .01]) (Figure 1B). To confirm that increased killing of leukemia targets was due to NK-cell–mediated cytotoxicity, we measured NK-cell activation. Dinaciclib treatment enhanced NK-cell degranulation compared with control treatment as measured by CD107a expression (17.7% vs 8.3%; P = .002) and inflammatory cytokine (IFNγ) production (17.3% vs 0.4%; P = .01) (Figure 1C).
To further investigate the translational potential for our findings, we evaluated the ability of dinaciclib to enhance NK-cell killing of primary patient-derived human AML targets. Consistent with the effects on human AML cell lines in vitro, dinaciclib also potently induced NK-cell degranulation in response to primary AML targets in vitro (Figure 1D). Furthermore, in a mouse xenograft model that recapitulates the key features of clinical NK immunotherapy for AML, dinaciclib enhanced the activity of primary human NK cells against human HL-60 AML targets (Figure 2B). Although NK cells failed to reduce the AML burden in untreated leukemic mice, NK-cell treatment in dinaciclib-treated mice significantly reduced the leukemic burden. The reduction in leukemic burden after dinaciclib was associated with a trend toward higher peripheral blood NK-cell numbers (Figure 2C), suggesting that dinaciclib-treated AML cells may promote expansion and/or survival of adoptively transferred NK cells. Together, these findings lend credence to the clinical potential for, and supports further investigation of, this novel combined targeted and immunotherapy approach.
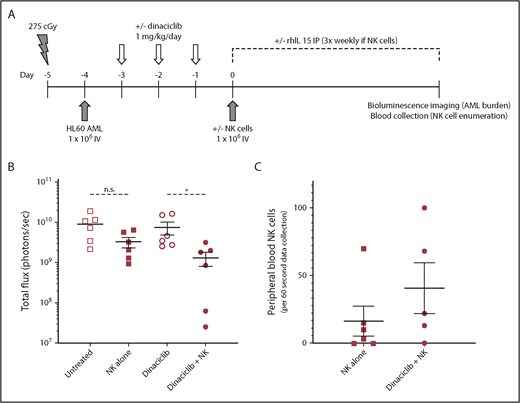
Dinaciclib enhances NK-cell killing of leukemic target cells in human AML cell line xenograft mice. NSG mice were conditioned, engrafted with luciferase-expressing HL60 AML cells, and then treated with dinaciclib and/or enriched primary human NK cells from healthy donors as outlined (A). Mice that received human NK cells also received 5 µg of recombinant human IL15 (rhIL15) 3 times per week. Leukemic burden was analyzed by bioluminescent imaging (B) and peripheral blood was obtained to enumerate human NK-cell numbers (human CD45+CD3−CD56+ cells) (C) at 28 days after NK-cell injection. Error bars represent mean ± standard error of the mean. *P < .05 using a paired Student t test corrected for multiple comparisons. IP, intraperitoneally; n.s., nonsignificant P value.
Dinaciclib enhances NK-cell killing of leukemic target cells in human AML cell line xenograft mice. NSG mice were conditioned, engrafted with luciferase-expressing HL60 AML cells, and then treated with dinaciclib and/or enriched primary human NK cells from healthy donors as outlined (A). Mice that received human NK cells also received 5 µg of recombinant human IL15 (rhIL15) 3 times per week. Leukemic burden was analyzed by bioluminescent imaging (B) and peripheral blood was obtained to enumerate human NK-cell numbers (human CD45+CD3−CD56+ cells) (C) at 28 days after NK-cell injection. Error bars represent mean ± standard error of the mean. *P < .05 using a paired Student t test corrected for multiple comparisons. IP, intraperitoneally; n.s., nonsignificant P value.
To explore potential mechanisms driving enhanced NK-cell activation against dinaciclib-treated AML targets, we evaluated the effects of dinaciclib on NK-cell ligand expression on leukemia cells. Dinaciclib treatment led to decreased expression of inhibitory ligands (Figure 3A), possibly through its inhibitory effects on CDK9-mediated transcription. Notably, HLA-E expression was markedly decreased on primary patient-derived AML blasts after dinaciclib treatment (Figure 3B). These data suggest that downregulation of HLA-E, which inhibits the NK-cell function through interaction with NKG2A on NK cells, contributes to enhanced NK-cell cytotoxicity. Therefore, we compared the effects of dinaciclib treatment to NGK2A blockade on NK-cell activation. Leukemic cells were preincubated with an NKG2A-blocking (z199 clone; Beckman Coulter, Brea, CA) or isotype control antibody at 1 µg/mL for 1 hour before coculture with effector cells. The NKG2A-blocking antibody enhanced NK-cell expression of CD107a and IFNγ after coculture with vehicle-treated leukemic cells but not dinaciclib-treated AML targets (Figure 3C). Furthermore, the NK-cell activation induced by dinaciclib was comparable to the effects of the NKG2A-blocking antibody. Taken together, these findings suggest a key role of dinaciclib-mediated downregulation of HLA-E on leukemia targets in enhancing NK-cell cytotoxicity.
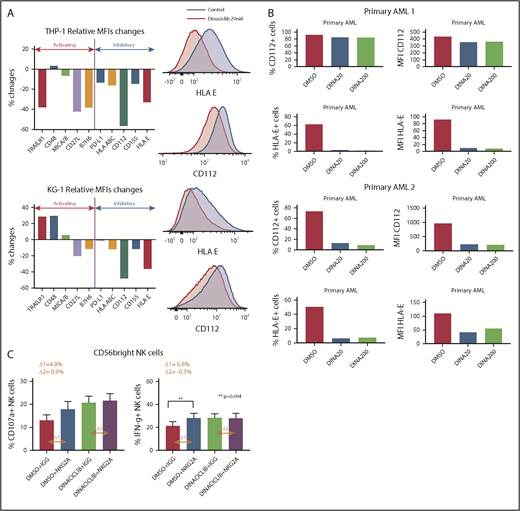
Dinaciclib modulates the expression of NK-cell ligands on AML cells and promotes NK-cell cytotoxicity against leukemic targets by disrupting the NK-cell–inhibitory HLA-E/NKG2A interaction. The expression of NK-cell ligands on leukemia targets were assessed by flow cytometry after leukemia cell lines (A) or primary patient-derived AML (B) targets were treated with dinaciclib or vehicle overnight. Percentage changes indicate changes of median fluorescence intensity (MFI) of NK ligands after dinaciclib treatment compared with vehicle treatment. The histograms are representative of 2 experiments. The ligand expression changes on THP-1 and KG-1 cell lines were examined in duplicates. (C) Healthy donors’ PBMCs were treated with 1 µg/mL NKG2A-blocking antibody (z199) or immunoglobulin G (IGG) isotype control for 1 hour and then incubated with KG-1 cells that had been treated with 20 nM dinaciclib or vehicle overnight. CD107a and IFNγ expression on NKG2A-expressing CD56-bright NK cells are shown. Error bars represent mean ± standard error of the mean. MICA/B, major histocompatibility class I chain-related protein A and B; PD-L, programmed death ligand; TRAILR, tumor necrosis factor–related apoptosis-inducing ligand receptor.
Dinaciclib modulates the expression of NK-cell ligands on AML cells and promotes NK-cell cytotoxicity against leukemic targets by disrupting the NK-cell–inhibitory HLA-E/NKG2A interaction. The expression of NK-cell ligands on leukemia targets were assessed by flow cytometry after leukemia cell lines (A) or primary patient-derived AML (B) targets were treated with dinaciclib or vehicle overnight. Percentage changes indicate changes of median fluorescence intensity (MFI) of NK ligands after dinaciclib treatment compared with vehicle treatment. The histograms are representative of 2 experiments. The ligand expression changes on THP-1 and KG-1 cell lines were examined in duplicates. (C) Healthy donors’ PBMCs were treated with 1 µg/mL NKG2A-blocking antibody (z199) or immunoglobulin G (IGG) isotype control for 1 hour and then incubated with KG-1 cells that had been treated with 20 nM dinaciclib or vehicle overnight. CD107a and IFNγ expression on NKG2A-expressing CD56-bright NK cells are shown. Error bars represent mean ± standard error of the mean. MICA/B, major histocompatibility class I chain-related protein A and B; PD-L, programmed death ligand; TRAILR, tumor necrosis factor–related apoptosis-inducing ligand receptor.
To our knowledge, this is the first report supporting the therapeutic potential for targeting oncogenic signaling to alter the expression of NK ligands on AML cells and thereby sensitize leukemic targets to NK immunotherapy. The use of primary leukemic blasts and NK cells as well as a mouse AML xenograft model that closely mimics the clinical use of adoptive NK-cell therapy for AML patients supports the translational relevance of our findings. By demonstrating that dinaciclib can sensitize AML targets to NK-cell–mediated cytotoxicity, our study introduces a new therapeutic paradigm for AML and supports further investigation into this promising new treatment approach.
Acknowledgments
This work was supported by the University of Minnesota Masonic Cancer Center and Academic Health Center Flow Cytometry Resource, which are supported by the National Institutes of Health, National Cancer Institute (P30CA77598), Minnesota Masonic Charities, and the Killebrew-Thompson Memorial Fund. C.E.E. was supported by the Masonic Cancer Center, University of Minnesota, and an American Cancer Society Mentored Research Scholar Grant (MRSG-18-223-01-LIB). H.D.Y. was supported by a National Institutes of Health, National Heart, Lung, and Blood Institute Hematology Research Training Grant (T32HL007062).
Authorship
Contribution: H.D.Y., J.S.M., and C.E.E. designed the study; H.D.Y. and D.K.S. performed the experiments; H.D.Y., M.F., J.S.M., and C.E.E. analyzed the data; H.D.Y. and C.E.E. wrote the manuscript; and all authors edited and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Craig E. Eckfeldt, University of Minnesota, MMC 480, 420 Delaware St SE, Minneapolis, MN 55455; e-mail: eckf0002@umn.edu.