

Key Points
In persons of AA, a polygenic risk score in the top 10% (compared with 25%-75%) is associated with an 80% increased multiple myeloma risk.
Common genetic variation contributes to the risk of multiple myeloma in men and women of AA.

Abstract
Persons of African ancestry (AA) have a twofold higher risk for multiple myeloma (MM) compared with persons of European ancestry (EA). Genome-wide association studies (GWASs) support a genetic contribution to MM etiology in individuals of EA. Little is known about genetic risk factors for MM in individuals of AA. We performed a meta-analysis of 2 GWASs of MM in 1813 cases and 8871 controls and conducted an admixture mapping scan to identify risk alleles. We fine-mapped the 23 known susceptibility loci to find markers that could better capture MM risk in individuals of AA and constructed a polygenic risk score (PRS) to assess the aggregated effect of known MM risk alleles. In GWAS meta-analysis, we identified 2 suggestive novel loci located at 9p24.3 and 9p13.1 at P < 1 × 10−6; however, no genome-wide significant association was noted. In admixture mapping, we observed a genome-wide significant inverse association between local AA at 2p24.1-23.1 and MM risk in AA individuals. Of the 23 known EA risk variants, 20 showed directional consistency, and 9 replicated at P < .05 in AA individuals. In 8 regions, we identified markers that better capture MM risk in persons with AA. AA individuals with a PRS in the top 10% had a 1.82-fold (95% confidence interval, 1.56-2.11) increased MM risk compared with those with average risk (25%-75%). The strongest functional association was between the risk allele for variant rs56219066 at 5q15 and lower ELL2 expression (P = 5.1 × 10−12). Our study shows that common genetic variation contributes to MM risk in individuals with AA.
Introduction
Multiple myeloma (MM) originates from a malignant clone of plasma cells, the terminally differentiated B-lymphocytes that produce antibody upon antigen recognition. It is the second most common hematologic malignancy in the United States, with ∼160 000 new cases in 2018,1 and remains largely incurable, with a 50% 5-year survival rate. Older age, male sex, African ancestry (AA), family history, and obesity, especially in young adulthood, are factors consistently associated with MM risk.2-4 In the United States, the incidence rate of MM is twice as high in men and women of AA compared with those of European ancestry (EA), for unknown reasons.2 Case reports of familial clustering5 and a two- to threefold increased risk among first-degree relatives6,7 suggest a genetic contribution to the etiology of MM. We previously showed that 5 of 8 risk loci identified in persons of EA also contribute to risk in persons of AA.8-10 Fifteen new MM risk loci have been identified in EA populations,11-13 but these have not been examined in populations of AA. Moreover, no genome-wide significant associations have been identified that explain MM risk specifically for AA individuals. Here, we added a second genome-wide association study (GWAS) in a meta-analysis, for a total of 1813 cases and 8871 controls, to assess the association between common genetic variation and MM risk in the largest study of cases and controls of AA conducted to date.
Methods
All studies contributing DNA samples had approval from their Institutional Review Boards according to the Declaration of Helsinki Ethical Principles for Medical Research for Human Subjects (1964). Signed informed consent was obtained from all participants at the time of specimen collection.
Study participants, genotyping, and quality control
There were 2 sets of study participants. Set 1 was a GWAS case-control study that was previously conducted in 1179 AA MM patients identified from 11 National Cancer Institute (NCI) comprehensive cancer centers and nonprofit hospitals and 4 NCI Surveillance, Epidemiology, and End Results cancer registries participating in the African American Multiple Myeloma Study in 2010 to 2015. In addition, DNA samples from the Multiethnic Cohort Study (n = 43), the University of California at San Francisco study (n = 32), and the Multiple Myeloma Research Consortium (n = 84) were included (total N = 1338 cases), as described elsewhere.8 Controls were AA subjects unaffected by MM and with existing GWAS data, including 2631 female controls from the African Ancestry Breast Cancer Consortium14 and 4447 male controls from the African Ancestry Prostate Cancer Consortium.15 Cases were genotyped using the Illumina HumanCore GWAS array, whereas controls were genotyped using the Illumina 1M-Duo. Quality control (QC) measures for cases and controls were conducted separately. Cases with call rate < 0.98 (n = 11), unexpected replicates (n = 14), first- or second-degree relatives (n = 2), and those who were sex discordant based on X chromosome genotypes (n = 6) were excluded.8 Single nucleotide polymorphisms (SNPs) with a call rate < 0.98 or replicate concordance < 1 based on 100 QC replicate samples were removed. QC procedures among controls were reported previously.14,15 Only SNPs that passed QC measures directly genotyped in both cases and controls were included in the imputation (n = 188 376).
Set 2 consisted of DNA samples from 421 AA MM patients from the Myeloma Center, University of Arkansas for Medical Sciences and 132 additional samples from continued enrollment in the African American Multiple Myeloma Study in 2016 (primarily from the MD Anderson Cancer Center) (total N = 553 cases). Controls were 2398 unaffected AA participants from the Multiethnic Cohort Study with existing GWAS data. Both cases and controls in Set 2 were genotyped using the Illumina MEGA array (total genotyped SNPs ∼ 1.7 million). Cases and controls were subjected to the same QC procedures. One case and 1 control were removed because of cross-set replication. Additional individuals were removed based on call rate < 0.95 (n = 3), sex mismatch (n = 13), ancestry outlier (estimated African [AFR] global ancestry < 0.1 by STRUCTURE16 ; n = 4), and 1 in a first-degree relative pair (estimated sharing identity by descent ≥ 0.375 by PLINK; n = 10), leaving 529 cases and 2389 controls for imputation. Control data were cleaned previously combined with the National Human Genome Research Institute Population Architecture Using Genomics and Epidemiology Consortium (NHGRI PAGE) samples17 ; thus, we first excluded SNPs that were low quality in these samples. Further SNP QC included removing monomorphic SNPs, variants with a call rate < 0.98, replication concordance < 1 based on 10 replicate pairs in cases, cross-platform replication concordance < 100% based on 8 replicate pairs in cases and controls, and SNPs with poor clustering by visual inspection. Additional removal criteria included SNPs with minor allele frequency (MAF) that deviated from the AFR individuals in the phase 3 1000 Genome Project (1KGP) data and indels not identified within 1KGP. SNPs found in both cases and controls were used for imputation (N = 1 046 801).
Shared identity by descent for Set 1 and Set 2 was calculated using PLINK to remove duplicate samples across sets. We excluded 21 cases and 571 controls from Set 1 that were included in Set 2, leaving a final analysis sample size for Set 1 of 7766 (1284 cases and 6482 controls). Genotyping, QC, and imputation procedures are described in supplemental Figure 1.
Imputation
For this analysis, Set 1 and Set 2 data were imputed using the same protocol. Genotype data were phased using SHAPEIT18 and then imputed to a cosmopolitan reference panel from the Haplotype Reference Consortium release 1 (N = 32 488 in total; n = 661 AFR)19 using the Michigan Imputation Server. We excluded SNPs with imputation quality score r2 < 0.5 and MAF < 0.01 in each dataset, leaving a total of 12 683 648 overlapping SNPs in Set 1 and Set 2 for statistical analyses.
Statistical analyses
GWAS analysis.
Principal components (PCs) were calculated using EIGENSTRAT.20 Risk allele frequencies (RAFs) were calculated by taking the average frequencies in Set 1 and Set 2 controls. Per-allele odds ratios (ORs) and standard errors were estimated using unconditional logistic regression, adjusted for age, sex, and the first 10 PCs, separately for Set 1 and Set 2. A fixed-effect meta-analysis with inverse variance weights was used to obtain the combined effects for each SNP. The genome-wide significance level was α = 5.0 × 10−8.
Admixture analysis.
We estimated local ancestry separately in Set 1 and Set 2 by RFMix v1.5.4,21 using European (EUR) and AFR populations in phase 1 1KGP as reference. Genotyped SNPs that passed our QC and were present in both sets were included in the analysis. We calculated individual global ancestry by averaging local AA values across the 22 autosomal chromosomes. For local ancestry at each locus, we performed case-only and case-control analyses22 using regression models adjusted for age and sex, separately in each of the 2 GWAS datasets. The case-only analyses compared local ancestry with global ancestry, whereas in case-control analyses we tested whether the average deviation of local ancestry from global ancestry was the same between cases and controls. A fixed-effect meta-analysis was then conducted, using P < 1 × 10−5 to define genome-wide significance. Continuous regions (adjacent regions with P < 1 × 10−4) that were significant in both case-only and case-control comparisons were considered suggestive risk regions for MM.
To identify the set of independent SNPs within the local ancestry signal region, we selected SNPs with marginal P < .001 and conducted forward-selection logistic regression using inclusion criteria of .001, adjusting for age, sex, and global AFR ancestry. To examine whether the detected local ancestry signal could be explained by variant dosage, we compared the marginal and conditional P values and OR percentage changes in local ancestry using logistic regression, adjusting for age, sex, and the first 10 PCs, with and without additional adjusting for allele dosage. We adjusted for both known risk variants and the independent-risk SNPs within the local ancestry signal region.
Association testing of known risk regions.
We were able to directly genotype or impute 23 known MM risk variants.9-13 rs34229995 was excluded because it is uncommon in the AA population (<1%); thus, the following analyses of known risk regions included 22 of the 23 reported EA risk alleles. Directional consistency of effect was defined as ORs in the AA MM meta-analysis that were in the same direction of effect (ie, >1) as those reported in the EA population. A nominal P value of .05 was used to determine statistical significance. We also examined the t(11;14) translocation MM-specific risk allele rs603965 at 11q13.3 in a pooled subset of our AA cases with that translocation (102 and 45 cases from Set 1 and Set 2, respectively), and in all 8871 controls, using logistic regression and adjusting for global AFR ancestry, age, sex, and set.
We used the 22 reported EA risk alleles as index markers to search for other markers within a ±250-kb region that could better capture MM risk in the AA participants (defined as a “better AA marker”). To minimize measurement error, we retained only those SNPs with high imputation quality (r2 ≥ 0.8). In each region, we examined SNPs with pairwise correlation (r2) ≥ 0.2 with the index variant in the EA population in 1KGP likely to capture the functional allele. To reduce false-positive associations, only SNPs with a P value that was smaller by 1 order of magnitude compared with the index variant were defined as a putative “better” marker of risk in AA individuals. A secondary marker within the known risk region was defined as a marker that had weak correlation (r2 < 0.2) with the index SNP among the EUR and AFR populations in 1KGP and P < 1 × 10−6 after conditioning on the index SNP.
Polygenic risk score analysis.
The aggregate effect of known risk alleles was examined using a weighted polygenic risk score (PRS) for each individual: PRSi =∑cm=1 βmgim, where gim is the risk allele dosage for individual i at SNP m, C defines risk SNPs at 22 known MM susceptibility loci, and βm is the weight for SNP m. We explored 2 sets of weights for the index SNPs: the marginal log ORs published from EA populations and log ORs in our AA MM meta-analysis. We also substituted better AA markers for 8 index SNPs and used weights of AA MM log ORs. The risk score in each set was categorized according to its percentile (<10%, 10%-25%, 25%-75%, 75%-90%, and ≥90%), and the risk associated with each category was estimated relative to the interquartile range of 25% to 75% using logistic regression and adjusting for the first 10 PCs, age, and sex. The PRS was constructed separately for each set, the results were combined in a fixed-effect meta-analysis, and the interaction between age at diagnosis and PRS scores was tested.
Expression quantitative trait loci analysis.
We performed an expression quantitative trait loci (eQTL) analysis using Affymetrix Human Genome U133 2.0 Plus Array data for CD138+ plasma cells isolated from bone marrow aspirates, as recently described,23 from 292 patients from the University of Arkansas for Medical Sciences included in the GWASs. Briefly, the expression data were preprocessed, and the probabilistic estimation of expression residuals method was applied to estimate nongenetic hidden confounders.24 Linear regression was used to test the association between the genotype of risk variants and gene expression of genes within 1 Mb, adjusting for probabilistic estimation of expression residuals factors. Risk variants, including suggestive novel signals, known markers identified from previous EA studies, and better AA markers, were examined in the eQTL analysis. A conservative Bonferroni-corrected P value of .0001 was used to determine statistical significance after correcting for 509 total tests.
Results
The GWAS meta-analysis indicated no evidence of overdispersion (λ = 1.03). We did not observe any genome-wide significant association (P < 5 × 10−8) between common alleles and MM risk (supplemental Figure 2; supplemental Table 1). Seven regions harbored signals with P < 1 × 10−6; however, the signal in 4 of them disappeared when a more stringent criterion (r2 ≥ 0.8) was used to filter imputed SNPs (supplemental Table 1). The 2 remaining suggestive novel risk alleles were located at 9p24.3 (rs13296848; OR, 1.25; Wald P = 3.44 × 10−7; RAF, 0.33 [cases] and RAF, 0.28 [controls]) and 9p13.1 (rs7034061; OR, 1.32; Wald P = 9.17 × 10−7; RAF, 0.15 [cases] and RAF, 0.13 [controls]) (supplemental Figure 3).
In admixture mapping analysis, we found a region on chromosome 2 where lower local AA was statistically significantly associated with MM risk in both case-only and case-control comparisons (P < 1 × 10−5), ranging from 23.1 to 29.8 Mb (2p24.1-2p23.1), which covered the known risk region at 2p23.3 (supplemental Figure 4; supplemental Table 2). The strongest local ancestry–MM association was observed at 28.8 to 29.2 Mb (2p23.2; OR per AFR chromosome, 0.79; 95% confidence interval [CI]: 0.72-0.88; P = 9.4 × 10−6). The known risk allele rs6746082 in that region, which is more common in the EA population (RAF, 0.79) than in the AA population (RAF, 0.56), was not significantly associated with MM risk in AA individuals (Table 1). When conditioning on rs6746082, the local ancestry–MM association became only slightly less significant, implying that the known risk allele does not explain the admixture signal. However, when conditioning on our AA MM better marker rs10180663 (identified in fine-mapping), the signal was attenuated by >2 orders of magnitude. In the forward-selection logistic regression, 3 independent SNPs, rs10180663, rs10169985, and rs6734496, were identified (conditional P < .001) as being associated with MM risk. When adjusting for these 3 SNPs, the admixture signal was no longer even nominally significant (OR per AFR chromosome, 0.94; 95% CI, 0.83-1.06; P = .3) (supplemental Figures 5 and 6).
Next, we focused on the 22 known MM risk variants, previously identified in EA populations with MAF ≥ 0.01, in AA individuals to assess their generalizability in the AA population (Figure 1; Table 1). Our study had 80% statistical power to detect the reported effect sizes at a significance level of α = 0.05 for 18 of the 22 loci (Table 1). Directional consistency was noted for 20 variants, 9 of which were nominally statistically significantly associated with MM risk (P < .05; Table 1). The average effect size for the AA population (ORAA, 1.09) was significantly smaller (P = 1.7 × 10−4, Student t test) compared with the reported values for the EA population (OREA, 1.20). The average RAF was slightly larger in the AA controls, but this difference was not statistically significant (RAFAA, 0.463; RAFEA, 0.451; P =.87; Student t test); however, 5 alleles had RAFs that differed greatly between the 2 populations (>0.2) (Figure 1; Table 1). Two alleles, rs6746082 at 2p23.3 and rs2811710 at 9p21.3, were more common among persons of EA, and 3 alleles, rs1052501 at 3p22.1, rs4487645 at 7p15.3, and rs1948915 at 8q24.21, were more common among persons of AA. In fine-mapping of the 22 risk regions, 8 were found to harbor a better marker for MM risk in persons of AA than the index variant by our criteria stated in Methods (Figure 2; Table 1; supplemental Figure 7). We found no significant evidence of secondary association signals in these regions in persons of AA. The association between rs603965 and t(11;14) MM risk replicated in the subset of our AA cases with translocation information at the nominal significance level (OR, 2.04; 95% CI, 1.41-2.95; P = 1.4 × 10−4).
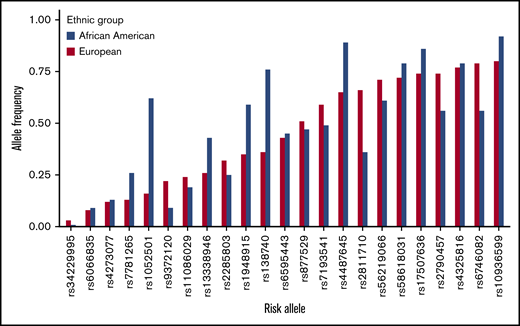
RAF of the 23 known risk alleles in 8871 unaffected controls of AA and in the EA population from the phase 3 1KGP.
RAF of the 23 known risk alleles in 8871 unaffected controls of AA and in the EA population from the phase 3 1KGP.

Regional association plot of the 2p23.3 risk region (25.4-25.9 Mb) in persons of AA. SNPs are plotted by position (x-axis) and −log10P value (y-axis). Linkage disequilibriums are estimated from the EUR population in phase 3 1KGP using r2 statistics. The index SNP (bottom red arrow to purple circle) is rs6746082. The surrounding SNPs are colored to indicate pairwise correlation with the index SNP. The most associated SNP in the AA population in this region is rs10180663 (top red arrow).
Regional association plot of the 2p23.3 risk region (25.4-25.9 Mb) in persons of AA. SNPs are plotted by position (x-axis) and −log10P value (y-axis). Linkage disequilibriums are estimated from the EUR population in phase 3 1KGP using r2 statistics. The index SNP (bottom red arrow to purple circle) is rs6746082. The surrounding SNPs are colored to indicate pairwise correlation with the index SNP. The most associated SNP in the AA population in this region is rs10180663 (top red arrow).
In PRS analyses with weights (ie, log ORs) from studies in EA populations, AA individuals in the top 10% PRS stratum had a 1.61-fold (95% CI, 1.38-1.88; P = 1.4 × 10−9) increased MM risk compared with those with average risk (PRS in the 25th-75th percentiles). Using weights from our AA MM meta-analysis, this OR was 1.66 (95% CI, 1.43-1.94; P = 7.5 × 10−11); when substituting the 8 index EA SNPs with their corresponding better AA markers, this association became slightly stronger (OR, 1.82; 95% CI, 1.56-2.11; P = 9.4 × 10−15) (Table 2). The results of an unweighted PRS were similar (supplemental Table 4). We did not detect any significant interaction between PRS and age at diagnosis on MM risk (data not shown).
In an eQTL analysis (supplemental Table 3), of the 2 suggestive novel variants from the GWAS meta-analysis, only rs7034061 at 9p13.1 was found to be marginally associated (P = .046) with nearby gene expression (EXOSC3); however, it did not remain significant after correcting for multiple comparisons. Of the 22 known risk variants, the strongest association was observed for risk variant rs56219066 at 5q15, with the risk allele being associated with lower ELL2 expression (P = 5.1 × 10−12). We also identified a significant association between the risk allele of rs2790457 at 10p12.1 and decreased expression of WAC (P = 2.29 × 10−11). Both eQTLs were recently demonstrated in malignant plasma cells from EA patients.13,25 The recently identified association between rs4487645 at 7p5.13 and CDCA7L expression in EA patients23 was also evident at a borderline statistically significant level in malignant plasma cells from AA patients (P = .067). Of the 8 better AA MM markers, 3 showed nominally significant associations with gene expression that were not significant after correction for multiple testing: rs10180663 at 2p23.3 with HADHA, rs9290375 at 3q26.2 with GPR160, and rs879882 at 6p21.33 with VARS2 and HCG27.
We further explored the potential overlap between suggestive novel signals and their correlated SNPs (r2 > 0.2, AFR population in 1KGP) with the genome regulatory domains and eQTL regions using the publicly available databases through HaploReg,26 UCSC Genome Browser,27 and GTEx Portal.28 Neither of the 2 suggestive novel risk alleles showed overlap with regulatory elements. However, 2 correlated variants of rs13296848 (rs7854502, r2 = 0.58; rs13285101, r2 = 0.51; supplemental Figure 8) displayed enrichments in promoter and enhancer histone markers in multiple tissues,29 and both were associated with KN motif and ankyrin repeat domains 1 (KANK1) expression in Epstein-Barr virus–transformed lymphocytes, spleen, and whole blood.28
Discussion
In this largest genetic study of MM in individuals of AA, we did not identify any novel locus for MM risk. Of the 22 reported MM EA risk alleles that we were able to examine, 20 were directionally consistent and 9 achieved nominal statistical significance in AA individuals, suggesting a common shared underlying risk variant in these regions. Although most of the reported risk alleles had a modest association (OR < 1.2) with MM risk in AA individuals, in aggregate, we observed that those in the top 10% risk stratum had a 1.6- to 1.8-fold increase in MM risk compared with the population average risk (25th-75th percentile of the PRS distribution).
Overall, the effect sizes among persons of AAs were smaller than in the corresponding discovery reports among patients of EAs. The possible explanations for the smaller effect sizes include bias caused by “winner’s curse” in the EA discovery set, random errors from sampling across different studies, modification by environmental factors, and different linkage disequilibrium structure across ethnic groups between the index risk alleles and the functional causal SNPs. Although our study has sufficient statistical power (>80%) to replicate 18 of the risk alleles at a significance level of 0.05, more than half of them did not achieve nominal significance, implying that the index SNP in EA populations may not be a valid proxy for the causal variant in AA populations; thus, our fine-mapping of those risk regions in persons of AA was warranted.
Our previous study identified 5 better AA markers among 7 of the reported EA MM risk variants.8 Here, we identified better AA markers in 8 of the 22 regions that we examined. Our current study had greater power in characterizing AA MM risk variants as the result of improved imputation coverage by using the Haplotype Reference Consortium reference panel and increased AA sample size. Notably, we identified a better marker, rs10180663, in 2p23.3 with a much stronger association in the AA population than in our previous study.8 In this region, previous GWAS studies in the EA population have reported 2 MM risk variants, rs67460829 and rs7577599,11 and a pleiotropic risk variant rs6546149 for B-cell malignancies, including chronic lymphocytic leukemia, Hodgkin lymphoma, and MM.30 Our better AA marker rs10180663 is correlated with all 3 of the aforementioned risk variants in the EA population (r2 > 0.25 in EUR population in 1KGP) and displayed the strongest association among AA populations. Variant rs10180663 was also previously identified as a suggestive risk allele for the t(11;14)(q13;q32) MM molecular subtype among the EA population (P = 2 × 10−6).31 Moreover, it overlapped with H3K4Me1 enrichment in hematopoietic stem cells and B cells26 and was predicted to be located at an enhancer region,29 suggesting that it might be a better surrogate for the causal MM variant in the 2p23.3 region in AA individuals.
Although no genome-wide novel variants were found, we did observe suggestive associations with rs13296848 located at 9p24.3, with a RAF of 0.33 among AA cases and a RAF of 0.28 among AA controls. This variant is located in the intron of the KANK1 gene, a candidate tumor suppressor for renal cell carcinoma.32 KANK1 may have a role in KPβ-associated thrombocythemia,33 which was found to be occasionally associated with MM.34 The correlated alleles of rs13296848 displayed enrichments in promoter and enhancer histone markers and were associated with KANK1 expression in Epstein-Barr virus–transformed lymphocytes, spleen, and whole blood.28 The other suggestive novel MM risk allele, rs7034061, was located at 19 kb 5′ of the gene insulin like growth factor binding protein like 1 (IGFBPL1), whose epigenetic inaction was reported as being involved in breast cancer pathogenesis.35 Moreover, serum fasting IGFBP1 concentration was associated with MM risk in a nested case-control study.36 However, no overlap of genome regulatory elements or IGFBPL1 expression enrichment was detected for rs7034061 or its correlated SNPs in publicly available databases. Fine-mapping in larger samples of AA cases and controls is needed to determine whether this region harbors a true risk variant that is associated with MM risk.
Our study is the first to examine the association between local ancestry and MM risk. Compared with GWASs, admixture mapping has enhanced power (fewer tests requiring correction) but a lower resolution for discovering disease risk regions.37 We found that the level of local AA in a continuous region that covered the known risk region 2p23.3 was inversely associated with MM risk (P < 1 × 10−5). The signal could not be explained by the known risk marker rs6746082, but it was completely explained by conditioning on the 3 independent SNPs, including rs10180663. Thus, the previously noted signal in this region was detected in a different population using admixture mapping.
Unlike prostate cancer, in which variants at a single locus (8q24) were discovered by admixture mapping in men of AA,38 we did not identify a single locus that explains the excess risk for MM in AA individuals, although our study was well powered (80% power to detect a single locus with an OR of 1.7). It is possible that multiple loci with small effects, rather than a single region with a large effect, contribute to MM risk in AA individuals, which would require a much larger sample size than we have here. It is also possible that there is etiologic heterogeneity by molecular subtype and that, by combining them, we diluted the signal.
From the gene expression studies in malignant plasma cells from AA MM patients, the only risk variants that significantly impacted gene expression after correcting for multiple tests were rs2790457 at 10p12.1 (WAC) and rs1423269 at 5q15 (ELL2), 2 eQTLs that were recently shown in EA MM patients.11,25 The functional basis of the majority of MM risk variants remains elusive, and further studies, perhaps in circulating B-memory lymphocytes from healthy individuals, are required to understand the underlying biology.
A possible limitation of our study is that the AA MM cases and controls in Set 1 were genotyped on different platforms. To minimize this bias, we conducted stringent pre- and postimputation QCs, including eliminating SNPs with low cross-platform replicate concordance and imputation to the same reference panel. There were only 188 376 genotyped SNPs overlapping between cases and controls in Set 1, because of the low density on the Illumina Human Core (298 930 SNPs) used for the cases. As a result, our imputation-based coverage of common variation was suboptimal for fine-mapping. A denser genotyping array may help to better localize causal variants in the AA population. In addition, given the low risk associated with these signals in some regions, a larger sample size is required to differentiate the causal variant from the index SNP. Moreover, because MM is characterized clinically by molecular subgroups,39 it is possible that genetic susceptibility may vary across these subtypes.12,31 The prevalence of each molecular subtype is <50%, and we did not have complete records of clinical molecular subtypes for all patients; thus, we could not evaluate subtype-specific associations. Although we had adequate power to examine most of the known risk variants at P < .05, the sample size was still not large enough for discovery of novel AA-specific genetic markers at a genome-wide significance level. Obtaining patient samples for such an uncommon cancer among a minority population is challenging. Nevertheless, this is the largest study in AA individuals to examine associations between common genetic variation and MM risk; our results show a role for genetic susceptibility in AA MM.
Acknowledgments
The work was supported by NCI, National Institutes of Health (NIH) grants 1R01CA134786 (W.C. and C. A. Haiman), R01 CA84979 (S.A.I.), 5U01CA164973 (L.L.M. and C. A. Haiman), P30CA01409 and P50CA100707 (K.C.A.), R21 CA1918896 (E.Z.), and R01 CA092447 (L.S.). The collection of incident MM patients used in this study was supported by the California Department of Health Services as part of the statewide cancer-reporting program mandated by California Health and Safety Code Section 103885 and by a grant from the Centers for Disease Control and Prevention to support cancer patient registration and ascertainment (1U58DP000807-01). Patient identification was made possible by federal funds from the NCI Surveillance Epidemiology and End Results Population-based Registry Program, NIH, Department of Health and Human Services. Genotyping of cases and some controls was performed at the University of Southern California Norris Comprehensive Cancer Center Genomics Core, which is supported by NCI Comprehensive Cancer Center Core grant P30 CA014089. Patient accrual and sample processing at Johns Hopkins Medical Center was supported by the NCI Sidney Kimmel Comprehensive Cancer Center core grant P30 CA006973.
K.A.R. was a postdoctoral fellow at the Keck School of Medicine of USC when she performed the work for this study. G.C.S. was a doctoral student when he performed the work for this study.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Authorship
Contribution: W.C., D.V.C., and C. A. Haiman designed the study, supported analyses, and critically reviewed and edited the manuscript; Z.D. performed genetic analyses and wrote the manuscript; N.W. performed eQTL analyses and cowrote the manuscript; G.C.S. and K.A.R. performed genetic analysis and QC; A.E.H. and X.S. managed data and performed genetic QC; V.H. and A.S. imputed genetic data; S.A., A.K.N., S.S., A.M., J.M., G.A.C., J.W., T.G.M., M.T., M.A.F., H.T., N.J., L.K., L.L.M. D.A., E.Z., B.C.-H.C., R.V., L.B.-M., G.J.M., J.A.Z., C. A. Huff, S.L., and R.Z.O. provided patient samples and clinical data, along with input on biological and clinical aspects of myeloma and molecular subtypes for the study; K.P. E.S.P., A.M.S., and C.B. provided patient samples and demographic data and input on epidemiologic aspects of the study; D.J.V.D.B. conducted the GWAS and provided the genotyping calling; K.C.A. provided input on etiologic and molecular aspects of myeloma; D.S. and E.Z. provided additional input on genetic ancestry analysis and admixture mapping; N.W., O.W.S., M.Z., and F.V.R. conducted eQTL analyses; S.I.B., W.J.B., G.C., V.L.S., R.K., P.J.G., W.R.D., A.H., B.N., E.A.K., B.A.R., J.L.S., J.S.W., L.S., E.M.J., L.B., A.O., W.Z., J.J.H., R.Z., S.J.N., E.Z., S.A.I., M.F.P., J.D.C., and S.J.C. contributed GWAS data from controls and provided guidance on genetic analysis methods; and all authors provided important intellectual content and reviewed and edited the manuscript.
Conflict-of-interest disclosure: C. A. Huff has acted as a consultant for Karyopharm Therapeutics, Sanofi, and miDiagnostics and is a member of the Safety Monitoring Board for Johnson and Johnson. T.G.M. has acted as a consultant for Roche and Juno Therapeutics and has received research funding from Amgen, Sanofi, and Seattle Genetics. J.M. is a member of the Speakers Bureau for Takeda Pharmacauticals and Celgene and owns stock in Celgene, Bristol-Myers Squibb, and bluebird bio. S.S. is a member of the Speakers Bureau for Takeda Pharmaceuticals, Celgene, and Janssen Pharmaceuticals and owns stock in Celgene, Bristol-Myers Squibb, and bluebird bio. S.L. has acted as a consultant for Janssen Pharmaceuticals, Takeda Pharmaceuticals, Celgene, Novartis, Bristol-Myers Squibb, Merck, and GlaxoSmithKline and has received research funding from Celgene, Takeda Pharmaceuticals, and Janssen Pharmaceuticals. K.C.A. is a consultant for Celgene, Jansen, Bristol-Myers Squibb and Sanofi, and a scientific founder of OncoPep and C4 Therapeutics. S.A. has acted as a consultant for Novartis, Amgen, and Takeda Pharmaceuticals, and has received research funding from Pharmacyclics. A.K.N. has acted as a consultant for Amgen, Novartis, Spectrum Pharmaceuticals, and Adaptive Biotechnologies. R.V. has received honoraria and research funding from Takeda Pharmaceuticals and Amgen and received honoraria from Celgene, Bristol-Myers Squibb, Janssen Pharmaceuticals, AbbVie, Jazz Pharmaceuticals, and Konypharma. J.A.Z. has acted as a consultant for Prothena and Janssen Pharmaceuticals; has acted as a consultant and received research funding from Bristol-Myers Squibb, Celgene, and Takeda Pharmaceuticals; and is a member of the Data Safety Monitoring Committee for Pharmacyclics. G.J.M. has acted as a consultant for Celgene, Takeda Phamaceuticals, and Bristol-Myers Squibb; has received research funding from Celgene; and has received honoraria from Celgene, Takeda Pharmaceuticals, and Bristol-Myers Squibb. R.Z.O. is a member of the Advisory Board for Amgen, Celgene, Forma Therapeutics, GlaxoSmithKline Biologicals, Ionis Pharmaceuticals, Janssen Biotech, Juno Therapeutics, Kite Pharma, Legend Biotech, Sanofi, Servier, and Takeda Pharmaceuticals; has acted as a consultant for Molecular Partners; and has received research funding from BioTheryX. The remaining authors declare no competing financial interests.
The current affiliation for G.C.S. is Millennium Pharmaceuticals Inc., a subsidiary of Takeda Pharmaceutical Company Ltd., Cambridge, MA.
The current affiliation for K.A.R. is Ancestry.com, San Francisco, CA.
The current affiliation for A.S. is Genomic Health, Inc., Redwood City, CA.
Correspondence: Wendy Cozen, Center for Genetic Epidemiology, Department of Preventive Medicine, Keck School of Medicine, University of Southern California, 1441 Eastlake Ave, MC 9175, Los Angeles, CA 90089-9175; e-mail: wcozen@med.usc.edu.



