Key Points
c-KIT activating mutations cause resistance to PARP inhibitor in AML1-ETO–positive leukemias.
c-KIT inhibitor avapritinib downregulates BRCA1/2 and DNA-PK catalytic subunit to restore the sensitivity to PARP inhibitor.
Introduction
Numerous reports indicate that acute myeloid leukemia (AML) cells accumulate high levels of spontaneous and genotoxic agent–induced DNA lesions, but they are able to survive because of enhanced/altered DNA repair activities.1-5 Because DNA damage may constrain survival and proliferation of leukemia cells, transformed cells need to be protected from the lethal effects of DNA damage, such as DNA double-strand breaks (DSBs).6 Thus, leukemia cells may be highly dependent on specific DSB repair mechanisms, and targeting these pathways could sensitize them to DNA-damaging agents.7
DSBs, the most lethal DNA lesions, are repaired by 2 major mechanisms: homologous recombination (HR; major proteins: BRCA1, BRCA2, PALB2, RAD51B, RAD51C, RAD51D, XRCC2, XRCC3, RAD54, and RAD51) and DNA-dependent protein kinase (DNA-PK)–mediated nonhomologous end-joining (D-NHEJ; major proteins: DNA-PK catalytic subunit, Ku70, Ku80, NHEJ1, Artemis, LIG4, and XRCC4).8 PARP1-dependent alternative NHEJ (Alt-NHEJ; major proteins: PARP1 and LIG3) serves as a back-up pathway.9,10
Chromosomal translocations involving the core binding factor (CBF) family members, such as AML1-ETO (RUNX1-RUNX1T1) and CBFB-MYH11, are among the most frequent cytogenetic aberrations found in AML.11 We and other investigators have shown that AML1-ETO–positive cells display BRCA1/2 deficiency, which diminishes HR activity and predisposes leukemia cells to synthetic lethality triggered by DNA repair inhibitors, such as the PARP inhibitor (PARPi) olaparib.12,13 These data suggested that PARPi’s, which are approved by the US Food and Drug Administration for the treatment of BRCA1/2-mutated breast and ovarian cancers,14 can be used to treat AML1-ETO–positive AMLs.
Additional mutations (eg, in c-KIT and NRAS) often accompany AML1-ETO+ and CBFB-MYH11+ AMLs.15,16 c-KIT mutations (c-KITmuts) in AMLs harboring AML1-ETO or CBFB-MYH11 are associated with poor disease outcome,17 warranting novel therapeutic approaches. In this study, we tested whether c-KITmuts can modulate the response of AML1-ETO– or CBFB-MYH11–positive AML cells to PARPi.
Methods
Primary AML cells and cell lines
Genetic aberrations in diagnostic primary AML samples collected for the Eastern Cooperative Oncology Group and the American College of Radiology Imaging Network (ECOG-ACRIN) E1900 clinical trial18 are described in supplemental Table 1. Samples of normal hematopoietic cells were purchased from STEMCELL Technologies (Vancouver, BC, Canada). Lin−CD34+ cells were obtained from mononuclear fractions by magnetic sorting using EasySep negative selection human progenitor cell enrichment cocktail, followed by human CD34 positive selection cocktail (STEMCELL Technologies), as described previously.12 The Kasumi-1 AML cell line harboring AML1-ETO + c-KIT(N822K) was purchased from the American Type Culture Collection.
Clonogenic assay
Cells (104 per 0.1 mL) were treated with the PARPi olaparib, the c-KIT inhibitor (c-KITi) avapritinib, and/or doxorubicin (all from Selleckchem) for 72 hours, followed by plating in methylcellulose, as described previously.12 Colonies were counted after 7 to 10 days.
Western blot
Kasumi-1 cells were left untreated or were treated with the c-KITi avapritinib (5 μM) for 48 hours. Total cell lysates and nuclear lysates were examined by western blot, as described previously.12
DSB repair
HR, D-NHEJ, and Alt-NHEJ were measured in Kasumi-1 cells that were treated or not with the c-KITi avapritinib (5 μM) for 72 hours using DR-GFP (HR), EJ2-GFP (D-NHEJ), and EJ5-GFP (Alt-NHEJ) reporter cassettes, as described previously.12
Results and discussion
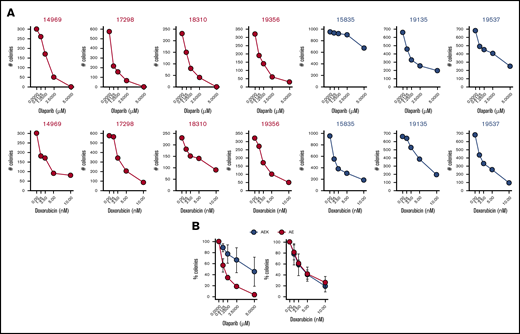
To test whether a c-KITmut (D816V or ΔY418+D419; supplemental Table 1) modulates the sensitivity of individual AMLs harboring AML1-ETO to PARPi, Lin−CD34+ cells were incubated with increasing concentrations of the PARPi olaparib or the cytotoxic drug doxorubicin, followed by clonogenic testing. Results clearly show that the presence of c-KITmut is accompanied by reduced sensitivity to olaparib but not to doxorubicin (Figure 1). This observation is supported by another report that c-KIT(N822K) rescued BRCA2 expression and HR activity in AML1-ETO–positive cells.19 In addition, although CBFB-MYH11–positive Lin−CD34+ cells appeared less sensitive to olaparib compared with their AML1-ETO counterparts, c-KITmut further diminished their sensitivity to the drug, whereas mutated NRAS exerted the opposite effect, without affecting their response to doxorubicin (supplemental Figure 1). Altogether, these results suggest that c-KITmut was associated with resistance to the PARPi olaparib without affecting the sensitivity to doxorubicin.
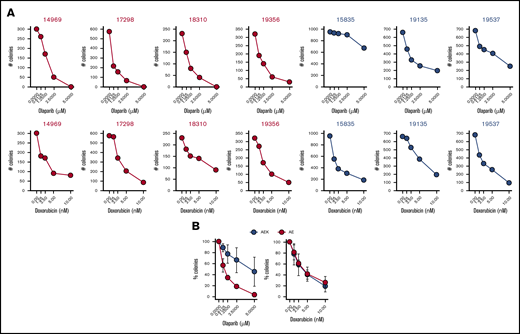
c-KITmut is associated with resistance to the PARPi olaparib, but not to doxorubicin, in AML1-ETO–positiveAMLs. Clonogenic potential of Lin−CD34+ cells from AML patients harboring AML1-ETO (AE) or AML1-ETO + c-KITmut (AEK) and treated with the indicated concentrations of olaparib or doxorubicin. (A) Mean number of colonies from individual samples tested in triplicates. (B) Mean percentage ± standard deviation of colonies compared with untreated counterparts from the samples harboring the same mutations.
c-KITmut is associated with resistance to the PARPi olaparib, but not to doxorubicin, in AML1-ETO–positiveAMLs. Clonogenic potential of Lin−CD34+ cells from AML patients harboring AML1-ETO (AE) or AML1-ETO + c-KITmut (AEK) and treated with the indicated concentrations of olaparib or doxorubicin. (A) Mean number of colonies from individual samples tested in triplicates. (B) Mean percentage ± standard deviation of colonies compared with untreated counterparts from the samples harboring the same mutations.
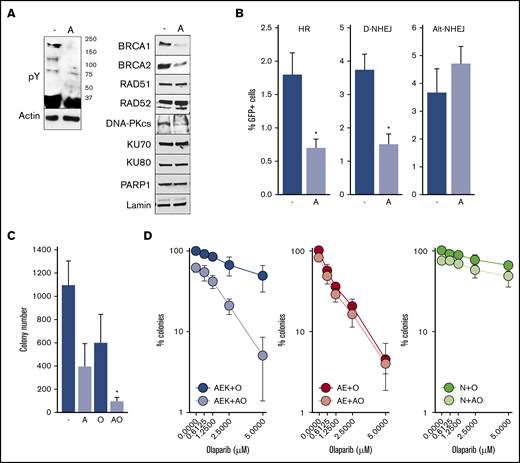
To determine whether constitutive activation of c-KITmut tyrosine kinase was responsible for olaparib resistance, avapritinib was used to inhibit the mutated kinase in AML1-ETO + c-KIT(N822K)–positive Kasumi-1 cells (Figure 2A, left panel).20 Inhibition of c-KIT(N822K) kinase by avapritinib (Figure 2A, left panel) was associated with downregulation of BRCA1 and BRCA2 (HR pathway) and the DNA-PK catalytic subunit (D-NHEJ pathway), but not PARP1 (Alt-NHEJ pathway) (Figure 2A, right panel). In concordance, avapritinib inhibited HR and D-NHEJ activity, but not Alt-NHEJ activity, in Kasumi-1 cells (Figure 2B) and restored their sensitivity to olaparib (Figure 2C). Moreover, avapritinib enhanced the sensitivity of primary AML1-ETO + c-KITmut–positive Lin−CD34+ cells to olaparib (Figure 2D, left panel) but did not affect the sensitivity of AML-ETO–positive cells bearing wild-type cKIT (Figure 2D, middle panel). In addition, avapritinib + olaparib exerted only a modest effect against Lin−CD34+ cells from healthy donors (Figure 2D, right panel). Although it has been reported that avapritinib inhibits mutated cKIT and PDGFRA oncogenic kinases,20 our results strongly support the role of cKITmut in modulation of the sensitivity of AML cells to PARPi.
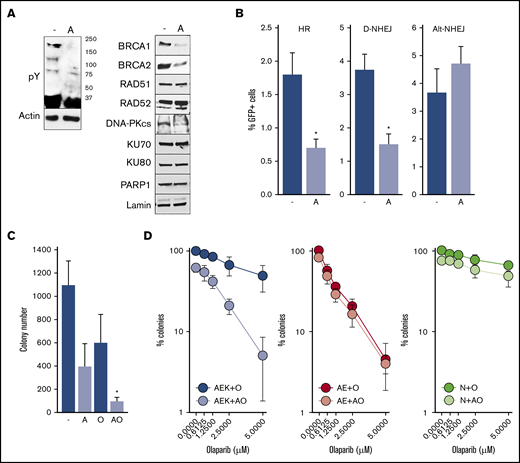
Inhibition of c-KITmut kinase causes DSB repair defects and restores sensitivity of AML1-ETO–positiveAML cells to the PARPi olaparib. (A) Tyrosine-phosphorylated proteins (pY) and indicated DSB repair proteins were detected by western blot in total cell lysates (left panel) and nuclear cell lysates (right panel) from Kasumi-1 cells treated (designated by “A”) or not (-) with avapritinib. Actin and lamin served as loading controls. (B) HR, D-NHEJ, and Alt-NHEJ activities in Kasumi-1 cells treated (designated by “A”) or not (-) with avapritinib. Results represent mean percentage ± standard deviation (SD) of GFP+ cells in DsRed+ population from 3 experiments. *P < .001, Student t test. (C) Kasumi-1 cells were left untreated (-) or treated with 5-μM avapritinib (designated by “A”), 5-μM olaparib (O), or avapritinib + olaparib (AO). Mean ± SD number of colonies from 3 experiments. *P < .05, Mann-Whitney rank sum test. (D) Clonogenic potential of Lin−CD34+ cells from 3 AML patients harboring AML1-ETO + c-KITmut (AEK), 3 AML1-ETO (AE)–positive AMLs, and from 3 healthy donors (N) treated with the indicated concentrations of olaparib (O) or 5-μM avapritinib + olaparib (AO). Mean percentage ± SD of colonies compared with untreated counterparts.
Inhibition of c-KITmut kinase causes DSB repair defects and restores sensitivity of AML1-ETO–positiveAML cells to the PARPi olaparib. (A) Tyrosine-phosphorylated proteins (pY) and indicated DSB repair proteins were detected by western blot in total cell lysates (left panel) and nuclear cell lysates (right panel) from Kasumi-1 cells treated (designated by “A”) or not (-) with avapritinib. Actin and lamin served as loading controls. (B) HR, D-NHEJ, and Alt-NHEJ activities in Kasumi-1 cells treated (designated by “A”) or not (-) with avapritinib. Results represent mean percentage ± standard deviation (SD) of GFP+ cells in DsRed+ population from 3 experiments. *P < .001, Student t test. (C) Kasumi-1 cells were left untreated (-) or treated with 5-μM avapritinib (designated by “A”), 5-μM olaparib (O), or avapritinib + olaparib (AO). Mean ± SD number of colonies from 3 experiments. *P < .05, Mann-Whitney rank sum test. (D) Clonogenic potential of Lin−CD34+ cells from 3 AML patients harboring AML1-ETO + c-KITmut (AEK), 3 AML1-ETO (AE)–positive AMLs, and from 3 healthy donors (N) treated with the indicated concentrations of olaparib (O) or 5-μM avapritinib + olaparib (AO). Mean percentage ± SD of colonies compared with untreated counterparts.
In agreement with our observation presented here, oncogenic tyrosine kinase inhibitor–induced dual deficiency in HR and D-NHEJ was also associated with highly effective elimination of FLT3(ITD)-positive AML cells and JAK2(V617F)-positive myeloproliferative neoplasm cells by PARPi.21,22 In conclusion, AML1-ETO–positive AML cells harboring c-KITmut (N822K, D816V, and ΔY418+D419) were resistant to the PARPi olaparib but not to doxorubicin. Inhibition of c-KITmut kinase activity by avapritinib inhibited HR and D-NHEJ and restored the sensitivity of AML cells to olaparib. We postulate that PARPi combined with c-KITi can be effective against c-KITmut–positive AMLs, especially those harboring AML1-ETO. Moreover, PARPi may be useful for eliminating CBFB-MYH11 + mutated NRAS–positive AMLs.
Acknowledgments
This work was supported by Leukemia and Lymphoma Society Translational Research Program award 6565-19; National Institutes of Health, National Cancer Institute grant R01 CA186238; and the When Everyone Survives Foundation (T.S.).
Authorship
Contribution: M.N.-S. performed experiments and contributed to the writing of the manuscript; E.M.P. provided genetically characterized AML samples from E1900; R.L.L. performed the somatic sequencing studies of E1900 samples; H.F.F. led trial E1900; M.S.T. was ECOG-ACRIN Leukemia Committee chair when E1900 was activated; M.R.L. is the current ECOG-ACRIN Leukemia Committee chair; and T.S. designed the studies, supervised the experiments, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Tomasz Skorski, Temple University School of Medicine, Sol Sherry Thrombosis Research Center and Fels Institute for Cancer Research and Molecular Biology, 3400 N Broad St, MRB 548, Philadelphia, PA 19140; e-mail: tskorski@temple.edu.