Key Points
Genome-wide 5hmC loci can be profiled in 1 to 2 ng of cfDNA from blood plasma and correlate with clinical features of DLBCL.
5hmC in cfDNA collected at the time of DLBCL diagnosis is associated with EFS and OS, independent of established prognostic factors.

Abstract
An elevated level of circulating cell-free DNA (cfDNA) has been associated with tumor bulk and poor prognosis in diffuse large B-cell lymphoma (DLBCL), but the tumor-specific molecular alterations in cfDNA with prognostic significance remain unclear. We investigated the association between 5-hydroxymethylcytosines (5hmC), a mark of active demethylation and gene activation, in cfDNA from blood plasma and prognosis in newly diagnosed DLBCL patients. We used 5hmC-Seal, a highly sensitive chemical labeling technique, to profile genome-wide 5hmC in plasma cfDNA from 48 DLBCL patients at the University of Chicago Medical Center between 2010 and 2013. Patients were followed through 31 December 2017. We found a distinct genomic distribution of 5hmC in cfDNA marking tissue-specific enhancers, consistent with their putative roles in gene regulation. The 5hmC profiles in cfDNA differed by cell of origin and were associated with clinical prognostic factors, including stage and the International Prognostic Index. We developed a 29 gene–based weighted prognostic score (wp-score) for predicting event-free survival (EFS) and overall survival (OS) by applying the elastic net regularization on the Cox proportional-hazards model. The wp-scores outperformed (eg, prognostic accuracy, sensitivity, specificity) established prognostic factors in predicting EFS and OS. In multivariate Cox models, patients with high wp-scores had worse EFS (hazard ratio, 9.17; 95% confidence interval, 2.01-41.89; P = .004) compared with those in the low-risk group. Our findings suggest that the 5hmC signatures in cfDNA at the time of diagnosis are associated with clinical outcomes and may provide a novel minimally invasive prognostic approach for DLBCL.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is a heterogeneous group of malignancies with distinct genetic abnormalities, molecular alterations, clinical features, and prognosis.1 Despite improved chemoimmunotherapies, ∼20% to 40% of patients will experience disease recurrence or mortality.2,3 Emerging evidence suggests that elevated levels of tumor-derived circulating cell-free DNA (cfDNA) in DLBCL correlate with poor prognosis4 and detect relapse months prior to clinically detectable disease by imaging.5,6 However, the tumor-specific molecular targets in cfDNA with prognostic value remain largely unknown.
The pathogenesis of DLBCL is strongly linked to perturbation of epigenetic mechanisms. Greater epigenetic heterogeneity,7,8 global hypomethylation,9 and aberrant gene-specific promoter methylation10,-12 have been linked with poorer survival and relapse. However, previous studies have only investigated 5-methylcytosines (5mC) or interpreted all modified cytosines as 5mC. In the human genome, 5mC can be oxidized by the human TET enzymes to 5-hydroxymethylcytosines (5hmC) in an active DNA-demethylation process.13,14 Although 5mC is typically associated with suppressed gene expression,15 5hmC is particularly enriched in gene bodies and enhancers that mark for specific gene/locus activation in the chromatin.16,17 The 5hmC levels change in tumors, and sustained loss has been associated with prognosis.18,-20 Because 5mC represses protein-coding genes, as well as a vast amount of transposons in the human genome, targeting 5hmC for prognostication could better reflect gene-activation changes and a greater specificity. However, because of the low abundance of 5hmC loci in the genome (∼0.5%-1% of CpG sites are hydroxymethylated vs 2%-8% for that of 5mC) and difficulties in distinguishing 5hmC from 5mC using conventional bisulfite conversion approaches,21 no study has evaluated 5hmC in cfDNA for its prognostic value in DLBCL.
In this study, we applied the 5hmC-Seal, a highly sensitive chemical labeling–based sequencing technology, to profile genome-wide 5hmC in cfDNA from blood plasma of 48 patients with newly diagnosed DLBCL. The 5hmC-Seal technology has been shown to be a robust profiling approach for enriching and quantifying 5hmC-modified DNA fragments with as little as 1 to 2 ng of cfDNA from <5 mL of plasma.22,-24 We tested the hypothesis that 5hmC profiles in cfDNA at the time of diagnosis reflect the clinical characteristics of DLBCL and are associated with survival.
Materials and methods
Study subjects
The overall study design is shown in Figure 1. We prospectively enrolled patients aged 20 years and older who were newly diagnosed with non-Hodgkin lymphoma at the University of Chicago Medical Center from 2010 to 2013. All diagnoses were confirmed by hematopathologists according to the 2008 World Health Organization criteria.25 Blood samples were drawn from consented patients and processed immediately to separate plasma. For this study, we included only DLBCL patients with blood plasma available. We excluded DLBCL patients with primary central nervous system lymphoma, posttransplantation lymphoproliferative disorder, transformation of a previously diagnosed indolent lymphoma, or with HIV infection. After exclusion, a total of 48 DLBCL patients was included in the cfDNA analysis. This study was approved by the Institutional Review Board at the University of Chicago.
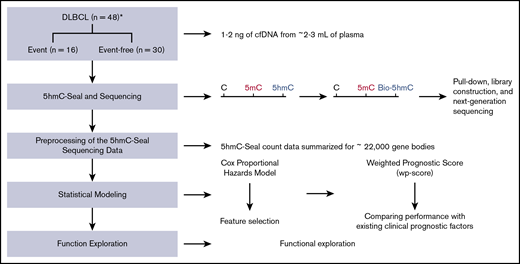
Study design and an overview of the 5hmC-Seal assay. A total of 48 cfDNA samples collected at the time of diagnosis from patients with DLBCL is included in this study. A weighted prognostic score based on the 5hmC marker genes is developed to evaluate prognosis after treatment. The 5hmC-Seal technique uses a chemical labeling strategy to sensitively profile 5hmC in cfDNA from nanogram-level DNA materials. *Clinical outcomes, including the development of clinical events (ie, relapse, death) are missing for 2 patients.
Study design and an overview of the 5hmC-Seal assay. A total of 48 cfDNA samples collected at the time of diagnosis from patients with DLBCL is included in this study. A weighted prognostic score based on the 5hmC marker genes is developed to evaluate prognosis after treatment. The 5hmC-Seal technique uses a chemical labeling strategy to sensitively profile 5hmC in cfDNA from nanogram-level DNA materials. *Clinical outcomes, including the development of clinical events (ie, relapse, death) are missing for 2 patients.
Sample preparation and the 5hmC-Seal profiling
Approximately 2 to 3 mL of frozen plasma from each subject was processed by centrifuging at 1350g for 12 minutes twice and at 13 500g for 12 minutes once, followed by cfDNA extraction (1-2 ng per sample) using the QIAamp Circulating Nucleic Acid Kit (Qiagen). Genomic DNA from cfDNA-paired tumor blocks for 7 patients was isolated (30-50 ng per sample) using a DNeasy Blood & Tissue Kit (Qiagen) and fragmented by sonication. We constructed 5hmC-Seal libraries according to an established protocol.22 DNA samples were first repaired and ligated with adaptors. Next, the T4 bacteriophage enzyme β-glucosyltransferase was used to transfer an engineered glucose moiety containing an azide-group to 5hmC in duplex DNA. A biotin tag was added to the azide group using Huisgen cycloaddition (“Click”) chemistry. Finally, the 5hmC-containing DNA fragments with biotin tags were captured by avidin beads. The 5hmC-Seal libraries were constructed through polymerase chain reaction amplification and sequenced using an Illumina NextSeq 500 platform (PE38) at the University of Chicago Genomics Core Facility. We randomly labeled the cfDNA samples for the 5hmC-Seal library constructions and sequencing. Technicians were blinded to clinical outcomes. Technical robustness, including reproducibility, of the 5hmC-Seal was demonstrated in our previous study.22
Processing of the 5hmC-Seal data
Bioinformatics processing of the 5hmC-Seal data from cfDNA was described in detail in our previous report.22 Briefly, raw sequencing reads were trimmed for adaptor sequences using Trimmomatic.26 Low-quality bases were also trimmed to a minimum length of 30 bp, followed by alignment to the human genome reference (hg19) using Bowtie 2 with the end-to-end alignment mode.27 Read pairs were concordantly aligned with fragment length ≤500 bp and with average ≤1 ambiguous base and up to 4 mismatched bases per 100-bp length. Alignments with Mapping Quality Score ≥10 were counted for gene bodies, according to the gene start and gene end annotations by the GENCODE Project (release 19),28 using featureCounts29 without strand information. The 5hmC-Seal libraries were sequenced to produce a median of ∼25 million reads in each sample, and a median number of ∼13.5 million unique reads (ie, >50%) mapped to ∼22 000 gene bodies. The raw count data summarized for the gene bodies were then normalized using DESeq230 and corrected for library size for statistical analysis. To explore gene regulatory relevance of 5hmC in cfDNA, we also summarized the 5hmC-Seal data according to the genomic peaks of H3K4me1, a tissue-specific marker for enhancers,31 as provided by the Roadmap Epigenomics Project32 for the B cell and other tissues for comparison.
Linking 5hmC in cfDNA with cell of origin and clinical characteristics
We examined whether the 5hmC-Seal data reflected the cell of origin (ie, germinal center B-cell–like [GCB] and activated B-cell–like [ABC] DLBCLs), as determined by the Han’s algorithm with immunohistochemistry staining,33 or were associated with standard prognostic factors, such as Ann Arbor stage (3/4 vs 1/2), serum lactate dehydrogenase (LDH) levels (elevated vs normal), and the International Prognostic Index (IPI; high = 3/4/5 vs low = 0/1/2). For each comparison, the top differential 5hmC marker genes (P < .05) from logistic models adjusting for age and sex were retained as candidates for further feature selection based on the elastic net regularization, using the glmnet library for the R statistical package.34 This feature-selection process was repeated 100 times, and a panel of 5hmC marker genes that were selected from ≥80% iterations was kept as final feature genes.
Developing a weighted prognostic model for DLBCL
We collected baseline clinical, laboratory, and treatment data, disease progression or relapse, and retreatment from electronic medical records. Deaths were ascertained using the National Death Index. We considered unplanned consolidative radiation therapy, but not radiation therapy as part of the initial treatment plan, as a retreatment. Event-free survival (EFS) was defined as time from diagnosis until relapse or progression, unplanned retreatment of lymphoma after initial immunochemotherapy, or death.35 Overall survival (OS) was defined as time from diagnosis until death from any cause. Follow-up was through 31 December 2017.
βk is the coefficient from the multivariate logistic model for gene k, and Gk is the normalized count of kth marker gene in the final panel. Kaplan-Meier curves were used to display survival curves based on the wp-scores (ie, risk scores). We then compared the prognostic accuracy, sensitivity, and specificity of the wp-score (high risk vs low risk) associated with clinical events with those using the established prognostic factors, including the serum LDH level (elevated vs normal), cell of origin (ABC vs GCB), Ann Arbor stages (1/2 vs 3/4), and the IPI (low = 0/1/2 vs high = 3/4/5). Multivariate Cox models were used to assess the association between the wp-scores and EFS or OS, controlling for age, sex, and standard prognostic factors. Log-rank P values were used to evaluate statistical significance for the Cox models.
Pathway analysis and exploration of tissue relevance
The TiGER (Tissue-specific Gene Expression and Regulation) database36 for tissue-specific expression was used to evaluate potential gene expression relevance of the 5hmC-Seal data derived from patient cfDNA. H3K4me1, a tissue-specific enhancer marker, peaks derived from various tissues from the Roadmap Epigenomics Project32 (accessed on 15 December 2018) were used to explore the relationships between 5hmC-Seal profiles from DLBCL patients and cis-regulatory elements. To explore the underlying biological connections of the candidate marker genes, we conducted Kyoto Encyclopedia of Genes and Genomes37 pathway enrichment analysis using the National Institutes of Health/DAVID tool.38 We used the Reactome Functional Interaction (FI)39 plug-in to explore FIs across the candidate marker genes associated with clinical events. Hubs of the Reactome FI networks were estimated based on the betweenness centrality, which detects the amount of influence that a node (ie, gene) has over the flow of information in a gene network.
Results
Patient characteristics
A total of 48 patients with newly diagnosed DLBCL was included in the study (Table 1). Median age at diagnosis was 59.5 years (range, 24-82 years), 63% (n = 30) were males, 50% were stage 1/2 based on the Ann Arbor staging system for lymphomas, 27% had an IPI score ≥ 3, and 68% had GCB-type DLBCL. In addition, most patients (67%) received R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone), followed by EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin plus rituximab; 17%), as the front-line treatment. Outcomes for 2 subjects cannot be determined. At the end of the follow-up, 16 patients had a clinical event, and 30 did not (Figure 1).
Distinct distributions of 5hmC in patient-derived cfDNA
The 5hmC-Seal sequencing reads obtained from patient-derived cfDNA showed distinct genomic distributions (Figure 2A-B). The 5hmC-Seal sequencing reads in cfDNA were enriched in gene bodies, whereas they were depleted in the flanking regions relative to the transcription start sites and transcription end sites (Figure 2A). The distribution of 5hmC in cfDNA was consistent with their putative roles in gene activation and significantly overlapped with the B-cell–derived Roadmap Epigenomics Project H3K4me1 peaks (Figure 2B). We also found that DLBCL patient-derived cfDNA samples were more enriched with the H3K4me1 peaks derived from the B cell than from other tissue types (eg, lung, pancreas, liver, and brain) using the Roadmap Epigenomics Project data (Student t test, P < .001, Figure 2B), suggesting tissue relevance of the profiled 5hmC-Seal data in patients with DLBCL.
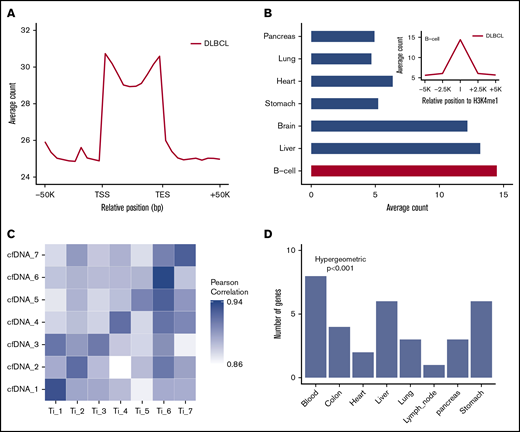
Distinct distributions of 5hmC in patient-derived cfDNA and tissue relevance. (A) The median and range of average counts are plotted against the relative genomic positions. The 5hmC-Seal data are enriched in gene bodies (split into 10 bins for all genes) relative to the flanking regions. (B) The 5hmC-Seal data are enriched in the B-cell–derived H3K4me1 loci compared with other tissue types (Student t test P < .001). H3K4me1 loci are obtained from the Roadmap Epigenomics Project data. (C) The top 100 highly variable (ie, most informative) 5hmC marker genes in cfDNA are highly correlated with those identified in the cfDNA-paired tissue samples from the same subject (n = 7). (D) The top 500 highly variable 5hmC marker genes in cfDNA are enriched within genes specific to blood compared with other tissues, based on the TiGER database for tissue-specific genes. TES, transcription end site; TSS, transcription start site.
Distinct distributions of 5hmC in patient-derived cfDNA and tissue relevance. (A) The median and range of average counts are plotted against the relative genomic positions. The 5hmC-Seal data are enriched in gene bodies (split into 10 bins for all genes) relative to the flanking regions. (B) The 5hmC-Seal data are enriched in the B-cell–derived H3K4me1 loci compared with other tissue types (Student t test P < .001). H3K4me1 loci are obtained from the Roadmap Epigenomics Project data. (C) The top 100 highly variable (ie, most informative) 5hmC marker genes in cfDNA are highly correlated with those identified in the cfDNA-paired tissue samples from the same subject (n = 7). (D) The top 500 highly variable 5hmC marker genes in cfDNA are enriched within genes specific to blood compared with other tissues, based on the TiGER database for tissue-specific genes. TES, transcription end site; TSS, transcription start site.
Next, in 7 patients with cfDNA-paired tissue samples, we compared 5hmC distributions between cfDNA samples and paired tumor tissue samples from the same patients (Figure 2C). We found that ∼16 000 gene bodies contained ≥30 sequencing reads in cfDNA and paired tissue samples. The top-ranking most variable genes (ie, most informative) in cfDNA showed higher correlation in paired tissue samples from the same individuals (mean Pearson’s r = 0.91) than from different patients (mean Pearson’s r = 0.88) (Figure 2C), supporting the tumor origin of a patient’s 5hmC profile in cfDNA. The most variable genes in cfDNA were also primarily enriched with genes specifically expressed in blood compared with other tissue types (hypergeometric P < .001), based on the TiGER database for tissue-specific gene expression (Figure 2D).
5hmC-Seal data reflect cell of origin and clinical characteristics
To evaluate the potential clinical utility and interpretation of cfDNA-based 5hmC prognostic markers for DLBCL, we compared the 5hmC profiles in DLBCL patient-derived cfDNA with the clinical characteristics of patients. We found that 5hmC marker genes detected in cfDNA differed by cell of origin and clinical characteristics of patients at diagnosis (supplemental Table 1). We found that 5hmC-Seal profiles in cfDNA distinguished GCB-type DLBCL from ABC-type DLBCL (Figure 3A), including genes involved in the glycosaminoglycan biosynthesis pathway (eg, EXTL1 encoding exostosin-like glycosyltransferase 1) that are related to the subtypes and aggressiveness of B-cell lymphoma.40 We also found that 5hmC-Seal profiles in cfDNA differed by Ann Arbor stage (1/2 vs 3/4) (Figure 3B), LDH level (elevated vs normal) (Figure 3C), and the IPI (Figure 3D).
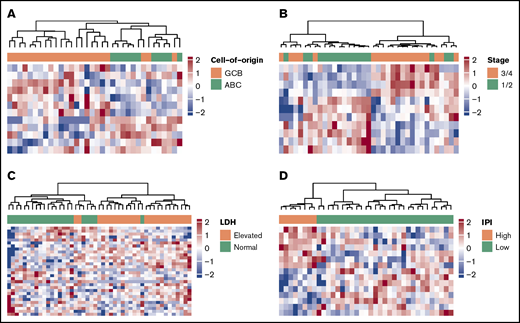
5hmC in patient-derived cfDNA differs by cell of origin and clinical characteristics. The heat maps are plotted using the final selected genes in each comparison. The 5hmC profiles in cfDNA are shown to be associated with cell of origin (GCB vs ABC, 12 genes) (A), Ann Arbor stages (1/2 vs 3/4, 11 genes) (B), LDH level (elevated vs normal, 18 genes) (C), and the IPI (low = 0/1/2 vs high = 3/4/5, 15 genes) (D).
5hmC in patient-derived cfDNA differs by cell of origin and clinical characteristics. The heat maps are plotted using the final selected genes in each comparison. The 5hmC profiles in cfDNA are shown to be associated with cell of origin (GCB vs ABC, 12 genes) (A), Ann Arbor stages (1/2 vs 3/4, 11 genes) (B), LDH level (elevated vs normal, 18 genes) (C), and the IPI (low = 0/1/2 vs high = 3/4/5, 15 genes) (D).
Prognostic value of 5hmC in cfDNA for DLBCL
Among the 46 DLBCL patients with available outcome data, 34 were alive at the end of the follow-up (ie, 31 December 2017). We identified 214 candidate marker genes potentially associated with clinical events (supplemental Table 2). We also explored functional annotations using these 214 candidate genes because the feature selection procedure that followed considered statistical significance, not biological relevance. Pathway analysis suggested that these 214 genes were involved in the Kyoto Encyclopedia of Genes and Genomes pathways (supplemental Table 3). Results from Reactome FI analysis suggested some functional interaction hubs important in the gene network among the candidate marker genes (supplemental Figure 1), such as HIST1H2BC (encoding histone cluster 1 H2B family member C) that was among the enriched pathways, as described above, TBP (encoding TATA-Box binding protein), and E2F1,41 GATA-3,42 and MLH1,43 which have been associated with the prognosis of DLBCL.
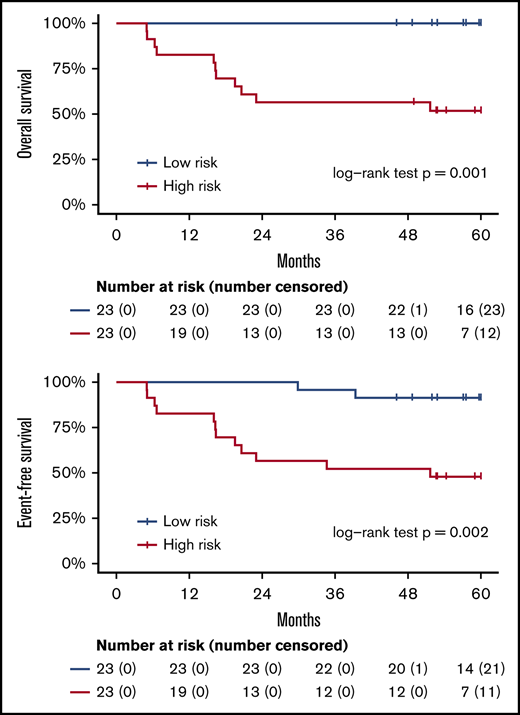
These 214 candidate genes were trimmed to 29 final marker genes after feature selection for predicting patient outcomes (Figure 4A). A wp-score was then computed for each patient based on the 29 marker genes. Compared with patients in the low-risk group (ie, low wp-score), patients in the high-risk group had worse OS (Figure 4B, log-rank P = .001) or worse EFS (Figure 4C, log-rank P = .002). Specifically, in the multivariate analysis controlling for age and sex, high-risk scores (wp-scores) were associated with poorer EFS (hazard ratio, 9.17; 95% confidence interval [CI], 2.01-41.89; P = .004) compared with low-risk scores (Table 2). Moreover, the wp-scores remained significantly associated with EFS after additional adjustment for standard prognostic factors, suggesting that the wp-scores are an independent prognostic factor for DLBCL (Table 2). Importantly, results for overall accuracy, sensitivity, and specificity showed that the wp-scores had an overall superior performance for predicting, at diagnosis, patients at risk for having a clinical event during the follow-up compared with standard clinical prognostic factors, such as elevated LDH level, advanced stages (3/4), ABC-type DLBCL, and high IPI (≥3) (Figure 4D). Data on MYC, BCL2, and BCL6 expression determined by immunohistochemistry were available for 14 patients. In exploratory analyses, the 5hmC-based wp-scores also performed better than double or triple expression (ie, MYC and BCL2 and/or BCL6) in predicting prognosis (data not shown). However, these results should be interpreted with caution, given the small sample size and exploratory nature of the analysis.
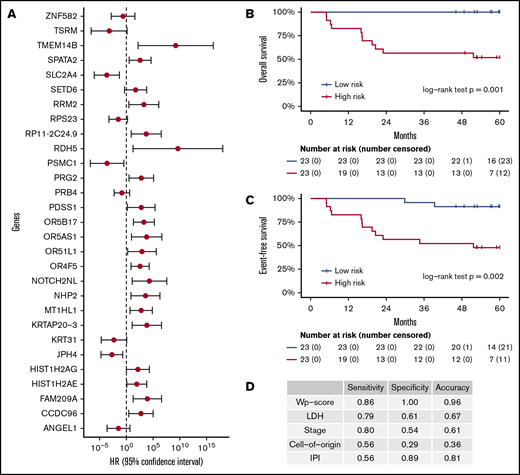
Prognostic implications of cfDNA-based 5hmC for DLBCL. The 5hmC marker genes are associated with the occurrence of clinical events (relapse, retreatment, or death) and can predict patient survival. (A) A weighted prognostic model consists of 29 marker genes that are associated with clinical events. The wp-scores are computed using these final marker genes for all patients with DLBCL. The wp-scores predict OS (B) and EFS (C) at the time of diagnosis. (D) The wp-scores show superior predictive performance (ie, sensitivity and specificity that maximized the Youden’s index) for the risk of developing clinical events compared with standard clinical prognostic factors, including serum LDH level, Ann Arbor stages, cell of origin, and the IPI.
Prognostic implications of cfDNA-based 5hmC for DLBCL. The 5hmC marker genes are associated with the occurrence of clinical events (relapse, retreatment, or death) and can predict patient survival. (A) A weighted prognostic model consists of 29 marker genes that are associated with clinical events. The wp-scores are computed using these final marker genes for all patients with DLBCL. The wp-scores predict OS (B) and EFS (C) at the time of diagnosis. (D) The wp-scores show superior predictive performance (ie, sensitivity and specificity that maximized the Youden’s index) for the risk of developing clinical events compared with standard clinical prognostic factors, including serum LDH level, Ann Arbor stages, cell of origin, and the IPI.
Discussion
In this prospective study of newly diagnosed patients with DLBCL, we profiled genome-wide 5hmC in cfDNA from blood plasma and investigated its association with prognosis and known prognostic markers. We found distinct genomic distributions of 5hmC in cfDNA and demonstrated the relevance of cfDNA-based 5hmC to tumor origin. In addition, 5hmC marker genes differed by cell of origin and clinical characteristics of patients at diagnosis. We identified a panel of 29 marker genes that were associated with the probability of having a clinical event. The wp-scores based on these 29 marker genes were associated with OS and EFS, independent of established prognostic factors. To our knowledge, this is the first study to profile genome-wide 5hmC in cfDNA and provide suggestive evidence of the prognostic value of these epigenetic markers in DLBCL.
Despite convincing evidence that supports 5hmC as a novel class of epigenetic biomarkers for various solid tumors and hematological malignancies,18 it remains technically challenging to profile 5hmC in cfDNA because of the scarcity of 5hmC. To address the gap, we applied the 5hmC-Seal, a highly sensitive and robust technique based on covalent chemical linkage44 and requiring as little as ∼1 to 2 ng of DNA from ∼2 to 3 mL of plasma.22 To our knowledge, the 5hmC-Seal is the only method that allows mapping genome-wide 5hmC, and it is highly sensitive for clinically feasible amounts of cfDNA samples. The assay has been validated and implemented for several cancers in our laboratories22,45 and those of other investigators.23,46
Our findings that genome-wide 5hmC signatures in cfDNA correlated with established prognostic factors and were associated with the prognosis of DLBCL suggested that cfDNA-based 5hmC signatures could complement current biopsy-based clinical practice for DLBCL prognostication. Delineating cell of origin33,47 or determining genetic alterations48,-50 is clinically important amid the rapid development of novel molecular targeted regimens for DLBCL.2,51 The major limitation is that these approaches require tissue biopsies, which are invasive and are prone to sampling bias as a result of intratumoral and spatial heterogeneity.52,-54 Accumulating evidence suggests that circulating cfDNA from blood plasma contains epigenetic information released from the tumor/tumor microenvironment into the blood and reflects tumor pathobiology.54,-56 As such, cfDNA offers transformative opportunities to overcome some of the limitations of tissue-based approaches. Two recent studies reported that global hypomethylation9 and aberrant DAPK1 methylation12 of cfDNA predicted poor outcomes in DLBCL. Our findings of the prognostic significance of 5hmC in cfDNA for DLBCL suggest that 5hmC may also play an important role in the progression of DLBCL, and it warrants further evaluation.
In our study, a weighted model consisting of 29 gene markers is associated with EFS and OS independent of standard prognostic factors, such as age, stage, LDH, and IPI. Some of these genes have been implicated in lymphoma, such as PDSS1 (encoding prenyl [decaprenyl] diphosphate synthase, subunit 1), NHP2 (encoding NHP2 ribonucleoprotein), and ANGEL1 (encoding angel homolog). We also found that the wp-score based on 5hmC markers outperformed (eg, overall accuracy, sensitivity, and/or specificity) existing prognostic factors in predicting a clinical event. For example, cell of origin is a well-established prognostic factor in DLBCL and is a potential biomarker for future personalized therapies.51,57 In this study, the sensitivity, specificity, and overall predictive accuracy of cell of origin for a clinical event is <50%: 0.56, 0.29, and 0.36, respectively. In contrast, the corresponding values for wp-score are 0.86, 1.00, and 0.96 (Figure 4D). LDH, one of the most commonly used biomarkers for DLBCL during scheduled clinical visits, also does not perform as well as the wp-score. These findings suggest that the 5hmC profiles in cfDNA hold the promise to be a convenient alternative that could supplement the current clinical practice to provide relevant clinical information and risk stratification for DLBCL.
The current study has several strengths, including the confirmation of DLBCL diagnosis and outcomes, the prospective study design, and the use of 5hmC-Seal, a state-of-the-art technique. There are also limitations. First, the relatively small sample size does not allow us to control for treatment approaches or to validate the marker panel in independent samples. Although the majority of patients (67%) received R-CHOP as the standard front-line treatment, EPOCH-R and other regimens accounted for 33%. The wp-score was slightly higher for the EPOCH-R group than for the R-CHOP group, but the difference was not statistically significant. Second, we have limited data on MYC, BCL2, and BCL6 expression and tumor burden. Comparing the prognostic significance of 5hmC-based wp-score with these prognostic factors is warranted in future work. Third, similar to other studies of DLBCL in European countries and North America, we were not able to evaluate the association between 5hmC in cfDNA and prognosis by race/ethnicity or population. Future studies with a large minority patient population are warranted to evaluate the generalizability of our results.
In conclusion, our findings suggest that 5hmC in patient-derived cfDNA profiled using the 5hmC-Seal, a highly robust and sensitive technique, has the potential to be a clinically convenient and minimally invasive prognostic approach for DLBCL. Future epigenetic studies of prognosis for DLBCL should include 5hmC as a stable and important epigenetic marker.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the Epidemiology and Research Recruitment Core of the University of Chicago Comprehensive Cancer Center for coordinating the subject recruitment and sample collection. C.H. thanks the University of Chicago Ludwig Center for partial support.
This work was supported in part by National Institutes of Health grants R21 MD011439 from the National Institute on Minority Health and Health Disparities (B.C.-H.C. and W.Z.), P30 CA060553 Career Development Fund from the National Cancer Institute (W.Z.), and UL1TR002389 from the National Center for Advancing Translational Sciences (B.C.-H.C.). C.H. is a Howard Hughes Medical Institute Investigator.
Authorship
Contribution: B.C.-H.C., C.H., and W.Z. designed the research and provided oversight; Q.Y. and K.Y. performed the 5hmC-Seal experiment; Z.Z., C.Z., and W.Z. analyzed the 5hmC-Seal data and performed statistical analyses; E.S. coordinated sample collection; G.V. was the study hematopathologist; P.M.B. and S.M.S. provided clinical advice and helped to interpret the data; B.C.-H.C., Z.Z., and W.Z. drafted the manuscript with input from all authors; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: C.H. is a scientific founder of Accent Therapeutics, Inc. and a member of its scientific advisory board. C.H. and W.Z. are shareholders of Epican Genetech Co. Ltd. The remaining authors declare no competing financial interests.
Correspondence: Brian C.-H. Chiu, University of Chicago, 5841 S Maryland Ave, MC200, Chicago, IL 60637; e-mail: bchiu@uchicago.edu; or Wei Zhang, Northwestern University, 680 N Lake Shore Dr, Suite 1400, Chicago, IL 60611; e-mail: wei.zhang1@northwestern.edu.