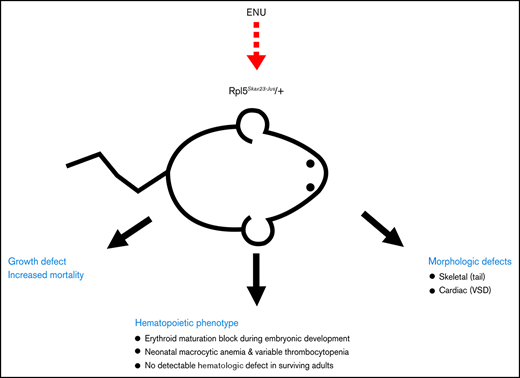
Key Points
This Rpl5Skax23-Jus/+ mouse model accurately recapitulates the variable anemia phenotype in DBA.

Abstract
Ribosome dysfunction is implicated in multiple abnormal developmental and disease states in humans. Heterozygous germline mutations in genes encoding ribosomal proteins are found in most individuals with Diamond-Blackfan anemia (DBA), whereas somatic mutations have been implicated in a variety of cancers and other disorders. Ribosomal protein-deficient animal models show variable phenotypes and penetrance, similar to human patients with DBA. In this study, we characterized a novel ENU mouse mutant (Skax23m1Jus) with growth and skeletal defects, cardiac malformations, and increased mortality. After genetic mapping and whole-exome sequencing, we identified an intronic Rpl5 mutation, which segregated with all affected mice. This mutation was associated with decreased ribosome generation, consistent with Rpl5 haploinsufficiency. Rpl5Skax23-Jus/+ animals had a profound delay in erythroid maturation and increased mortality at embryonic day (E) 12.5, which improved by E14.5. Surviving mutant animals had macrocytic anemia at birth, as well as evidence of ventricular septal defect (VSD). Surviving adult and aged mice exhibited no hematopoietic defect or VSD. We propose that this novel Rpl5Skax23-Jus/+ mutant mouse will be useful in studying the factors influencing the variable penetrance that is observed in DBA.
Introduction
The ribosomopathies, disorders that result from disruptions in various structural and functional elements of the ribosome, have a wide range of clinical manifestations.1-3 Among the ribosomopathies, Diamond-Blackfan anemia (DBA) is the disorder most often linked to anemia. However, many nonhematopoietic phenotypes, particularly in the skeleton and heart, are also found in DBA, as is an increased risk of developing malignancies at a young age.4-8 Inherited heterozygous mutations in genes encoding ribosomal proteins (RPs) lead to most cases of DBA.7 Several mechanisms for the development of anemia in DBA have been proposed, including nucleolar stress, heme toxicity, reduced translation of key erythroid differentiation proteins (eg, GATA1), and aberrant innate immune activation.9-15
DBA classically presents as severe macrocytic anemia before the first year of life, often requiring red blood cell transfusions.16 The penetrance is variable, with some patients developing failure of erythropoiesis before birth (ie, hydrops fetalis), whereas others exhibit no phenotype, even into adulthood.17 Some individuals can cycle through periods of anemia remission and relapse for unknown reasons. Remission is defined by the DBA registry as “an adequate hemoglobin level without any treatment, lasting 6 months, independent of prior therapy.”16 There are no known genetic modifiers, sex, or treatment differences between patients who attain remission and those who remain symptomatic.18 Approximately 20% of patients enter remission by age 25, but, for unknown reasons, relapses occur after viral illnesses or pregnancy and are more common in patients with RPL15 (eL15) variants.19,20 This dynamic anemia presentation has never been adequately represented in an animal model, and therefore the etiology of this variable presentation and spontaneous remission from anemia remains obscure.
The existing DBA animal models include several murine and zebrafish mutants that variably mimic some DBA disease phenotypes.21-23RPS19 (eS19), the first identified mutated gene in DBA, is also the most common, accounting for ∼25% of cases of DBA.7,24 Animal models of Rps19 haploinsufficiency exhibit a wide variety of manifestations, ranging from no phenotype to anemia, growth retardation, and variable decreases in other hematopoietic lineages (platelets and white blood cells [WBCs]).25-27 In our study, we focused on RPL5 (uL18), which is the second most commonly mutated RP gene in DBA. Patients with RPL5 mutations often have more severe presentations with increased presence of developmental defects and a lower chance of attaining spontaneous remission.7,28-31 Somatic mutations in RPL5 have also been linked to various malignancies.32 Two previous studies, one describing Rpl5 heterozygous mice and one describing Rpl5 depletion by short hairpin RNA, have been reported.33,34 Rpl5 haploinsufficient mice exhibited soft tissue sarcoma, but no erythroid defect, whereas mice with Rpl5 depletion by shRNA demonstrated mild anemia. Zebrafish with Rpl5 knockdown using an antisense morpholino oligonucleotide have also been reported to exhibit various developmental and hematopoietic defects.35
In this report, we generate and describe what, to the best of our knowledge, is the first murine Rpl5 mutant mouse model of DBA to demonstrate variably penetrant anemia. We propose that these mice mimic this aspect of the human disease and will be instructive in future studies to better understand this elusive phenomenon.
Materials and methods
Generation of the Skax23m1Jus mouse mutant
The Skax23m1Jus mouse mutant (MGI: 3046775), with a kinked tail and small size, was generated and identified as part of a chromosome 11 balancer mutagenesis screening at the Mouse Mutagenesis and Phenotyping Center for Developmental Defects at Baylor College of Medicine (Houston, TX). The study design of this mutagenesis screening has been described in detail elsewhere.36 In brief, mice heterozygous for the balancer chromosome 11 were crossed with ENU-treated C57BL/6J mice. To generate individual lines of mice that inherit the same mutation, G1 animals were crossed with the generated G3 offspring, which were analyzed for linkage to the chromosome 11 balancer. Skax23m1Jus was identified as a dominant mouse mutant with a kinked tail and a small size that failed to segregate to chromosome 11. Skax23m1Jus G3 mutants were outcrossed onto a 129S6/SvEvTac background, and the resulting N1 progeny were archived as frozen sperm and subsequently recovered at The Jackson Laboratory (https://www.jax.org/jax-mice-and-services) by in vitro fertilization of C57BL/6J oocytes. The Skax23m1Jus mutant stock was generated and maintained by matings of presumed heterozygous mice that displayed the phenotype to 129S6/SvEv mice. For genetic mapping studies, Skax23m1Jus/+ mutants with a kinked tail and small size were backcrossed to 129S6/SvEv mice for 5 consecutive generations. Mice were examined macroscopically for the presence of a severely kinked tail and small size (Figure 1A-B). The protocols relevant to this study were approved by the Institutional Committee for Animal Care in Research at Baylor College of Medicine, the Research Center of Sainte Justine Hospital (Western Michigan University School of Medicine), and the University of Michigan.
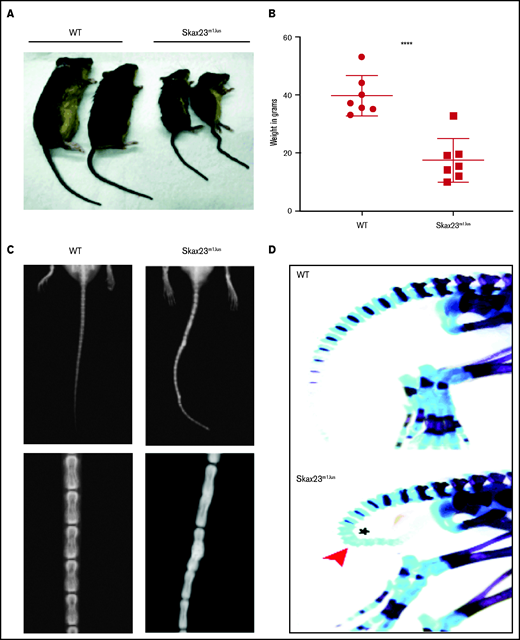
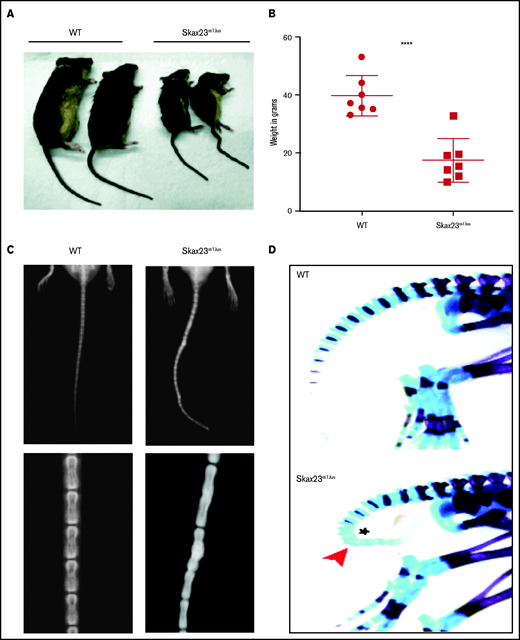
Skax23m1Jus mice grow poorly and exhibit a kinky tail defect caused by delayed endochondral ossification. (A) The Skax23m1Jus mouse mutant was generated and identified as part of the chromosome 11 balancer mutagenesis screening at the Mouse Mutagenesis and Phenotyping Center for Developmental Defects at Baylor College of Medicine in Houston (http://www.mouse-genome.bcm.tmc.edu). Skax23m1Jus was identified as a dominant mutant with a kinky tail phenotype and poor growth. (B) Mutant adult mice (n = 7) were significantly smaller than WT (n = 7) ****P < .0001, WT vs Skax23m1Jus mice. (C) Radiographic analyses of adult mice show abnormal fusion and hypertrophy of tail bone joints in Skax23m1Jus mice when compared with WT littermates (low-power view, 1.5-2×; high-power view, 3×). (D) Wholemount staining of newborn mice using Alcian blue (for cartilage) or Alizarin red (for bone) was performed. Skax23m1Jus newborn mice had kinky tails (arrowhead) with delayed endochondral ossification (asterisk).
Skax23m1Jus mice grow poorly and exhibit a kinky tail defect caused by delayed endochondral ossification. (A) The Skax23m1Jus mouse mutant was generated and identified as part of the chromosome 11 balancer mutagenesis screening at the Mouse Mutagenesis and Phenotyping Center for Developmental Defects at Baylor College of Medicine in Houston (http://www.mouse-genome.bcm.tmc.edu). Skax23m1Jus was identified as a dominant mutant with a kinky tail phenotype and poor growth. (B) Mutant adult mice (n = 7) were significantly smaller than WT (n = 7) ****P < .0001, WT vs Skax23m1Jus mice. (C) Radiographic analyses of adult mice show abnormal fusion and hypertrophy of tail bone joints in Skax23m1Jus mice when compared with WT littermates (low-power view, 1.5-2×; high-power view, 3×). (D) Wholemount staining of newborn mice using Alcian blue (for cartilage) or Alizarin red (for bone) was performed. Skax23m1Jus newborn mice had kinky tails (arrowhead) with delayed endochondral ossification (asterisk).
Genetic mapping
For genetic mapping studies, 21 mice with a kinked tail phenotype from 4 backcrosses (4 N1F1, 5 N2F1, 4 N3F1, and 8 N4F1) were genotyped with 180 single-nucleotide polymorphisms (SNPs) distributed randomly across the genome. The markers were informative for both C57BL/6J and 129S6/SvEv strains, and the average intermarker distance was 15 Mb. Of these, 26 markers failed to genotype and 5 were not informative in both strains, resulting in 149 markers for genotyping data analyses. Candidate regions were prioritized by the percentage of C57BL/6J heterozygosity in affected mice. Fine mapping of the candidate region on chromosome 5 was performed by genotyping 20 affected mice included in the original whole-genome scan (4 N1F1, 5 N2F1, 3 N3F1, and 8 N4F1) and 10 affected mice obtained from a fifth backcross to 129S6/SvEv animals (10 N5F1) with 14 SNPs selected from within this region.
Whole-exome sequencing
Whole exome sequencing (WES) was conducted in 2 affected mice (1 N1F1 and 1 N1F4) and 1 unaffected 129S6/SvEv mouse. WES capture was performed with the Sureselect Mouse All Exon 50 Mb (Agilent Technologies). Paired-end sequencing was performed on an AB Hiseq 2000. Raw data were aligned to the C57BL/6 NCBI37/mm9 genome reference build on the Burrows-Wheeler Aligner. Duplicate reads were removed, remaining reads were realigned locally, and variants were called, using GATK 2.6.4. Variant annotation was performed with Annovar (http://www.openbioinformatics.org/annovar/). This process resulted in variant coverage 15 times the average. Mutations mapping to the candidate region on chromosome 5 that were heterozygous in both affected mice and absent from the C57BL/6 reference genome and from the 129S6/SvEv mice were prioritized for validation by Sanger sequencing. Genotyping protocols and polymerase chain reaction (PCR) primers are described in detail in supplemental Methods.
Cell culture
Spleens were isolated, crushed to generate a single-cell suspension, and strained, followed by red blood cell lysis performed using ACK Lysing Buffer (Lonza). Total spleen cells were washed and resuspended in complete medium (RPMI, 10% fetal bovine serum, 2 mM l-glutamine, 1% penicillin/streptomycin, 50 μM 2-mercaptoethanol, and 10 mM HEPES) at a concentration of 2 × 106 cells/ml. LPS was added to the culture medium at 10 μg/mL for 24 to 72 hours. The number and viability of cells were assessed with an ADAM MC Auto Cell Counter.
Western blot analysis
Total spleen cells were harvested at 48 hours of culture, and protein extracts were prepared for western blots, as previously described, using the antibodies listed in supplemental Table 1.37
Polysome profiling
Total spleen cells grown in complete medium supplemented with LPS (10 μg/mL) for 36 hours were processed for polysome profiling, as previously described.31
Statistics
The Student t test in GraphPad Prism 9 was used to determine statistical significance. Error bars signify the standard error of the mean. Statistics for Mendelian ratios were calculated with Chi squared (χ2)analysis.
Results
Description of the Skax23m1Jus kinky tail dominant mutant
Skax23m1Jus mutant mice can be easily identified at birth because of their small size and kinked tail (Figure 1A; supplemental Figure 1). Mice that survived the neonatal period continued to grow poorly and surviving adult mutant mice were about half the weight of wild-type (WT) mice (Figure 1B). Examination of the skeleton via radiography of adult mice showed abnormal hypertrophy and fusion of tail bone joints (Figure 1C). Additional skeletal analysis of newborn Skax23m1Jus mice demonstrated reduced skull and bone lengths, although only femur lengths were significantly decreased (supplemental Figure 1B-C). However, all newborn Skax23m1Jus mice exhibited kinky tails with no evidence of endochondral ossification in the lower caudal vertebrae (Figure 1D).
Identification of Rpl5 (uL18) as the gene defective in Skax23m1Jus
To map the genetic defect in Skax23m1Jus mice, those with a kinky tail phenotype were subjected to 5 consecutive backcrosses to 129S6/SvEvTac animals. A whole-genome scan using 149 SNPs informative for both C57BL/6J and 129S6/SvEv was conducted on 21 N1F1, N2F1, N3F1, and N4F1 mice with a kinky tail phenotype. Because the ENU-induced mutation was generated on the C57BL/6J background, regions with the highest percentage of heterozygosity for C57BL/6J were prioritized. This analysis identified a ∼41-Mb region on chromosome 5 that had the highest percentage of heterozygosity in C57BL/6J animals (supplemental Figure 2A). Analysis of 13 SNPs from the whole-genome scan that covered this region identified a haplotype consisting of 3 SNPs (rs13478451, rs32103915, and rs29534493) that was heterozygous in the C57BL/6J genome in all 21 mice analyzed (supplemental Figure 2B). Fine mapping within this candidate region was performed by genotyping 20 affected mice included in the original whole-genome scan and 10 additional affected mice obtained from a fifth backcross with 14 SNPs selected from this region. This fine mapping reduced this region to 11 Mb from 107112543 bp (rs13478444) to 118405516 bp (rs13478483) on the NCBI37/mm9 assembly (supplemental Figure 2C).
To identify the gene defective in Skax23m1Jus mice, we conducted WES on 2 affected mice and 1 unaffected 129S6/SvEv mouse. Mutations present within the candidate region on chromosome 5 in the 2 affected mice and absent from both parental strains and the reference genome were prioritized for study, and 4 mutations obeying these criteria were identified (data not shown). After validation by Sanger sequencing, only 1 intronic mutation in Rpl5 (aka uL18), c.3 + 6C>T, was confirmed (Figure 2A).20 The RPL5 gene is mutated in some forms of DBA and a different RP mutation (RPS7 or eS7) found in patients with DBA produces a kinked tail and a smaller size in another mouse model, making this mutation an excellent candidate.38 The Rpl5c.3 + 6T forms part of a highly conserved and frequent intron consensus sequence.39 It is absolutely conserved across the 9 species analyzed (supplemental Table 3) and is predicted to be probably pathogenic, according to the Transcript-Inferred Pathogenicity Score of 0.898).40 For genotype-phenotype studies, genotyping was performed on 228 mice with straight tails and 95 with kinky tails. The c.3 + 6C>T heterozygous mutation was absent from all mice with straight tails and segregated with the kinky tail phenotype in all 95 mice analyzed, indicating complete penetrance. No homozygous mice bearing this variant were identified, suggesting embryonic lethality in the homozygous state. This variant was also absent in 28 other inbred strains (data not shown), confirming that it is not a rare variant and that it is specific to the phenotype.
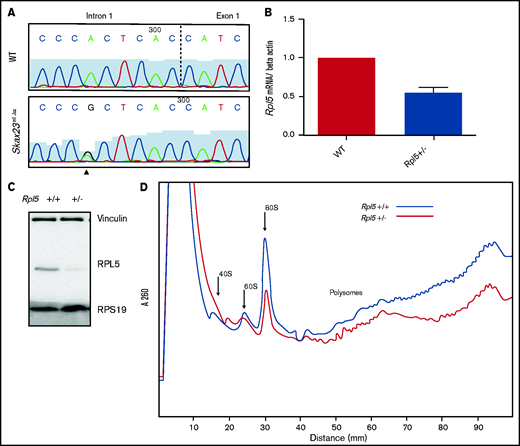
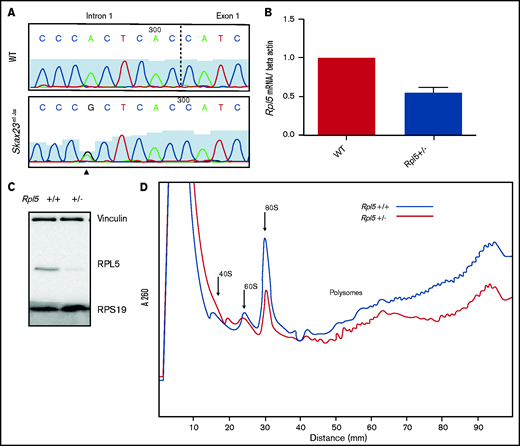
Rpl5 intronic mutation identified in Skax23m1Jus mice leads to decreased mRNA, protein levels, and polysome defects. (A) Chromatogram of reverse-strand partial sequences of exon 1 and intron 1 of Rpl5 showing the heterozygous mutation (triangle) at the sixth nucleotide in intron 1. The exon-intron junction is indicated by the dotted line. (B) RT-qPCR showed a ∼50% reduction in Rpl5 mRNA in the Rpl5+/− mice when compared with WT ** P< .01. (C) Western blot analysis with vinculin and RPS19 used as loading controls showed reduced levels of RPL5 protein in Rpl5+/− mice when compared with WT. (D) Results from 3 separate experiments indicate Rpl5+/− mice have reduced 60S subunits, 80S ribosomes, and polysomes, consistent with Rpl5 haploinsufficiency.
Rpl5 intronic mutation identified in Skax23m1Jus mice leads to decreased mRNA, protein levels, and polysome defects. (A) Chromatogram of reverse-strand partial sequences of exon 1 and intron 1 of Rpl5 showing the heterozygous mutation (triangle) at the sixth nucleotide in intron 1. The exon-intron junction is indicated by the dotted line. (B) RT-qPCR showed a ∼50% reduction in Rpl5 mRNA in the Rpl5+/− mice when compared with WT ** P< .01. (C) Western blot analysis with vinculin and RPS19 used as loading controls showed reduced levels of RPL5 protein in Rpl5+/− mice when compared with WT. (D) Results from 3 separate experiments indicate Rpl5+/− mice have reduced 60S subunits, 80S ribosomes, and polysomes, consistent with Rpl5 haploinsufficiency.
The c.3 + 6C>T intronic mutation results in Rpl5 haploinsufficiency
Quantitative reverse transcription-PCR analysis demonstrated a ∼50% reduction in Rpl5 mRNA in Skax23m1Jus mutants compared with that in WT mice (Figure 2B). Western blot analysis (in triplicate) showed reduced RPL5 protein in mutant mice (Figure 2C). Analysis of 3 separate polysome experiments demonstrated reduced 60S subunit, 80S ribosomes, and polysomes, consistent with Rpl5 haploinsufficiency (Figure 2D). We refer to the Rpl5Skax23-Jus/+ mutants as Rpl5+/− throughout the article.
Neonatal Rpl5+/− mice have macrocytic anemia, cardiac defect, and increased mortality
Newborn mice from WT × Rpl5+/− matings were genotyped (14 litters; n = 58). The percentage of Rpl5+/− offspring was significantly lower than expected by Mendelian predictions (Table 1). Three litters were observed over time to determine the percentage of newborn pups that died after birth and before weaning, (WT, n = 8; Rpl5+/−, n = 5). Three mutant pups (60%) died after birth: 2 on day 1 and 1 on day 21 (Table 1). The pup that died 21 days after birth was found to have severe pancytopenia (WBC count, 1 × 109/L; hemoglobin (Hb), 3.5 g/dL; platelets, 11000 × 109/L). Two pups from additional litters were runted and appeared lethargic and therefore had to be euthanized 14 to 15 days after birth. Complete blood count analyses were performed at the time of euthanasia and demonstrated Hb 6.6 and 7.3 g/dL, which were significantly lower than the Hb of their WT littermate control (Hb, 14.7g/dL). One of the 2 latter pups euthanized also had thrombocytopenia with a platelet count of 14000, whereas the other had normal platelet counts.
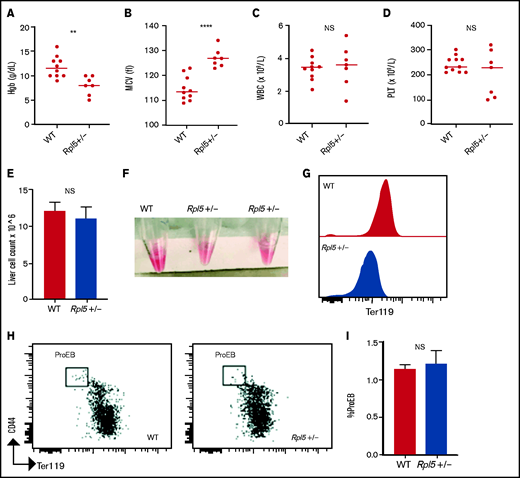
We next analyzed complete blood counts of newborn mice at days 1 to 3 after birth (WT, n = 10; Rpl5+/−, n = 7) and found that Rpl5+/− mice had macrocytic anemia (Figures 3A-B) but normal WBC and platelet counts (Figures 3C-D). Newborn Rpl5+/− livers had normal cellularity (Figure 3E), but cell suspensions from these livers appeared paler compared with those from WT livers. In addition, the Ter119 mean fluorescence intensity, as determined by flow cytometry, was reduced in Rpl5+/− compared with that in WT livers (Figures 3F-G). Further flow analysis of erythroid differentiation showed no significant difference in the proerythroblast (CD44hi Ter119low) population between mutant and WT livers (WT, n = 3; Rpl5+/−, n = 3; Figure 3H-I).41
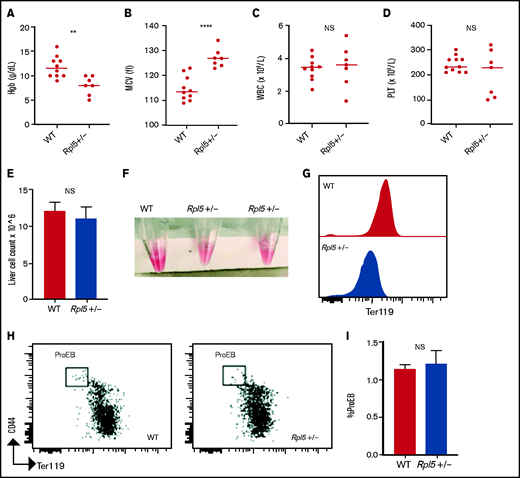
Rpl5+/− newborn mice have macrocytic anemia. Newborn mice (days 1-3) were euthanized and the blood counts analyzed (WT, n = 10; Rpl5+/−, n = 7). Rpl5+/− mice exhibited evidence of anemia (A) with significantly lower hemoglobin (Hb) levels and (B) macrocytosis, as demonstrated by an elevated mean corpuscular volume (MCV). There was no significant (NS) difference in WBC (C) or platelet (PLT) counts (D) between Rpl5+/− and WT mice. The horizontal line represents the median value for each group. **P < .01; ****P < .0001. Newborn livers (days 1 and 2) were isolated, and flow cytometry was performed to analyze erythroid differentiation. There was no difference in liver cellularity (E) but all Rpl5+/− mice had paler livers (F) with decreased Ter119 mean fluorescence intensity (G). (H-I) There was no difference in the number of proerythroblasts (WT, n = 3; Rpl5+/−, n = 3).
Rpl5+/− newborn mice have macrocytic anemia. Newborn mice (days 1-3) were euthanized and the blood counts analyzed (WT, n = 10; Rpl5+/−, n = 7). Rpl5+/− mice exhibited evidence of anemia (A) with significantly lower hemoglobin (Hb) levels and (B) macrocytosis, as demonstrated by an elevated mean corpuscular volume (MCV). There was no significant (NS) difference in WBC (C) or platelet (PLT) counts (D) between Rpl5+/− and WT mice. The horizontal line represents the median value for each group. **P < .01; ****P < .0001. Newborn livers (days 1 and 2) were isolated, and flow cytometry was performed to analyze erythroid differentiation. There was no difference in liver cellularity (E) but all Rpl5+/− mice had paler livers (F) with decreased Ter119 mean fluorescence intensity (G). (H-I) There was no difference in the number of proerythroblasts (WT, n = 3; Rpl5+/−, n = 3).
Newborn mice were also analyzed for the presence of craniofacial abnormalities (common in patients with DBA who have heterozygous RPL5 mutations) and cardiac defects. Histologically, of the 6 Rpl5+/− mice evaluated, 5 showed evidence of VSD (supplemental Figure 3A), and 1 had a double-outlet right ventricle in addition to VSD ( data not shown). There was no evidence of VSD by histology in surviving adult Rpl5+/− mice (WT, n = 2; mutant, n = 7). These results suggest that the presence of a VSD was associated with increased risk of death of Rpl5+/− newborn animals. There was no evidence of a craniofacial defect by micro computed tomography or histology in Rpl5+/− mice (supplemental Figure 3B-C).42
Rpl5+/− mice exhibit a severe erythroid maturation defect at E12.5
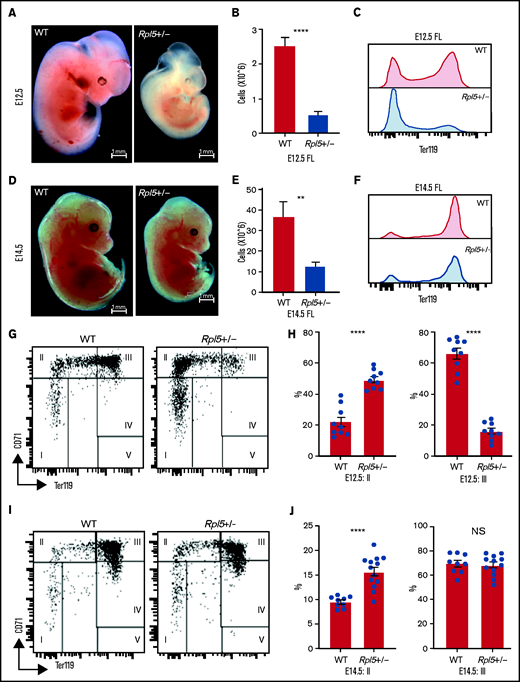
To determine whether the onset of the anemia in Rpl5+/− mice was before or after birth, we analyzed embryonic day (E) 12.5 and E14.5 embryos generated from timed matings of WT females x Rpl5+/− males. Genotyping of 5 litters at E12.5 (n = 38) revealed 28 live embryos with no significant difference between observed and expected genotypes (Table 2). However, 10 dead embryos were observed at different stages of reabsorption, all of which had the Rpl5+/− genotype, indicating that death in some mutants occurred before E12.5 (Table 2). E12.5 Rpl5+/− embryos were significantly growth retarded and pale, with 2 mutant embryos showing signs of intracranial bleeding (Figure 4A; supplemental Figure 4: embryos 20-5 and 20-6). Dead embryos were found at both E12.5 (supplemental Figure 4B: embryo 3-5) and E14.5 (supplemental Figure 5: embryo 19-3). At E12.5, compared with WT fetal livers (FLs), Rpl5+/− FLs had a significantly lower number of total live cells and, notably, a smaller proportion of Ter119+ cells. (Figure 4B-C). Mutant embryos at E14.5 had less prominent growth retardation and pallor compared with E12.5 Rpl5+/− embryos (Figure 4D; supplemental Figure 5). At E14.5, the FL cellularity remained significantly diminished in Rpl5+/− embryos; however, the percentage of Ter119+ cells had clearly increased (compared with E12.5), although it remained lower than normal (Figures 4E-F).
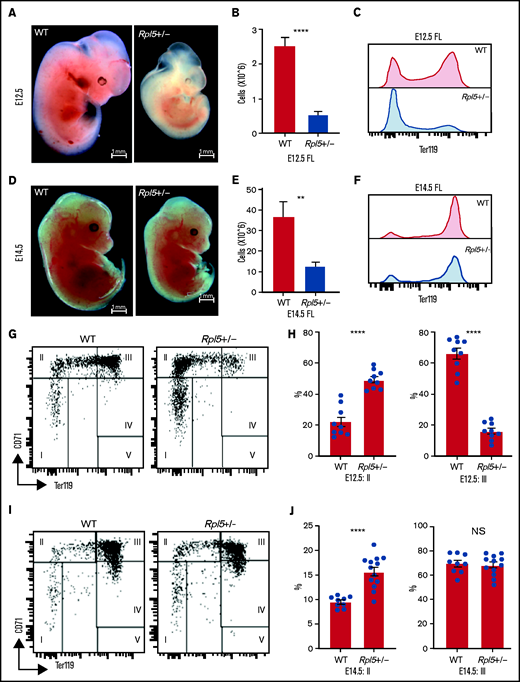
Rpl5+/− mice have a severe block in erythroid differentiation at E12.5 that improves by E14.5. (A) Timed WT x Rpl5+/− matings were set up to collect E12.5 and E14.5 embryos. E12.5 Rpl5+/− embryos were generally growth retarded and pale. (B) Mutant E12.5 embryos displayed a marked reduction in FL cellularity, with fewer FL Ter119+ cells (C). (D) E14.5 mutant embryos had improved growth, pallor, and FL cellularity (E) and increased Ter119+ cells (F). (G) Analysis of red cell differentiation by CD71 and Ter119 showed a significant delay in the transition from population 2 to 3 in the FL of Rpl5+/− mice at E12.5 (quantified in panel H). A similar analysis conducted on E14.5 FL cells showed an improvement in the E12.5 differentiation block (I), with no difference in population 3 at this stage (J). E12.5 WT n = 9, Rpl5+/− n = 9 (3 separate litters); E14.5 WT n = 9, Rpl5+/− n = 12 (4 separate litters). **P < .01; ****P < .0001, WT vs Rpl5+/− mice.
Rpl5+/− mice have a severe block in erythroid differentiation at E12.5 that improves by E14.5. (A) Timed WT x Rpl5+/− matings were set up to collect E12.5 and E14.5 embryos. E12.5 Rpl5+/− embryos were generally growth retarded and pale. (B) Mutant E12.5 embryos displayed a marked reduction in FL cellularity, with fewer FL Ter119+ cells (C). (D) E14.5 mutant embryos had improved growth, pallor, and FL cellularity (E) and increased Ter119+ cells (F). (G) Analysis of red cell differentiation by CD71 and Ter119 showed a significant delay in the transition from population 2 to 3 in the FL of Rpl5+/− mice at E12.5 (quantified in panel H). A similar analysis conducted on E14.5 FL cells showed an improvement in the E12.5 differentiation block (I), with no difference in population 3 at this stage (J). E12.5 WT n = 9, Rpl5+/− n = 9 (3 separate litters); E14.5 WT n = 9, Rpl5+/− n = 12 (4 separate litters). **P < .01; ****P < .0001, WT vs Rpl5+/− mice.
Further analysis of FL erythropoiesis was performed at E12.5 and E14.5 by examining CD71 and Ter119 expression.43,44 At E12.5, erythroid cells at stage 2 of differentiation (CD71+/Ter119−/low) were increased in Rpl5+/− FL, whereas erythroid cells at stage 3 of differentiation (CD71+/Ter119+) were reduced, suggesting a delay in erythroid differentiation resulting from Rpl5 haploinsufficiency (Figures 4G-H). Additional analysis using CD44 and Ter119 showed a significant (threefold) increase in the proerythroblast (proEB) population in mutant animals, again indicative of an erythroid differentiation block (supplemental Figure 6A-B; E12.5 WT, n = 9; Rpl5+/−, n = 9; 3 litters). A similar analysis of E14.5 FL cells showed an improvement in the erythroid differentiation block, with no difference in the percentage of erythroid cells at stage 3 of differentiation between Rpl5+/− and WT FL (Figures 4I-J). Additional analysis demonstrated that the proportion of proEB in the E14.5 livers remained elevated (albeit improved) at 1.5-fold compared with WT livers (supplemental Figure 6C-D; E14.5: WT, n = 9; Rpl5+/−, n = 12; 4 litters).
E12.5 FL cells were next analyzed to assess whether there was evidence of increased apoptosis of Rpl5+/−erythroid cells.45 Populations 2 and 3 were analyzed for apoptosis by staining with annexin V and DAPI (supplemental Figure 7). There was no significant difference in the percentage of population 2 or 3 erythroid cells undergoing early or late apoptosis between Rpl5+/− and WT embryos (WT, n = 6; Rpl5+/−, n = 6; 2 litters).
Adult Rpl5+/− mice exhibit no anemia or hematopoietic defects
Analysis of Rpl5+/− mice at 7 weeks of age showed no evidence of anemia or blood count abnormalities, when compared with WT controls (supplemental Figure 8A-F; WT, n = 10; Rpl5+/−, n = 10). A similar analysis of aged mice also did not demonstrate any blood count abnormality (supplemental Figure 8G-L; WT, n = 11; Rpl5+/−, n = 7). The bone marrow (BM) of 5-month-old mice was analyzed by flow cytometry for red cell markers (Ter119, CD44) after exclusion of nonerythroid cells (CD45+, Gr-1, CD11b, and CD3) and dead cells. No significant difference in Ter119 mean fluorescence intensity was found between the BM of Rpl5+/− and WT animals (supplemental Figure 9A). There was also no significant difference in the percentage of the proerythroblast population between Rpl5+/− and WT animals (supplemental Figure 9B-C; WT, n = 3; Rpl5+/−, n = 3). Further flow analysis of the BMs of adult mice did not reveal significant differences in BM cellularity and the percentage of early stem/progenitor cells, mature myeloid cells, or mature lymphocyte subpopulations (supplemental Figure 10).46,47
Discussion
In this study, we identified and characterized Skax23m1Jus mice with a novel intronic ENU-induced mutation in the Rpl5 gene, affecting the sixth nucleotide in intron 1 (c.3 + 6C>T mutation). This mutation maps to the candidate region on chromosome 5 and segregates with the kinky tail phenotype with 100% penetrance. It was absent from the parental strains and 28 other inbred strains, suggesting that it was the cause of the phenotype. Our best hypothesis at present is that the c.3 + 6C>T mutation affects splicing, possibly causing intron 1 retention and leading to nonsense-mediated mRNA decay. Alternatively, it may affect intronic transcription regulatory elements of an unknown nature. Additional in vitro studies are needed to elucidate the mechanism by which this mutation affects Rpl5 mRNA and protein expression levels. This mutation leads to a stable decrease in Rpl5 mRNA and protein levels and to polysome defects, making it distinct from the other published Rpl5 zebrafish and mouse models. The morphological defects (small size and kinky tail) in this model mouse are similar to the previously described Rps7 mutant mouse and correspond to the increased incidence of congenital abnormalities in patients with RPL5 variants.7,28,29,38
Our initial characterization of adult Rpl5+/− mice demonstrated a surprising lack of anemia, without detectable defects in stem and progenitor cells. However, the observation of increased mortality of newborn mutant pups led to the discovery of macrocytic anemia at birth that worsened in some pups, possibly explaining their demise. However, the anemia observed in Rpl5+/− mice at birth resolved completely in adult animals that survived to 7 weeks of age. Examination of FL cells revealed a more severe erythroid differentiation defect at E12.5 that slowly resolved by E14.5 in the surviving animals. These variable observations, seen at different developmental time points in Rpl5+/− mice, were reminiscent of the variable anemia and spontaneous unexplained remissions that can occur in some patients with DBA. The mechanism underlying this phenomenon is unknown, but environmental and/or cell-extrinsic factors such as elevated reactive oxygen species have been proposed to play a role.9 As the inbred mice examined in this study have been backcrossed >7 times with no change in phenotype (now >98% 129/SvJ congenic by Jax genome scan), genetic polymorphisms are unlikely to influence this variably penetrant anemia. The mice are also housed in a specific-pathogen–free environment and monitored frequently for invasive pathogens, so it is equally unlikely that viral or other infections have contributed to the anemia. Furthermore, as the variation in presentation occurs within the same litter in the same cage (ie, some mutants succumb before weaning, whereas other mutant littermates survive to adulthood), the variability in phenotype is unlikely to be related to major environmental changes, but this certainly requires further study. In our current work, we did not examine whether somatic reversion of Rpl5 occurs and leads to selection of more fit clones that can rescue the hematopoietic defect, which has been described previously in both DBA and Fanconi anemia.48-52
A question raised by this DBA model is whether the variable anemia phenotype is strain dependent. Our mice are currently maintained on a 129/SvJ background, but we have not yet studied this erythroid phenotype in other common inbred backgrounds, such as C57BL/6. Also, it is unclear whether the variable anemia observed in our model is also present in other DBA animal models resulting from mutations in other DBA genes or this phenotype is specific to Rpl5 or to this particular Rpl5 intronic mutation. Of note, variants in noncoding and splice site regions of genes encoding RPs have been reported in patients with DBA.7,53,54 As the variation in phenotype is present in patients with DBA of all genotypes, it is unlikely that the findings reported here are specific for Rpl5, but determining the specificity requires further study.
The nonerythroid (skeletal or cardiac) manifestations of DBA have not been as extensively studied as the erythroid phenotype. We showed that the kinky tail in Rpl5+/− mutant mice is caused by delayed endochondral ossification. Why RP haploinsufficiency affects certain tissues and not others is an area that requires further study but is possibly explained by selectively decreased translation of vulnerable mRNAs. In a previous study describing a Rpl38 haploinsufficient mouse, skeletal defects were concluded to be related to decreased translation of specific Hox mRNAs, whereas global translation appeared unaffected.55 As the RPL5 genotype is strongly associated with craniofacial malformations, we initially extensively analyzed mice for such defects that could result in poor feeding and growth retardation. However, we were unable to find conclusive evidence of craniofacial malformations by histology or by micro computed tomography. The presence of the cardiac defects (including VSD) was not surprising, as a subset of patients with DBA also have such defects. In addition, neural crest cells have been shown to play a major role both in both craniofacial and cardiac morphogenesis, and neural crest defects can lead to septal defects such as VSD.56-58 Moreover, several genes involved in ribosome biogenesis are necessary for neural crest cell proliferation and survival, and defects in these genes result in severe craniofacial defects.59
The observation reported here that most newborn Rpl5+/− mice have VSDs, whereas surviving mice do not, requires some consideration. It is possible that the cardiac progenitor cells are subject to the same process that creates a range of severity in the erythroid lineage. Therefore, the presence of a VSD may be a way to track those mice that are on a trajectory for total erythroid failure. The finding of variable thrombocytopenia is consistent with the phenotype in patients with DBA and also warrants further study.60,61 We therefore propose that this Rpl5Ska23-Jus/+ mutant mouse model can be used to study the underlying mechanisms leading to variable penetrance and possibly remission of anemia in DBA.
We have described a murine model of DBA that appears to recapitulate several features of the human disease that were not found in other previously described mouse models, including the variable anemia. To date, to our knowledge, no other animal models have captured this dynamic erythroid presentation, which is a key feature of human DBA. We also propose using VSD as a possible way to track those mice whose trajectory may lead to a more severe erythroid defect so that the underlying mechanism(s) may be more fully explored. The results of this study suggest that the variation in penetrance in DBA may be stochastic without the influence of genetic modifiers or significant environmental influences. Future studies aimed at improving our understanding of the mechanism of this variable penetrance may reveal novel biological pathways relevant to RP deficiency states. This novel Rpl5Ska23-Jus/+ mutant strain presents a unique opportunity to study fundamental mechanisms involved in normal and disordered erythropoiesis and should be useful in testing new therapeutic approaches .
Acknowledgments
The authors thank Michael Clemente in the core flow cytometry facility at Western Michigan University School of Medicine; the members of the vivarium at Western Michigan University School of Medicine and the University of Michigan; the Physiology Phenotyping Core at the University of Michigan; Evan Keller for the use of radiography equipment; Jinlu Dai for assistance with imaging; and Kim-Chew Lim, Sowmya Balasubramanian, Mark Chiang, Jordan Shavit, Emily Philipp-Petrick, Selest Nashef, Stephan Owens, and the members of the Rothstein laboratories for support and assistance.
This work was supported by M-cubed, a research seed funding program for faculty at the University of Michigan (S.A.S., V.K., R.F., and R.K.); Canadian Institutes of Health Research 130411 (Z.K.); National Institutes of Health, National Heart, Lung and Blood Institute grants R01 HL148333 (R.K.) and T32 HL007622 (M.J.), National Institute of Child Health and Human Development U01 HD39372 (M.J.J.), and National Cancer Institute grant R01 CA115503 (M.J.J.) and the National Cancer Institute grant P30CA046592 grant to The University of Michigan Rogel Cancer Center (R.K.).
Authorship
Contribution: F.F.M., G.-A.S., C.P., and E.S. performed the experiments, interpreted the data and edited the manuscript; L.Y., P.L., A.L., M.-C.G., M.J., and M.J.J. designed and performed the experiments, interpreted the data, and edited the manuscript; Y.W., G.M., R.K., Q.L., R.F., T.L.R., J.D.E., and V.K. designed the experiments, interpreted the data, and edited the manuscript; Z.K. and S.A.S. designed and performed the experiments, interpreted the data, and wrote and edited the manuscript.
Conflicts of interest disclosure: J.D.E. has served as a consultant for, and is a shareholder in Imago Biosciences. The remaining authors declare no competing financial interests.
Correspondence: Sharon A. Singh, University of Michigan, 1500 E. Medical Center Dr, SPC 5718, Ann Arbor, MI; e-mail: singhsa@med.umich.edu.