

Key Points
S100A9 overexpression promotes gilteritinib resistance in FLT3-ITD+ AML cells.
Gilteritinib-induced upregulation of S100A9 is mediated through loss of BCL6 enrichment at the S100A9 promoter.
Abstract
Drug resistance and relapse are common challenges in acute myeloid leukemia (AML), particularly in an aggressive subset bearing internal tandem duplications (ITDs) of the FLT3 receptor (FLT3-ITD+). The tyrosine kinase inhibitor gilteritinib is approved for the treatment of relapse/refractory AML with FLT3 mutations, yet resistance to gilteritinib remains a clinical concern, and the underlying mechanisms remain incompletely understood. Using transcriptomic analyses and functional validation studies, we identified the calcium-binding proteins S100A8 and S100A9 (S100A8/A9) as contributors to gilteritinib resistance in FLT3-ITD+ AML. Exposure of FLT3-ITD+ AML cells to gilteritinib increased S100A8/A9 expression in vivo and in vitro and decreased free calcium levels, and genetic manipulation of S100A9 was associated with altered sensitivity to gilteritinib. Using a transcription factor screen, we identified the transcriptional corepressor BCL6, as a regulator of S100A9 expression and found that gilteritinib decreased BCL6 binding to the S100A9 promoter, thereby increasing S100A9 expression. Furthermore, pharmacological inhibition of BCL6 accelerated the growth rate of gilteritinib-resistant FLT3-ITD+ AML cells, suggesting that S100A9 is a functional target of BCL6. These findings shed light on mechanisms of resistance to gilteritinib through regulation of a target that can be therapeutically exploited to enhance the antileukemic effects of gilteritinib.
Introduction
An aggressive subset of acute myeloid leukemia (AML)–bearing internal tandem duplications (ITDs) of the FLT3 receptor (FLT3-ITD) occurs in ∼30% of adult patients and is associated with poor prognosis and low overall survival.1-3 Gilteritinib, a tyrosine kinase inhibitor of FLT3, AXL, and ALK,4,5 has been approved for the treatment of FLT3-mutated AML,6 yet drug resistance remains a clinical concern. We sought to understand gilteritinib-resistance mechanisms in FLT3-ITD+ AML, and identified the calcium-binding protein S100A9 as a potential therapeutic target mediating resistance.
S100A8 and S100A9 are constitutively expressed in myeloid cells and have roles in cell differentiation, autophagy, and apoptosis.7-10 Overexpression of S100A8 in AML is associated with poor survival,11 and altered expression of S100A8 and/or S100A9 confers resistance or reduced sensitivity to agents in AML therapy, including venetoclax, doxorubicin, vincristine, and etoposide,12-14 but they have yet to be studied in the context of gilteritinib resistance. Although changes in autophagy and apoptosis may play a role in drug response,12-14 the exact mechanisms of S100A8/A9 in drug resistance remain poorly understood. Furthermore, the transcription factors that govern S100A8/A9 expression in AML remain unknown. We identified a connection of gilteritinib-induced S100A9 expression with BCL6, a transcriptional corepressor, to promote drug resistance in FLT3-ITD+ AML.
Methods
Xenograft mouse models were generated and treated as previously described.15 Human FLT3-ITD+ AML cell lines (MOLM13 and MOLM13-RES), a murine cell line (BAF3) containing human FLT3-ITD or FLT3-ITD/D835Y, and human primary AML samples were treated with gilteritinib, PLX51107, tasquinimod, and BI-3802, separately or in combination, before sample collection or an assay was performed. Details are in the supplemental Methods. All animal studies were reviewed and approved by The Ohio State University (OSU) Institutional Animal Care and Use Committee. Deidentified, genomically annotated human primary AML samples were obtained after written informed consent was received under an OSU Institutional Review Board–approved protocol and in accordance with the Declaration of Helsinki.
Results and discussion
Gilteritinib induces expression of S100A8/A9 in vitro and in vivo
We used RNA sequencing (RNA-seq) analysis to identify alternative mechanisms of gilteritinib resistance. Two FLT3-ITD+ AML cell lines (MOLM13 and MOLM13-RES, a resistant cell line bearing the FLT3 D835Y mutation) were xenografted into NSG mice and treated with gilteritinib until leukemia progression.15,16 Leukemic cells were isolated from the bone marrow, and gene expression analyses indicated that S100A8 and S100A9 (S100A8/A9) were 2 of the top 25 genes upregulated after gilteritinib treatment in both models (S100A9 ranked 11th and 5th, and S100A8 ranked 14th and 20th in MOLM13 and MOLM13-RES xenografts, respectively; Figure 1A). We confirmed this transcriptional upregulation by reverse transcription-polymerase chain reaction (RT-PCR; Figure 1B), but only S100A9 protein was significantly increased in the MOLM13-RES model (Figure 1C; supplemental Figure 1A).
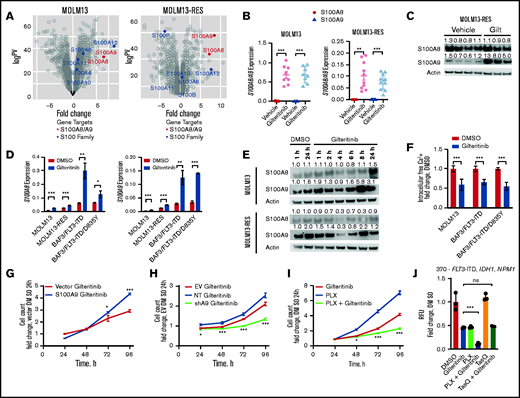
Gilteritinib induces S100A8/A9 expression and subsequently confers gilteritinib resistance. (A) Expression of S100 proteins in MOLM13 and MOLM13-RES xenograft models treated with vehicle or gilteritinib, 30 mg/kg once daily for 5 days per week (n = 8-10 mice per treatment cohort). AML cells were isolated from bone marrow at the study end point and analyzed by RNA-seq. (B) Expression of S100A8 and S100A9 by RT-PCR in cells obtained from the MOLM13 and MOLM13-RES xenograft models to validate the RNA-seq data. (C) Immunoblot of S100A8 and S100A9 expression in the MOLM13-RES xenograft model treated with vehicle or gilteritinib. Representative image of 3 to 4 samples per cohort. An immunoblot analysis was performed on Li-Cor Odyssey Fc, quantitated by Image Studio Software, and normalized to the actin control, as indicated above each blot. (D) Expression of S100A8 and S100A9 by RT-PCR in FLT3-ITD+ cells treated with 10 nM gilteritinib for 24 hours (n = 3). In all graphs, error bars represent the standard deviation. (E) Immunoblot of S100A8 and S100A9 expression in MOLM13 and MOLM13-RES cells treated with dimethyl sulfoxide (DMSO) or 10 nM gilteritinib for up to 24 hours. Representative blot of 3 separate experiments. An immunoblot was captured on Li-Cor Odyssey Fc, quantified by Image Studio Software, and normalized to actin control and then DMSO for 1 hour, as indicated above each blot. (F) Intracellular free-calcium assay of FLT3-ITD+ AML cell lines treated with 10 nM gilteritinib for 24 hours (n = 9). Cell growth assay of S100A9 overexpressed (G) or knocked down (H) in MOLM13 cells. One representative growth curve from 3 separate experiments (n = 3). Data were normalized to vector DMSO (G) or EV DMSO (H) at 24 hours. Statistical analysis was performed between the overexpression construct and vector or NT and shA9. (I) Cell growth assay of MOLM13 cells treated with 100 nM PLX51107 and 10 nM gilteritinib, separately or in combination. One representative growth curve from 3 separate experiments (n = 3). Data were normalized to DMSO at 24 hours. Statistical analysis was performed between the combination and gilteritinib-alone treatment groups. (J) Human primary AML cells (patient sample 370) with the indicated mutations were treated with DMSO, 150 nM gilteritinib, 500 nM PLX51107, and 2.5 µM tasquinimod, separately or in combination for 96 hours. Data were normalized to DMSO (CellTiter-Glo assay; n = 3 replicates). Statistical analysis was performed between the combination and gilteritinib-alone treatment groups. *P < 0.05; ** P < 0.01; *** P < 0.001; ns, not significant, by 2-tailed, unpaired Student t test. EV, empty vector; Gilt, gilteritinib; NT, non-targeting short hairpin RNA control; PLX, PLX51107; shA9, S100A9 short hairpin RNA; TasQ, tasquinimod; RFU, relative fluorescence units.
Gilteritinib induces S100A8/A9 expression and subsequently confers gilteritinib resistance. (A) Expression of S100 proteins in MOLM13 and MOLM13-RES xenograft models treated with vehicle or gilteritinib, 30 mg/kg once daily for 5 days per week (n = 8-10 mice per treatment cohort). AML cells were isolated from bone marrow at the study end point and analyzed by RNA-seq. (B) Expression of S100A8 and S100A9 by RT-PCR in cells obtained from the MOLM13 and MOLM13-RES xenograft models to validate the RNA-seq data. (C) Immunoblot of S100A8 and S100A9 expression in the MOLM13-RES xenograft model treated with vehicle or gilteritinib. Representative image of 3 to 4 samples per cohort. An immunoblot analysis was performed on Li-Cor Odyssey Fc, quantitated by Image Studio Software, and normalized to the actin control, as indicated above each blot. (D) Expression of S100A8 and S100A9 by RT-PCR in FLT3-ITD+ cells treated with 10 nM gilteritinib for 24 hours (n = 3). In all graphs, error bars represent the standard deviation. (E) Immunoblot of S100A8 and S100A9 expression in MOLM13 and MOLM13-RES cells treated with dimethyl sulfoxide (DMSO) or 10 nM gilteritinib for up to 24 hours. Representative blot of 3 separate experiments. An immunoblot was captured on Li-Cor Odyssey Fc, quantified by Image Studio Software, and normalized to actin control and then DMSO for 1 hour, as indicated above each blot. (F) Intracellular free-calcium assay of FLT3-ITD+ AML cell lines treated with 10 nM gilteritinib for 24 hours (n = 9). Cell growth assay of S100A9 overexpressed (G) or knocked down (H) in MOLM13 cells. One representative growth curve from 3 separate experiments (n = 3). Data were normalized to vector DMSO (G) or EV DMSO (H) at 24 hours. Statistical analysis was performed between the overexpression construct and vector or NT and shA9. (I) Cell growth assay of MOLM13 cells treated with 100 nM PLX51107 and 10 nM gilteritinib, separately or in combination. One representative growth curve from 3 separate experiments (n = 3). Data were normalized to DMSO at 24 hours. Statistical analysis was performed between the combination and gilteritinib-alone treatment groups. (J) Human primary AML cells (patient sample 370) with the indicated mutations were treated with DMSO, 150 nM gilteritinib, 500 nM PLX51107, and 2.5 µM tasquinimod, separately or in combination for 96 hours. Data were normalized to DMSO (CellTiter-Glo assay; n = 3 replicates). Statistical analysis was performed between the combination and gilteritinib-alone treatment groups. *P < 0.05; ** P < 0.01; *** P < 0.001; ns, not significant, by 2-tailed, unpaired Student t test. EV, empty vector; Gilt, gilteritinib; NT, non-targeting short hairpin RNA control; PLX, PLX51107; shA9, S100A9 short hairpin RNA; TasQ, tasquinimod; RFU, relative fluorescence units.
Because S100A8/A9 were significantly increased after drug treatment in vivo, we interrogated their response during gilteritinib treatment in vitro in 4 FLT3-ITD+ cell lines: MOLM13, MOLM13-RES, and murine BAF3 cells expressing human FLT3-ITD or FLT3-ITD with a D835Y mutation (FLT3-ITD/D835Y). S100A8 and S100A9 transcripts were significantly upregulated in all cell lines after acute gilteritinib exposure (Figure 1D). Furthermore, ex vivo gilteritinib treatment of a human primary AML sample with FLT3-ITD significantly increased both transcripts (supplemental Figure 1B), and serial primary AML samples collected from 2 patients treated with gilteritinib had a significant increase in S100A9 (supplemental Figure 1C). Interestingly, S100A9 protein expression was increased after gilteritinib treatment in MOLM13 and MOLM13-RES cells in a time-dependent manner, whereas S100A8 had minimal change (Figure 1E). Because these small, soluble proteins can form homodimers, heterodimers, and oligomers that bind and regulate calcium,17,18 we used an intracellular free-calcium assay to assess the calcium levels after gilteritinib treatment. Our results indicate that free calcium significantly decreased after acute exposure to gilteritinib (Figure 1F), presumptively because of sequestration by S100A8/A9. This observation is consistent with reports of decreased calcium levels or release after venetoclax (a BCL-2 inhibitor) treatment in AML12 or in prednisolone-resistant MLL-rearranged infant ALL when S100A8/A9 are overexpressed.19
Modulation of S100A9 alters sensitivity to gilteritinib in vitro and ex vivo
Because S100A9 expression was more consistently and significantly changed than S100A8 in our models, we focused our subsequent efforts on determining the functional significance of gilteritinib-induced S100A9 expression in FLT3-ITD+ AML. We assessed cell growth in the presence of gilteritinib after overexpression or knockdown of S100A9 in MOLM13 cells (Figure 1G-H; supplemental Figure 1D-G). Cells with overexpression of S100A9 were less sensitive to gilteritinib than those expressing the control vector (Figure 1G). Conversely, cells after knockdown of S100A9 were more sensitive to gilteritinib than were cells with empty vector or the non-targeting control (Figure 1H). In addition, pharmacological targeting of S100A9 with tasquinimod, a quinolone-3-carboxamide that binds and inhibits S100A9,10,20 partially sensitized MOLM13 cells to gilteritinib (supplemental Figure 1H). More recently, BET inhibition has been shown to suppress S100A8/A9,10,21 and in line with this observation, we found that cotreatment of the BET inhibitor PLX51107 with gilteritinib suppressed growth of MOLM13 cells when compared with either drug alone (Figure 1I; supplemental Figure 1I). Furthermore, PLX51107 sensitized human primary AML cells to gilteritinib, whereas tasquinimod did not enhance the antileukemic effect of gilteritinib in the patient samples (Figure 1J; supplemental Figure 1J). Collectively, the data indicate that overexpression of S100A9 confers resistance to gilteritinib. It should be noted that S100A8 has been found to enhance resistance to etoposide and vincristine by modulating apoptosis and autophagy pathways13,14 and can drive proliferation, whereas S100A9 promotes differentiation through TLR4 in AML.22 Our present results add to this prior knowledge and indicate an effect of S100A9 on cell growth that promotes a gilteritinib-resistance phenotype that can be targeted by genetic or pharmacological inhibition.
Gilteritinib-induced upregulation of S100A9 is mediated through loss of BCL6 enrichment at the S100A9 promoter
To identify the transcriptional regulators that govern gilteritinib-induced upregulation of S100A8/A9, we performed a transcription factor (TF) screen by cotransfecting HEK293 cells with expression vectors of 1623 transcriptional regulators (supplemental Figure 2A) and S100A8/A9 promoter luciferase constructs. TFs that increased or decreased promoter activity by greater than twofold were considered potential candidates (Figure 2A). Narrowing our focus to top TFs with overlapping effects on both S100A8 and S100A9 promoter activity revealed RUNX2 and BCL6 as 2 candidate transcriptional regulators (Figure 2A). Further validation indicated that RUNX2 enrichment at the S100A9 promoter was unchanged after gilteritinib treatment (supplemental Figure 2B), whereas BCL6 overexpression resulted in a significant reduction in both S100A8/A9 promoter activity (Figure 2B), as well as significantly decreased BCL6 enrichment at the S100A8/A9 promoters in gilteritinib-treated MOLM13 cells (Figure 2C; supplemental Figure 2C). Furthermore, accessibility at the S100A9 promoter significantly increased after gilteritinib treatment in vivo (supplemental Figure 3A), whereas there was no significant change at the S100A8 promotor (supplemental Figure 3B). These results indicate that gilteritinib decreases localization of BCL6 to the S100A9 promoter.

Gilteritinib-induced upregulation of S100A9 is mediated through loss ofBCL6 enrichment at the S100A9 promoter. (A) Putative hits with a greater than twofold change in the S100A8 promoter (left) and S100A9 promoter (middle) from the transcription factor screen. The overlapping hits with a greater than fivefold change resulted in RUNX2 and BCL6 (right). (B) Relative luciferase activity in a promoter assay with luciferase-expressing vectors containing S100A8/A9 promoters or empty vector and transfected with BCL6 (promoter assay; n = 4). In all graphs, error bars represent the standard deviation. (C) BCL6 enrichment at the S100A8 or S100A9 promoter in MOLM13 cells treated with 10 nM gilteritinib or dimethyl sulfoxide (DMSO) for 24 hours. Enrichment was normalized to input (chromatin immunoprecipitation assay; n = 4). (D) Relative luminescence in MOLM13 cells treated with 1 µM BI-3802 for 24 and 48 hours. Data were normalized to DMSO at 24 hours (CellTiter-Glo Assay; n = 3). (E) Expression of S100A8 and S100A9 by RT-PCR in MOLM13 cells treated with 10 nM gilteritinib or 1 µM BI-3802 for 24 hours (n = 3). (F) Immunoblot of BCL6 and S100A9 expression in MOLM13 cells treated with 10 nM gilteritinib ± 1 µM BI-3802 for 24 hours. Representative image of 2 separate experiments. The immunoblot was captured on film and normalized to the glyceraldehyde phosphate dehydrogenase (GAPDH) control. (G) Cell growth assay of MOLM13 cells pretreated with BI-3802 (1 µM; 24 hours) followed by cotreatment with gilteritinib (10 nM) for 48 hours. Data were normalized to DMSO at 24 hours (n = 9). *P < 0.05; **P < 0.01; ***P < 0.001, by 2-tailed, unpaired Student t test.
Gilteritinib-induced upregulation of S100A9 is mediated through loss ofBCL6 enrichment at the S100A9 promoter. (A) Putative hits with a greater than twofold change in the S100A8 promoter (left) and S100A9 promoter (middle) from the transcription factor screen. The overlapping hits with a greater than fivefold change resulted in RUNX2 and BCL6 (right). (B) Relative luciferase activity in a promoter assay with luciferase-expressing vectors containing S100A8/A9 promoters or empty vector and transfected with BCL6 (promoter assay; n = 4). In all graphs, error bars represent the standard deviation. (C) BCL6 enrichment at the S100A8 or S100A9 promoter in MOLM13 cells treated with 10 nM gilteritinib or dimethyl sulfoxide (DMSO) for 24 hours. Enrichment was normalized to input (chromatin immunoprecipitation assay; n = 4). (D) Relative luminescence in MOLM13 cells treated with 1 µM BI-3802 for 24 and 48 hours. Data were normalized to DMSO at 24 hours (CellTiter-Glo Assay; n = 3). (E) Expression of S100A8 and S100A9 by RT-PCR in MOLM13 cells treated with 10 nM gilteritinib or 1 µM BI-3802 for 24 hours (n = 3). (F) Immunoblot of BCL6 and S100A9 expression in MOLM13 cells treated with 10 nM gilteritinib ± 1 µM BI-3802 for 24 hours. Representative image of 2 separate experiments. The immunoblot was captured on film and normalized to the glyceraldehyde phosphate dehydrogenase (GAPDH) control. (G) Cell growth assay of MOLM13 cells pretreated with BI-3802 (1 µM; 24 hours) followed by cotreatment with gilteritinib (10 nM) for 48 hours. Data were normalized to DMSO at 24 hours (n = 9). *P < 0.05; **P < 0.01; ***P < 0.001, by 2-tailed, unpaired Student t test.
The BCL6 inhibitor BI-3802 promotes degradation of BCL6.23 BI-3802–mediated BCL6 degradation was not cytotoxic in MOLM13 cells at a concentration of 1 µM (Figure 2D), but increased S100A8/A9 transcripts and S100A9 protein (Figure 2E-F). Because BCL6 binds and represses the transcriptional activation of S100A9, we used BI-3802 to emulate the S100A9-overexpression model. Pretreatment followed by cotreatment of BI-3802 with gilteritinib caused MOLM13 cells to grow at a significantly accelerated rate, compared with gilteritinib treatment alone (Figure 2G), which may have been due to enhanced upregulation of S100A9. Collectively, these data indicate that BCL6 negatively regulates S100A9 expression, but gilteritinib promotes the dissociation of BCL6 from the S100A9 promoter.
In summary, our data support a novel mechanism of gilteritinib-induced upregulation of S100A9 that is mediated through BCL6. Through a currently unknown mechanism, gilteritinib promotes BCL6 dissociation from the S100A9 promoter, allowing for upregulation of S100A9 that promotes cell growth and a resistance phenotype. Our studies provide insight into the transcriptional regulation of S100A9 that can be further evaluated across a range of therapies and cancers and exploited for potential targeting in combinatorial treatment strategies to prevent or overcome drug resistance.
Acknowledgments
The authors thank The Ohio State University (OSU) Comprehensive Cancer Center (OSUCCC) for use of the following shared resources: Targeted Validation Shared Resource for acquisition of NSG mice, Genomics Shared Resource for RNA-seq and ATAC-seq data acquisition, and the Flow Cytometry Shared Resource for the use of the LSRFortessa; the patients who provided samples for the study and to the OSUCCC Leukemia Tissue Bank for sample procurement; the OSU University Laboratory Animal Resources (ULAR) for housing and care of animals; and Sofia Porter and Eugene Oltz for lending their technical expertise in preparation of the ATAC-seq library.
This work was supported by the National Institutes of Health, National Cancer Institute grant R01 CA138744 (S.D.B.), The Ohio State University Comprehensive Cancer Center Pelotonia Foundation (S.D.B., A.S.), and the Comprehensive Cancer Center at The Ohio State University using the Pelotonia Fellowship Program (M.E.Z.T.). The content is solely the responsibility of the authors and does not represent the official views of the funding agencies. The OSUCCC Leukemia Tissue Bank is supported by National Institutes of Health, National Cancer Institute grant NCIP30 CA016058.
Authorship
Contribution: M.E.Z.T. was responsible for the concept of the study, curated the data, and performed investigations, formal analyses, validation, and visualization; devised the methodology; and wrote, reviewed, and edited the original draft of the manuscript; J.Y.J. performed investigations and formal analyses; Z.T. and J.S. performed investigations; D.R.B. was responsible for the concept of the study, performed investigations, and reviewed and edited the manuscript; M.J.C. performed formal analyses, and provided resources; A.S. was responsible for the concept of the study, acquired the funding, and reviewed and edited the manuscript; N.P. was responsible for the concept of the study, curated the data, performed formal analyses and investigations, and reviewed and edited the manuscript; S.D.B. was responsible for the concept of the study, acquired the funding, served as project administrator and supervisor, and reviewed and edited the manuscript; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Sharyn D. Baker, The Ohio State University, 410 Biomedical Research Tower, 460 W 12th Ave, Columbus, OH 43210; e-mail: baker.2480@osu.edu.


