Key Points
When performed in accordance with modern ASH and ISTH guidelines, PA testing is sensitive and specific for ITP diagnosis.
More glycoproteins targeted by autoantibodies predicts for more severe disease, and autoantibodies resolve with clinical remission.

Abstract
Platelet autoantibody (PA) testing has previously shown poor sensitivity for immune thrombocytopenia (ITP) diagnosis, but no previous study used both 2011 American Society of Hematology (ASH) guidelines for ITP diagnosis and 2012 International Society on Thrombosis and Haemostasis (ISTH) PA testing recommendations. We therefore performed a comprehensive retrospective study of PA testing in adult patients with ITP strictly applying these criteria. Of 986 PA assays performed, 485 assays in 368 patients met criteria and were included. Sensitivity and specificity of a positive test result for diagnosis of active ITP (n = 228 patients) were 90% and 78%, respectively. Sensitivity and specificity of a negative test result for clinical remission (n = 61 assays) were 87% and 91%. Antibodies against both glycoprotein IIb (GPIIb)/IIIa and GPIb/IX were required for the presence of antibodies against GPIa/IIa in patients with ITP. Logistic regression analysis revealed that more positive autoantibodies predicted more severe disease (relative to nonsevere ITP, relative risk ratio for severe ITP and refractory ITP was 2.27 [P < .001] and 3.09 [P < .001], respectively, per additional autoantibody); however, serologic testing did not meaningfully predict treatment response to glucocorticoids, intravenous immunoglobulin, or thrombopoietin receptor agonists. Sixty-four patients with ITP had multiple PA assays performed longitudinally: all 10 patients achieving remission converted from positive to negative serologic results, and evidence for epitope spreading was observed in 35% of patients with ongoing active disease. In conclusion, glycoprotein-specific direct PA testing performed using ISTH recommendations in patients meeting ASH diagnostic criteria is sensitive and specific for ITP diagnosis and reliably confirms clinical remission. More glycoproteins targeted by autoantibodies predicts for more severe disease.
Introduction
Autoantibodies directed against platelet glycoproteins have long been accepted as a major pathophysiologic mechanism in immune thrombocytopenia (ITP). This was first reported in the now classic studies showing the ability of plasma from patients with ITP to cause thrombocytopenia in healthy subjects.1-3 Unfortunately, previous studies of platelet autoantibody (PA) testing have shown poor sensitivity for the diagnosis of ITP. As a result, current practice guidelines published by the American Society of Hematology (ASH) do not endorse routine PA testing for this purpose.4,5 Notably, many of the studies evaluating the diagnostic utility of PA testing used poorly defined ITP populations and did not use standardized criteria for ITP diagnosis, thereby likely including patients who did not have true ITP.6 Furthermore, many studies included both pediatric and adult patients despite the known clinical and pathophysiologic differences between these groups.7,8 These issues could considerably confound the assessment of a diagnostic test for ITP, a condition for which there is no validated standard diagnostic test.
Nonetheless, PA testing continues to remain attractive in the evaluation of the thrombocytopenic patient due to the lack of a known biomarker specific for ITP. Direct assays for platelet autoantibodies (which measure antibodies on platelets, as opposed to indirect assays, which measure free antibodies in plasma) that are capable of detecting glycoprotein-specific autoantibodies are considered optimal for PA testing.9 This includes a number of solid-phase enzyme-linked immunosorbent assays (ELISAs) such as the monoclonal antibody–specific immobilization of platelet antigen (MAIPA) assay10 and the monoclonal antigen capture ELISA (MACE).11 Flow cytometric techniques for detection of platelet-associated IgG12 are also available but are not recommended owing to poor specificity.9 Immunobead assays using both ELISA-based and flow cytometric techniques have also been developed.13,14 Previous studies of direct glycoprotein-specific PA testing have shown good specificity but poor sensitivity for ITP diagnosis.15
Because platelet autoantibodies are considered pathogenic, PA testing may provide information beyond diagnostic confirmation such as prediction of treatment response. Unfortunately, previous studies examining the impact of platelet autoantibodies on ITP prognostic features have been limited by sample size16 or use of indirect tests, resulting in poor assay performance.17 In consideration of a relation to treatment response, there is suggestive evidence that anti-glycoprotein Ib (GPIb)/IX antibodies may predict response to glucocorticoids18 or intravenous immunoglobulin (IVIG),19 although results of these studies have not been confirmed.
Given the limitations of many of the previous studies, we sought to investigate the utility of direct PA testing in ITP diagnosis, prediction of disease severity, and prediction of therapeutic response using a large sample of direct PA assays in adult patients with ITP. In addition, as we performed serial testing in many of our patients with ITP, we sought to examine the serologic evolution of these patients in a longitudinal fashion to assess for evidence of epitope spreading as well as resolution of autoantibodies with clinical remission, neither of which, to our knowledge, has been previously described. We therefore conducted a retrospective analysis of PA testing performed at our institution. In designing the protocol for this study, we aimed to optimize both the clinical diagnostic and laboratory assay validity by strictly following published evidence-based consensus guidelines for each, an approach not previously applied in studies of PA testing in ITP. The ASH 2011 evidence-based practice guidelines for ITP (hereafter referred to as ASH 2011 guidelines)4 offer standardized criteria relating to diagnosis, disease severity, and treatment response that were applied to optimize clinical validity. Of note, the 2019 update to the ASH 2011 guidelines5 did not modify these standardized criteria and so they remain unchanged in the ASH 2019 guidelines. Similarly, the recommendations of the Platelet Immunology Scientific Subcommittee of the International Society on Thrombosis and Haemostasis (ISTH) for implementation of PA testing in clinical trials of ITP9 were applied to optimize laboratory assay validity.
Methods
Patients and data collection
This study was approved by the institutional review board (approval 2018P000964/PHS) of the Massachusetts General Hospital. Between 1 January 2014, and 30 June 2018, all patients aged ≥18 years who had PA testing performed were identified via query of the laboratory information system at our institution.
To be included in the analysis, patients with ITP were required to fulfill the diagnostic criteria for ITP, as established by the ASH 2011 guidelines for ITP,4 at the time of initial clinical evaluation. Patients fulfilling these criteria were included irrespective of whether they had primary or secondary ITP. Two of the authors (H.A.-S. and D.J.K.) confirmed the diagnosis for each patient before collection of PA testing results. Patients with Evans syndrome were included if: (1) they clearly met ASH 2011 guidelines for ITP when no appreciable hemolysis was occurring; or (2) if hemolysis did not remit, the only perturbations to the complete blood cell count were those clearly secondary to hemolytic anemia. Thrombocytopenic patients with a confirmed non-ITP diagnosis who had PA testing performed were identified as control subjects. We excluded patients who never had a clinical diagnosis established to explain their thrombocytopenia at any point over the course of follow-up. Patient data collected included: age, sex, etiology of ITP (primary or secondary), date of ITP diagnosis, previous ITP therapies, PA testing dates and results, platelet count at time of PA testing, ITP disease severity at time of PA testing, and response (as defined by using the ASH 2011 guidelines) to glucocorticoids, IVIG, and thrombopoietin receptor agonists. To be considered evaluable, a treatment had to be administered before or within 6 months after PA assay.
ITP disease severity was defined according to ASH 2011 guidelines4 (these definitions were not updated in the 2019 guidelines), which classify active ITP into 3 severity categories: (1) patients with nonsevere ITP have not developed bleeding symptoms requiring treatment; (2) patients with severe ITP have developed bleeding symptoms requiring treatment; and (3) patients with refractory ITP have undergone splenectomy but subsequently develop bleeding symptoms requiring additional treatment. Clinical remission was defined as achieving a platelet count ≥100 × 109/L off all ITP-directed treatment of ≥12 months without relapse for the entire duration of available follow-up.
PA assay inclusion and exclusion criteria
Only PA assays that followed all 5 ISTH PA testing recommendations were included. These inclusion criteria were as follows: (1) collection of patient sample in acid citrate dextrose solution-A anticoagulant; (2) collection of a minimum sample volume of 30 mL; (3) use of a direct test measuring glycoprotein-specific autoantibodies; (4) testing utilizing antibodies to GPIIb/IIIa and GPIb/IX; and (5) centralization of processing and testing in a single laboratory. Exclusion criteria were receipt of a platelet transfusion within 96 hours before PA testing, receipt of IVIG ≤30 days before PA testing, and patient never being thrombocytopenic. Data logged in the laboratory information system and electronic medical record were used to assess these inclusion and exclusion criteria for each assay.
All PA assays meeting inclusion criteria were performed by using the commercially available PakAuto assay (Immucor, Brookfield, WI), a direct and indirect solid-phase ELISA-based test measuring antibodies against GPIIb/IIIa, GPIb/IX, and GPIa/IIa. This direct glycoprotein-specific PA assay measures antibodies eluted from the surface of platelets (it is not an MAIPA or MACE assay). Sample volume was 40 mL for all included specimens. A result was positive if optical density values were ≥2 times the value obtained for the mean of the negative controls for the corresponding glycoprotein. Additional details on the PakAuto can be found in the published assay instructions for use.20 All included assays were performed at a single central laboratory (BloodCenter of Southeast Wisconsin, Milwaukee, WI). Unless otherwise specified, only results of direct assays were considered in analyses.
Positive PA assay test results were defined as the presence of one or more glycoprotein-specific platelet autoantibodies on direct testing.
Data analysis and statistical testing
Test characteristics (sensitivity, specificity, positive predictive value, and negative predictive value) of a positive PA assay for a diagnosis of active ITP were determined by using the first PA assay obtained for each patient (both ITP and control patients). In addition, test characteristics of a negative PA assay for predicting remission in patients with previously active ITP in clinical remission at the time of testing were determined by using all included PA assays in patients with ITP.
Multinomial logistic regression was used to model the probability of ITP disease severity (nonsevere, severe, or refractory)4 in patients with active disease at the time of PA assay based on the number of positive glycoprotein-specific autoantibodies (0, 1, 2, or 3), age, sex, duration of disease (time from diagnosis to performance of assay), and platelet count at time of assay. The Hausman test was performed to confirm the assumption of independence of irrelevant alternatives with all multinomial logistic regression testing.21 The primary analysis included all assays from patients with active ITP, and as a check of internal validity (given the inclusion of multiple assays from some patients), an additional analysis was performed including only the first assay performed on each patient. Splenectomy status and number of previous treatments could not be included in this model because they are not independent of disease severity based on ASH 2011 guidelines (bleeding requiring treatment defines severe disease, and requirement for treatment after splenectomy defines refractory disease). Separate multiple logistic regression models using the first assay performed on each patient with active disease were used to model probability of response to treatment (glucocorticoids, IVIG, or thrombopoietin receptor agonists) based on presence of a certain glycoprotein-specific autoantibody (anti-GPIIb/IIIa, anti-GPIb/IX, or anti-GPIa/IIa), age, sex, duration of disease, number of previous treatments, and splenectomy status.
In ITP patients with multiple PA assays performed over time, patients were split into 2 groups for evaluation of longitudinal serologic evolution: those with no change in disease status and those who achieved clinical remission. Epitope spreading was defined as an increase in the number of positive autoantibodies over time.
Statistical analysis was performed and graphs for figures were prepared by using Stata version 14.2 (StataCorp, College Station, TX), GraphPad Prism 7 (GraphPad, Inc., San Diego, CA), and Microsoft Excel 360 (Microsoft Corporation, Seattle, WA).
Results
PA assays and patient characteristics
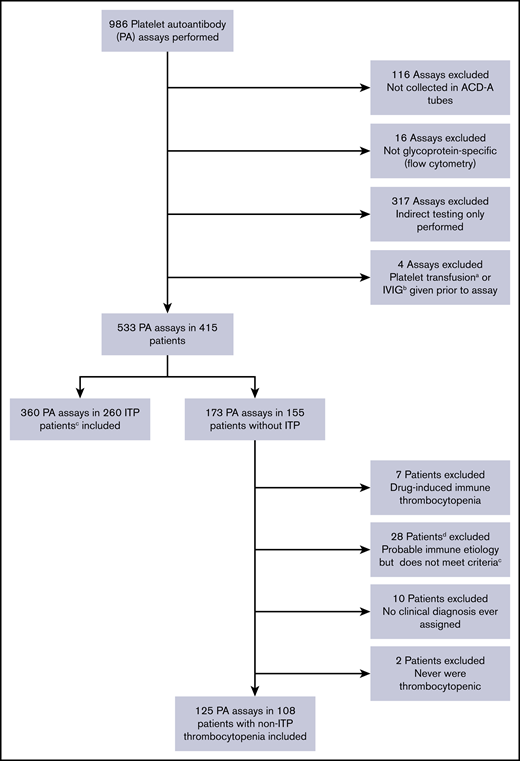
Of 986 PA assays performed, 533 performed in 415 patients satisfied all 5 ISTH recommendations; 29 additional assays were ordered but could not be performed by the laboratory due to inadequate platelet counts (nearly all with platelet counts ≤5 × 109/L). A total of 360 assays in 260 patients with ITP meeting ASH ITP diagnostic criteria and 125 assays in 108 control patients with thrombocytopenia and a confirmed non-ITP diagnosis were included (Figure 1). Baseline characteristics of patients with ITP are detailed in Table 1, and diagnoses of non-ITP control subjects are listed in supplemental Table 1.
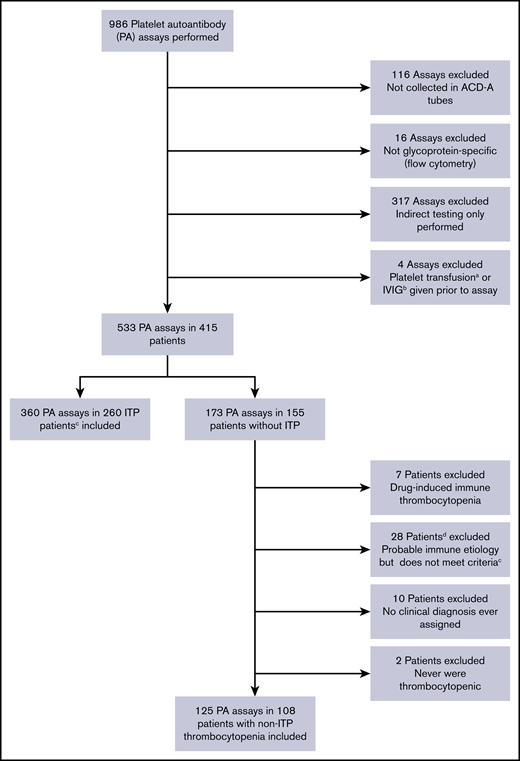
Flow diagram describing application of inclusion and exclusion criteria for the study.aWithin 4 days before PA assay. bWithin 30 days before PA assay. cPer ASH 2011 clinical practice guidelines. dOne of these 28 patients had 2 assays performed, and thus 29 assays were excluded according to this criterion. ACD-A, acid citrate dextrose solution A.
Flow diagram describing application of inclusion and exclusion criteria for the study.aWithin 4 days before PA assay. bWithin 30 days before PA assay. cPer ASH 2011 clinical practice guidelines. dOne of these 28 patients had 2 assays performed, and thus 29 assays were excluded according to this criterion. ACD-A, acid citrate dextrose solution A.
Test characteristics of direct PA assay
Of the 260 patients with ITP undergoing PA testing, 228 patients had the initial assay performed in the setting of active clinical disease. The sensitivity and specificity of a positive assay for diagnosis of active ITP were 90% (95% CI, 85-93) and 78% (95% CI, 69-85), respectively, with a positive assay having a positive likelihood ratio of 4.0 for a diagnosis of active ITP.
Sixty-one of the 360 assays performed in patients with ITP were obtained during clinical remission. Fifty-three were negative for all antibodies, and 8 were positive for at least 1 antibody. The sensitivity and specificity of a negative assay for clinical remission were 87% (95% CI, 76-93) and 91% (95% CI, 87-93), respectively, with a negative test result having a positive likelihood ratio of 9.3 for remission. The 8 positive assays were performed in 8 unique patients, and 1 of these patients (positive for anti-GPIIb/IIIa antibodies) subsequently relapsed 15 months after entering remission; the other 7 have not relapsed.
Additional test characteristics of PA testing for diagnosis of active ITP or clinical remission are listed in Table 2.
Serologic patterns of positive PA assays
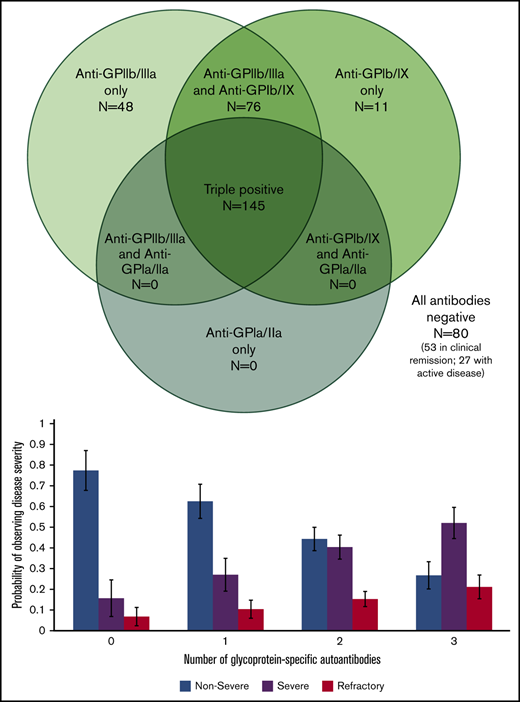
Of 7 possible serologic patterns, only 4 were observed. Figure 2 illustrates the number of positive assays with each pattern. Notably, autoantibodies against both GPIIb/IIIa and GPIb/IX were required for the presence of autoantibodies against GPIa/IIa.

Serologic patterns of platelet autoantibody assays in ITP patients. Results of PA assays in patients with ITP (360 assays total in 260 patients).
Serologic patterns of platelet autoantibody assays in ITP patients. Results of PA assays in patients with ITP (360 assays total in 260 patients).
Logistic regression models of disease severity and treatment response
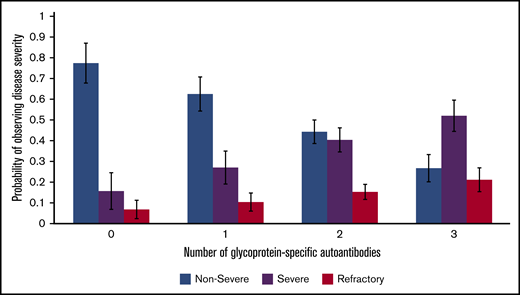
Our multinomial logistic regression model of patients with active disease found a statistically significant predictive relation between an increasing number of positive autoantibodies and disease severity. Relative to nonsevere ITP, the relative risk ratio for severe ITP and refractory ITP was 2.27 (P < .001) and 3.09 (P < .001), respectively, per 1 additional positive autoantibody (Figure 3). This relation was still present when only the first performed PA assay from each patient with active disease was included in the model: relative to nonsevere ITP, the relative risk ratio for severe ITP and refractory ITP was 2.73 (P <. 001) and 2.75 (P = .002) per 1 additional positive autoantibody.
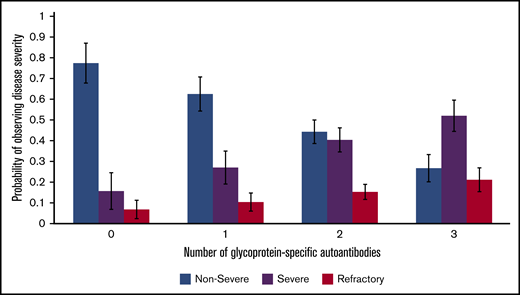
Relation of glycoprotein-specific autoantibody number with disease severity. Multinomial logistic regression model for probability of disease severity based on number of glycoprotein-specific autoantibodies (error bars represent 95% confidence intervals). Model only included patients with active disease at the time of PA testing.
Relation of glycoprotein-specific autoantibody number with disease severity. Multinomial logistic regression model for probability of disease severity based on number of glycoprotein-specific autoantibodies (error bars represent 95% confidence intervals). Model only included patients with active disease at the time of PA testing.
Multiple logistic regression models used to model probability of response to treatment (glucocorticoids, IVIG, or thrombopoietin receptor agonists) found a significant predictive relation between presence of anti-GPIa/IIa and nonresponse to corticosteroid treatment (odds ratio, 0.101; P = .002). No significant relation was found between an autoantibody and nonresponse to IVIG or thrombopoietin receptor agonists.
Longitudinal serologic evolution
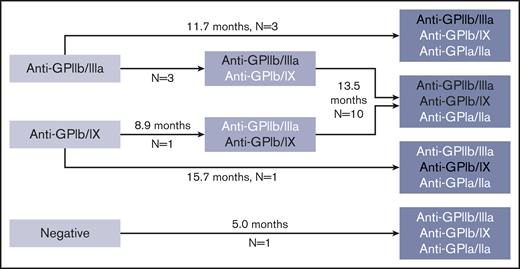
Sixty-four patients had multiple (2-5) PA assays performed over time. In evaluation of serologic evolution over time, 28 patients had stable findings (first and last positive PA assay results were identical; in 3 of these patients with ≥3 assays, there were intervening serologic changes between the first and last assays performed); 7 patients had a reduction in number of autoantibodies; and 19 patients exhibited evidence for epitope spreading, with an increase in the number of autoantibodies (Figure 4).
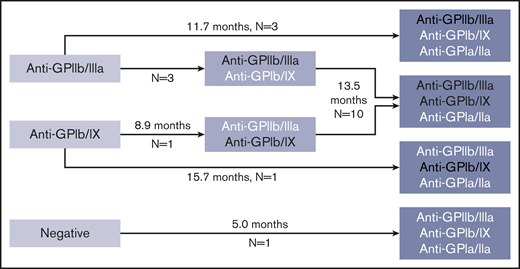
Pattern of possible epitope spreading observed in patients with increasing numbers of glycoprotein-specific autoantibodies on interval testing. Median durations between assays given. White text delineates new autoantibodies present in subsequent assay not present in previous assay. Platelet count at the time of testing was similar at first test compared with repeat test (88 × 109/L at first test and 71 × 109/L at repeat test, P = .17 by paired Student t test).
Pattern of possible epitope spreading observed in patients with increasing numbers of glycoprotein-specific autoantibodies on interval testing. Median durations between assays given. White text delineates new autoantibodies present in subsequent assay not present in previous assay. Platelet count at the time of testing was similar at first test compared with repeat test (88 × 109/L at first test and 71 × 109/L at repeat test, P = .17 by paired Student t test).
Ten patients with multiple assays had active disease at the time of their first assay and subsequently achieved clinical remission. All of these patients converted from a positive to a negative serologic finding after clinical remission (2 of whom first experienced an increase in the number of positive autoantibodies while disease was active and then subsequently achieved clinical remission with conversion of serologic results to negative).
Indirect PA testing
Indirect PA testing was performed with all direct tests. Test characteristics of indirect testing alone for the diagnosis of ITP were poor (supplemental Table 2). Results of indirect testing were positive in 33% of patients with ITP and correlated poorly with direct test positivity (of those with a positive direct test result, only one-third also had a positive indirect test result). Addition of indirect testing results to direct testing results (with a test result considered positive if either direct or indirect assays were positive) reduced performance of PA testing compared with direct testing alone.
Discussion
Recognizing the significant heterogeneity and limitations of the existing literature (Table 3), we sought to perform a reappraisal of PA testing in ITP. Specifically, now that modern evidence-based guidelines have emerged capable of guiding and standardizing both the clinical and laboratory aspects of this assessment, we aimed to use both sets of criteria to maximize the validity of our assessment as none of the previously published studies had done so. In addition, our goal was to describe for the first time the longitudinal serologic evolution of serial PA assays in ITP and the relation between clinical severity of disease as defined by using ASH 2011 criteria and serologic findings.
Using this approach, we found that the sensitivity of a positive direct PA assay for the presence of active ITP was 90%, much higher than the studies of glycoprotein-specific assays previously reported in the literature6,11,14,16,17,22-24,26-29 (Table 3); specificity was 78%, which is in line with some of these previous studies and slightly lower than others. The strength of our approach is emphasized when comparing our findings vs the results of the study performed by Davoren et al,24 which used the same assay but did not follow ASH 2011 criteria for diagnosis or all ISTH guidelines for PA testing; it also included a mixture of pediatric and adult patients, and the sensitivity for the assay in this study was only 53%. We found that a positive test result had a positive likelihood ratio of 4.0, which corresponds to an increase in the absolute probability of a diagnosis of ITP by ∼25%.30 Although it is never possible to know the pretest probability of an ITP diagnosis in a given patient, if, for example, a thrombocytopenic patient theoretically had a pretest probability of 60% for a diagnosis of ITP, a positive PA assay performed according to the criteria outlined in this study would increase that theoretical probability of an ITP diagnosis to 85%. For comparison, a D-dimer level of 2500 to 4999 ng/mL has a positive likelihood ratio of ∼4.0 for a diagnosis of acute pulmonary embolism.31 Indirect testing was not useful in ITP diagnosis (supplemental Table 2), however, and actually reduced the performance of direct testing alone when direct and indirect results were considered together. In addition, indirect testing was not useful in identifying patients missed by direct testing; only 3% of patients with ITP had false-negative direct testing results and positive indirect testing. Indirect testing has had similarly poor diagnostic performance in other studies.15
It is possible that the test characteristics of PA testing in previous studies were affected by poor diagnostic validity (many patients with nonimmune-mediated thrombocytopenia classified as ITP and vice versa). As a diagnosis of exclusion, adjudication of an ITP diagnosis can be challenging (even for a panel of international experts32 ), highlighting the need for a combination of objective evidence-based criteria and clinician judgment, as was performed in the current study. In addition, the test characteristics of PA testing in previous studies may have been affected by suboptimal laboratory practices. For example, most collected samples for testing in EDTA anticoagulant, which is known to result in platelet shape change, altered glycoprotein integrity and reduced platelet harvest9 ; many used only indirect tests; and most did not describe the volume of blood collected for testing, with inadequate volume resulting in inadequate platelet yield in severely thrombocytopenic patients, which can compromise assay validity.
We additionally evaluated the testing characteristics of PA assays in assessing remission in patients with known ITP, which has not previously been described. Utilizing a rigorous definition of clinical remission (platelet count ≥100 × 109/L off all ITP therapy for ≥12 months), we found the sensitivity and specificity of a negative assay for clinical remission to be 87% and 90%, respectively. With a positive likelihood ratio of 9.3, negative PA testing following our parameters increases the absolute probability of remission by 40% to 45%, effectively confirming remission for patients in whom it is clinically suspected.30 For many patients maintaining stable platelet counts for months to years on chronic thrombopoietin receptor agonist or immunosuppressive therapy, a negative PA assay can therefore reassure the patient and physician that discontinuation of treatment is likely to be safe.
In consideration of the relation between serology and clinical features, our logistic regression model found a significant predictive relation between disease severity as defined by using ASH 2011 guidelines and number of positive glycoprotein-specific autoantibodies (Figure 3). This result might be expected from a pathophysiologic perspective and has been reported in other autoimmune conditions, such as antiphospholipid antibody syndrome.33,34 In addition to being more likely to have fewer glycoprotein-specific autoantibodies, patients with nonsevere ITP were more likely to have no glycoprotein-specific autoantibodies (false-negative testing). Given that false-negative test results could occur in the setting of autoantibodies to platelet antigens not being tested by the assay, such as GPIV or GPV, this outcome is consistent with our general hypothesis that more autoantibodies increase the likelihood of more severe disease. Coupled with our finding that ∼35% of patients with active disease who had multiple assays performed exhibited evidence for epitope spreading, this hypothesis could explain why some patients develop more severe and treatment-refractory disease over time; the inconsistent interval between PA assays in these patients, however, prevents an assessment of the amount of time required for emergence of additional glycoprotein-specific autoantibodies. Given that all 145 assays positive for anti-GPIa/IIa antibodies in patients with ITP were “triple-positive” in that they also had anti-GPIIb/IIIa antibodies and anti-GPIb/IX antibodies, anti-GPIa/IIa seems to be the third autoantibody acquired of the 3 assessed by using this assay (Figures 2 and 4). Given the relation between number of autoantibodies present and disease severity, this scenario raises the question of whether anti-GPIa/IIa has distinct pathogenicity, which, to our knowledge, has not been explored in other studies.
Other studies have investigated the relation between serology and disease severity or prognosis, but none has explored the relation between number of glycoprotein-specific autoantibodies and disease severity. One study found that patients with positive indirect MAIPA test result had a significantly increased bleeding score, although the clinical relevance of this finding is questionable because only 15 of 108 patients in this study had a positive indirect MAIPA test result.17 Another study examined the likelihood of clinical worsening (need to start or modify therapy because of worsening thrombocytopenia or hospital admission) in 50 patients with ITP undergoing MACE testing; the study found an increased rate of clinical worsening in MACE-positive patients (18 of 25) vs MACE-negative patients (8 of 25).16 Although these findings are intriguing, the study was small, and only 25 patients had positive assays. A third study evaluating an immunobead assay in 282 patients with ITP found a higher degree of positivity for a given autoantibody in patients with the most severe disease; the authors defined their own disease severity categories for this assessment.14
In logistic regression models of treatment response, the presence of anti-GPIa/IIa antibodies was associated with a reduced likelihood of response to glucocorticoids, although we do not expect that this finding is clinically relevant. Although a majority (82%) of corticosteroid nonresponders were positive for GPIa/IIa, corticosteroid nonresponsiveness overall was rare (13% of patients), and 79% of patients with anti-GPIa/IIa did respond to glucocorticoids. We also found that the presence of anti-GPIb/IX antibodies did not predict nonresponse to glucocorticoids or IVIG, in contrast to 2 previously published retrospective studies. One study including 34 patients with anti-GPIb/IX antibodies examined response to glucocorticoids18 and the other including 66 patients with anti-GPIb/IX antibodies examined response to IVIG19 ; each study reported a significantly lower rate of treatment response in these patients. The current study evaluated the treatment response of 104 patients with anti-GPIb/IX antibodies receiving glucocorticoids and 59 such patients receiving IVIG. Some studies suggest distinct Fc-dependent vs Fc-independent effects of anti-GPIb/IX antibodies on platelets and associated implications regarding nonresponse to glucocorticoids or IVIG.35,36 It is possible that the disparity in detecting such a relation in our study compared with the previous studies is due to a difference in ability of the PA assays used to distinguish the differential effects of anti-GPIb/IX antibodies. Finally, although a possible relation between presence of certain glycoprotein-specific autoantibodies and nonresponse to thrombopoietin receptor agonists is possible given the action of platelet autoantibodies on megakaryocytes,37 we observed no such relation. These findings are consistent with a recent study that reported no relation between platelet autoantibodies and thrombopoietin levels in patients with ITP,38 which is in contrast with previous mouse studies suggesting such a relation.39 This area remains controversial; it is unclear whether PA specificity predicts relevant clinical disease characteristics (eg, platelet clearance patterns) in humans, as has been noted in studies of mice.40,41
The limitations of our study primarily derive from its retrospective nature. Because it was retrospective, we could not optimize the timing of PA assay performance. Although our center’s practice was to obtain PA testing on all suspected ITP patients over the duration of analyzed data in this study, we did not assess every patient with ITP at our center for whether PA testing was performed (the first step in our analysis was an assessment of circumstances surrounding performance of each assay, followed by assessment of patients in whom they were performed); thus, there could have been some unknown selection bias in a theoretical population of thrombocytopenic patients not receiving testing. Some patients were actively receiving therapy at the time of testing, which is potentially relevant because receipt of glucocorticoids and other immunosuppression has been shown to reduce levels of platelet autoantibodies.42-44 We suspect that any possible impact this may have had is minor, however, given that the sensitivity of positive PA testing for ITP was 90% in our study. In addition, assay timing is relevant in our assessment of PA impact on treatment response, given our findings describing evidence of epitope spreading; we selected a 6-month window after PA assay for evaluation of treatment response when the study was designed, and a shorter interval could have been used as well (although the number of patients included would have been more limited). It is noteworthy, however, that given the challenges in diagnosis of ITP and that final adjudication of an ITP diagnosis is often made over many years of caring for a patient, our retrospective approach may have had real advantages over a prospective one in terms of diagnostic validation. The PA testing used in this study did not assess for anti-GPV or anti-GPIV antibodies, which may be clinically relevant in ITP.45,46 Finally, as is a limitation of any study in this field, there is no gold standard for ITP diagnosis. We used ASH ITP diagnostic criteria and extended durations of clinical assessment to define ITP in our study, adhering to a very strict standard, but despite this approach, misdiagnosis of some patients is possible. Indeed, rates of ITP misdiagnosis can be significant even at major ITP referral centers.47
In conclusion, strict adherence to ASH 2011 clinical practice guidelines for ITP diagnosis and ISTH 2012 recommendations for PA testing was successful in considerably improving the sensitivity of glycoprotein-specific platelet autoantibodies for diagnosis of ITP. We found a predictive relation between a higher number of glycoprotein-specific autoantibodies and disease severity and did not find a clinically relevant relation between presence of a certain glycoprotein-specific autoantibody and response to glucocorticoids, IVIG, or thrombopoietin receptor agonists. We additionally found that anti-GPIa/IIa antibodies only occur in the setting of preexisting positivity for both anti-GPIIb/IIIa and anti-GPIb/IX antibodies, possibly due to a distinctive sequence of epitope spreading, and reported evidence for epitope spreading in ∼35% of patients with longitudinal serologic evaluation. Lastly, we found that serologic test results typically turn negative when a patient enters clinical remission.
Acknowledgments
The authors thank Brian Curtis, of the BloodCenter of Southeast Wisconsin, for his valuable consultation regarding the particulars of the PakAuto assay. They also thank Jason Baron, of Massachusetts General Hospital Pathology, for his assistance with the laboratory information system.
H.A.-S. is the recipient of the National Hemophilia Foundation-Shire Clinical Fellowship Award and Harvard KL2/Catalyst Medical Research Investigator Training Award, which provide salary support.
Authorship
Contribution: H.A.-S. designed the study, collected data, analyzed data, created tables and figures, and wrote and revised the manuscript; R.P.R., R.S.K.L., D. B. Smith, K.G., A.E.F., and D. B. Sykes collected data and revised the manuscript; and D.J.K. designed the study, critically revised the manuscript, and supervised the study.
Conflict-of-interest disclosure: H.A.-S. reports consultancies with Agios, Dova, and Moderna; and research funding from Agios and Dova. D.J.K. reports research funding from Protalex, Bristol-Myers Squibb, Rigel, Bioverativ, Agios, Syntimmune, Principia, and Alnylam; and consultancies with ONO, Pfizer, 3SBios, Eisai, GlaxoSmithKline, Genzyme, Shire, Amgen, Shionogi, Rigel, Syntimmune, MedImmune, Novartis, Alexion, Bioverativ, argenx, Zafgen, Fujifilm, Principia, Kyowa Kirin, Takeda, and Platelet Disorders Support Group. The remaining authors declare no competing financial interests.
Correspondence: Hanny Al-Samkari, Division of Hematology, Massachusetts General Hospital, Zero Emerson Pl, Suite 118, Room 112, Boston, MA 02114; e-mail: hal-samkari@mgh.harvard.edu.