Abstract
Since the US Food and Drug Administration (FDA) approvals of parenteral decitabine and azacitidine, DNA methyltransferase inhibitors, otherwise referred to as DNA hypomethylating agents (HMAs), have been a mainstay in the treatment of higher-risk myelodysplastic syndromes. The development of oral HMAs has been an area of active interest; however, oral bioavailability has been quite poor due to rapid metabolism by cytidine deaminase (CDA). This led to the development of the novel CDA inhibitor cedazuridine, which was combined with an oral formulation of decitabine. Preclinical work demonstrated a pharmacokinetic and pharmacodynamic profile approximate to parenteral decitabine, leading to early-phase clinical trials of oral cedazuridine-decitabine (C-DEC) in myelodysplastic syndromes and chronic myelomonocytic leukemia (CMML). A combination of oral decitabine 35 mg with oral cedazuridine 100 mg was established as the recommended phase 2 dose. Phase 2 data confirmed bioequivalence of C-DEC when compared with parenteral decitabine, and a larger phase 3 trial has demonstrated similar results, leading to the FDA approval of C-DEC for use in intermediate/high-risk myelodysplastic syndrome (MDS) and CMML. This review will focus upon the current role of HMA therapy in MDS/CMML, preclinical and clinical development of C-DEC, and potential roles of oral HMA therapy in myeloid malignancies moving forward.
Introduction
Myelodysplastic syndromes (MDSs) are a heterogeneous group of clonal hematopoietic stem cell disorders characterized by peripheral cytopenias and a variable risk of progression to acute myeloid leukemia (AML).1 Given the variable outcomes associated with these diseases, various prognostic scoring systems, including the International Prognostic Scoring System (IPSS) and the Revised IPSS (R-IPSS), have been developed as tools to risk-stratify patients with MDS.2,3 While treatment strategies for patients with lower-risk MDS focus primarily on improvement of cytopenias and quality of life, treatment modalities in higher-risk MDS focus on the prevention of disease progression and improvement of overall survival.4 Currently, the one treatment modality with curative potential is allogeneic hematopoietic stem cell transplantation (allo-HCT).5 Given the potential toxicities of allo-HCT, particularly in an older age group who often have multiple comorbidities, the parental DNA hypomethylating agents (HMAs) 5-aza-2′-deoxycytidine (decitabine) and 5-azacitidine (azacitidine), which have been demonstrated to have a modest impact on the natural history of these diseases, are currently the standard of care in higher-risk MDS.6 Over the last several years, there has been much interest in developing oral formulations of HMAs, given the potential ease of administration and convenience associated with oral agents. In July 2020, an oral combination of cedazuridine-decitabine (C-DEC or ASTX727) (INQOVI, Astex Pharmaceuticals) was approved by the US Food and Drug Administration (FDA) for the treatment of patients with IPSS intermediate/high-risk MDS and the following MDS French-American-British (FAB) subtypes: refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, and chronic myelomonocytic leukemia (CMML). In this article, we review the preclinical and clinical development of C-DEC, as well as the current and future applications of this drug in patients with myeloid malignancies.
HMA therapy
Development, drug metabolism, and mechanism of action of HMAs
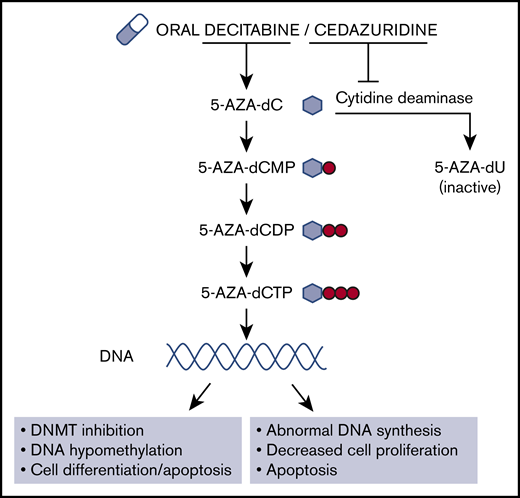
Decitabine is a nucleoside analog that requires incorporation into DNA to exert its anticancer effect, and it is an S-phase–specific agent.7,8 It is a prodrug that undergoes a 3- step phosphorylation process intracellularly to be converted to the active moiety, decitabine triphosphate (5-AZA-dCTP), which is then incorporated into DNA by DNA polymerases (Figure 1). The initial and rate-limiting step is catalyzed by deoxycytidine kinase, with subsequent steps being catalyzed by deoxycytidine monophosphokinase and nucleoside diphosphokinase, respectively. Upon incorporation into DNA, 5-AZA-dCTP forms a covalent complex with DNA methyltransferase (DNMT), which leads to trapping and degradation of the enzyme.9,10 DNMT catalyzes the process of DNA methylation, which involves the addition of a methyl group to the 5th carbon of 2-deoxycytosine within the 5′-cytosine-guanosine dinucleotides of DNA.

Oral cedazuridine 100 mg with decitabine 35 mg is given as a single pill. Decitabine is a nucleoside inhibitor that is incorporated into DNA after phosphorylation to 5-AZA-dCTP. Once incorporated into DNA, it inhibits DNMT, causing hypomethylation and altered gene expression, which is hypothesized to affect cellular differentiation and apoptosis in the treatment of MDS and AML. It may also cause abnormal DNA synthesis and replication, therefore decreasing cell proliferation and inducing apoptosis. Cedazuridine is a CDA inhibitor that prevents the degradation of decitabine when taken orally. 5-AZA-dC, 5-aza-2′deoxycytidine; 5-AZA-dCMP, 5-aza-2′deoxycytidine monophosphate; 5-AZA-dCDP, 5-aza-2′deoxycytidine diphosphate; 5-AZA-dU, deazuridine.
Oral cedazuridine 100 mg with decitabine 35 mg is given as a single pill. Decitabine is a nucleoside inhibitor that is incorporated into DNA after phosphorylation to 5-AZA-dCTP. Once incorporated into DNA, it inhibits DNMT, causing hypomethylation and altered gene expression, which is hypothesized to affect cellular differentiation and apoptosis in the treatment of MDS and AML. It may also cause abnormal DNA synthesis and replication, therefore decreasing cell proliferation and inducing apoptosis. Cedazuridine is a CDA inhibitor that prevents the degradation of decitabine when taken orally. 5-AZA-dC, 5-aza-2′deoxycytidine; 5-AZA-dCMP, 5-aza-2′deoxycytidine monophosphate; 5-AZA-dCDP, 5-aza-2′deoxycytidine diphosphate; 5-AZA-dU, deazuridine.
Aberrant DNA methylation and epigenetic dysregulation has been implicated in the pathogenesis of myeloid malignancies, including MDS, and has been linked to the transcriptional repression of a variety of genes, including tumor suppressor genes.11-15 Inhibition of DNMT by decitabine leads to DNA hypomethylation and is thought to allow for subsequent reactivation of these genes. This purported effect on the epigenome has been challenging to link definitively to responses observed with the agent in the clinical setting. The effects of decitabine are likely pleiotropic via additional mechanisms such as the induction of DNA damage.7,10 Azacitidine gets converted intracellularly to the same active moiety 5-AZA-dCTP prior to incorporation into DNA and thus has a similar mechanism of action to decitabine, but a significant proportion of azacitidine is incorporated into RNA as well.16,17
Both decitabine and azacitidine were initially studied as traditional cytotoxic agents in leukemia at much higher doses than those used in clinical practice today.18-20 These doses were found to be quite myelosuppressive and subsequent trial designs of lower-dose schedules, where the hypothesized effects on DNA methylation and the epigenome are thought to predominate, form the basis for the current use of HMAs in myeloid malignancies.20-22
HMA therapy in MDS, CMML, and AML
Both decitabine and azacitidine are FDA approved as parenteral therapies in the treatment of MDS, demonstrating objective evidence of efficacy, improved quality of life outcomes, and/or survival benefit as summarized in Table 1.23-26 Azacitidine specifically demonstrated a significantly improved median overall survival compared with conventional therapy in MDS patients. Of note, the conventional therapies in this trial were heterogenous and included the following: best supportive care, low-dose cytarabine, and intensive chemotherapy. This makes the overall survival benefit of azacitidine vs each of these specific therapies less clear.25 Regarding decitabine, a follow-up study by Steensma and colleagues established the efficacy of a 5-day dosing regimen every 28 days at a dose of 20 mg/m2 for decitabine, which is the dosing strategy most commonly used in practice today.27 Decitabine has been prospectively evaluated in CMML, with response rates of 48% to 69%.28,29 In addition, a Surveillance, Epidemiology, and End Results-Medicare analysis of older patients with CMML demonstrated a survival benefit in patients treated with HMAs.30 A recent phase 3 trial comparing decitabine to hydroxyurea in proliferative CMML demonstrated no survival benefit in the decitabine arm; however, ∼33% of patients on the hydroxyurea arm ultimately received an HMA.31 Azacitidine and decitabine have also been incorporated into the treatment of older adults with AML (Table 1).32,33 Of note, while azacitidine and decitabine have not been directly compared in a randomized trial, Surveillance, Epidemiology, and End Results-Medicare analyses in both the MDS and AML population have demonstrated similar efficacy and survival with both agents.30,34,35 While both parenteral azacitidine and decitabine have demonstrated efficacy in MDS and are the most commonly used initial therapy, the burden of parenteral therapies has led to the development of oral analogs.
C-DEC
Nonclinical and early-phase development of oral decitabine-cedazuridine therapy
The impetus to develop compounds with the potential to improve oral bioavailability of HMAs stemmed from initial experience derived from early-phase trials with oral HMAs. For example, in a phase 1 study of oral decitabine in 12 patients with MDS, pharmacokinetic parameters following oral doses showed significant variability between different patients and dose levels.36 The absolute bioavailability of the drug was 4% to 14%, which is similar to what has been observed with the oral formulation of azacitidine, CC-486.37 This variability is due to the extensive metabolism and first-pass elimination of oral HMAs by the enzyme cytidine deaminase (CDA) (Figure 1), which is highly expressed in the gut and liver. To overcome the CDA-mediated degradation of oral decitabine, tetrahydrouridine, a competitive inhibitor of CDA, was tested in preclinical models. Sequential treatment with tetrahydrouridine followed by decitabine extended decitabine exposure time and increased its bioavailability in mice and baboons.38 The pharmacokinetic profile of decitabine-tetrahydrouridine (DEC-THU) combination was subsequently evaluated in a phase 1 study of sickle cell disease patients. While there was clinical efficacy demonstrated by increased hemoglobin F, which was associated with decreased DNMT protein levels in treated patients, this study was not designed to provide a comparison between the kinetics of IV decitabine and oral DEC-THU.39 In addition, previous work has demonstrated instability of THU in an acidic environment like that of gastric fluid.40
Cedazuridine (E7727), a synthetic nucleoside analog derived from THU, was designed to overcome this instability (Figure 1).41 The combination of 3 mg/kg oral decitabine (corresponding to a human equivalent dose of 36 mg/m2) with escalating doses of 0.1 to 10 mg/kg oral cedazuridine was tested in cynomolgus monkeys.42 The concentration-time profile of oral E7727 + decitabine, given 30 minutes apart, resembled that achieved with IV decitabine over 1-hour infusion. In addition, this combination induced long interspersed nuclear element 1 (LINE-1) demethylation, which is a widely accepted pharmacodynamic marker of epigenetic modulation.43
The favorable pharmacokinetic and pharmacodynamic profile of C-DEC in preclinical models led to the further development of this combination by simultaneous (rather than sequential as studied in animal models) administration in a phase 1 study of patients with MDS and CMML.44 The study used a dose-escalation design in cohorts of 6 patients to establish a fixed-dose oral combination of decitabine and cedazuridine that can emulate the pharmacokinetics of a standard dose of 20 mg/m2 IV decitabine infusion. In cycle 1, each patient received a cohort-defined dose of oral decitabine on day −3, IV decitabine infusion on day 1, and cohort-defined doses of oral decitabine plus cedazuridine on days 2 to 5. Among 44 patients enrolled in this study, there was no evident increase in toxicity compared with what was previously reported for IV decitabine. Oral decitabine 30 mg and 40 mg combined with cedazuridine 100 mg produced day-5 area under the curve (AUC) (146 ng × h/mL and 221 ng × h/mL, respectively) closest to the day-1 IV decitabine (164 ng × h/mL). The study reported similar results with cumulative 5-day AUC assessment, which was performed to evaluate the accumulation with oral formulation, since IV formulation was shorter acting and known not to accumulate. In addition, the fixed-dose oral combination was effective in inducing LINE-1 demethylation. In summary, this trial demonstrated that the oral fixed-dose combination could emulate the pharmacokinetic profile of IV decitabine. Since the AUC of IV decitabine fell between AUCs for oral decitabine 30 mg and 40 mg, a combination of oral decitabine 35 mg plus cedazuridine 100 mg was suggested for further development in phase 2/3 studies.
Late-phase clinical development of C-DEC
A phase 2, multicenter, open-label, randomized, crossover study, was designed to compare systemic decitabine exposure, demethylation activity, and safety of oral C-DEC to IV decitabine.45 The primary end points were oral/IV decitabine exposure over 5 days, DNA demethylation of oral C-DEC vs IV decitabine, and overall response rate. Adult patients with MDS (IPSS low, intermediate-1, intermediate-2, or high risk) and CMML were included and allowed to have ≥1 prior cycle of HMA therapy (Table 2). Patients were randomized 1:1 to receive the recommended phase 2 dose of oral cedazuridine 100 mg/decitabine 35 mg for 5 days in cycle 1 followed by IV decitabine 20 mg/m2 per day for 5 days in cycle 2 or the reverse sequence (IV decitabine for 5 days in cycle 1 followed by 5 days of oral C-DEC in cycle 2). From cycle 3 onwards, all patients received oral therapy. In the dose-confirmation stage, patients received the cedazuridine and oral decitabine together as separate capsules. After showing similar decitabine exposure, a second cohort of patients received a fixed-dose combination capsule (ASTX727). Eighty-six patients were randomized, and 80 went on to receive treatment with 50 in the dose-confirmation cohort and 30 in the fixed-dose combination. The oral/IV ratios of geometric least square mean (LSM) 5-day AUC was 93.5% for the dose-confirmation cohort and 97.6% using the fixed oral drug dose combination and similar LINE-1 demethylation assays, thus demonstrating similar systemic exposure and pharmacodynamics of oral C-DEC compared with IV decitabine. Patients received a median of 7 cycles (1-29), and 35% of all treated patients achieved a response by 3 cycles. The overall response rate (ORR), including complete remission (CR), PR, marrow CR (mCR), and HI, was 60%. The CR rate was 21%, and median overall survival of the entire cohort was 18.3 months (Table 3).45 These markers of efficacy were similar to that seen in MDS patients treated with IV decitabine on a 5-day schedule.27 The most common grade 3+ adverse events were hematologic, with 46% of patients experiencing grade ≥3 neutropenia, 44% with grade ≥3 thrombocytopenia, and 20% with grade ≥3 neutropenic fever. The most common nonhematologic and noninfectious adverse events were fatigue, nausea, and diarrhea, with the vast majority of these being grade <3.45 This adverse event profile was similar to that seen in patients treated with IV decitabine.
Because only 30 of the 80 patients were treated with the fixed-dose capsule while the other 50 were treated with separate capsules during the dose-confirmation stage, a phase 3 trial (ASTX727-02 ASCERTAIN study) with a similar design was initiated to demonstrate bioequivalence and clinical activity of oral C-DEC and IV decitabine in a larger population, utilizing the fixed-dose capsule. It had similar inclusion criteria including patients with MDS (IPSS intermediate-1, intermediate-2, or high risk) and CMML (Table 2). The preliminary results of 133 patients have been reported with median follow-up of 12.6 months and median treatment of 8 cycles. Of the 138 patients who were randomized, 133 were treated on study, of whom 88% had MDS and 12% had CMML.46 The primary end point of oral/IV ratios of geometric LSM 5-day AUC was achieved with a ratio of 99% (90% confidence interval [CI], 93% to 106%).47 The ORR (CR+PR+mCR+HI) was 62% with a CR rate of 22% and a median CR duration of 14 months (Table 3). In addition, the median duration of best response was 12.7 months, and 26% of patients proceeded to allo-HCT. Median overall survival has not been reached with a median follow-up time of 24.7 months. These efficacy results are similar to those historically noted with IV decitabine, although it is important to note that some lower-risk MDS patients were enrolled in this study. Of the 57 patients who were transfusion dependent at baseline, 30 (53%) became transfusion independent for at least 8 weeks. The most common grade ≥3 adverse events were hematologic in nature including neutropenia (52%), thrombocytopenia (50%), and anemia (40%), similar to those reported with IV decitabine.46 Overall, these results show that oral C-DEC results in similar systemic exposure to IV decitabine, thus providing an orally bioavailable alternative to the parenteral formulation.
Future directions
The future of oral HMA therapies in myeloid malignancies goes beyond the demonstration of bioequivalence compared with parenteral options. Alternative dosing schedules, combination approaches, and other novel cedazuridine-based drug formulations are in active investigation as well (Table 4).
Alternative dosing regimens have been investigated with parenteral HMAs with the intent of maximizing their epigenetic effects. Saunthararajah and colleagues investigated the use of an increased-frequency dosing schedule of decitabine that composed of a 4-week induction phase of decitabine 0.2 mg/kg per day administered 2 days per week followed by a maintenance phase with titration of dosing based upon cytopenias and bone marrow cellularity. In addition to tolerability in this patient population, correlative work demonstrated DNMT1 depletion without cytotoxicity and significant reduction in levels of the oncoprotein MYC.48 Studies in patient-derived xenograft (PDX) models of AML in mice models have also demonstrated the development of HMA resistance due to lack of DNMT1 depletion. Utilization of the more frequent dosing schedule demonstrated more prolonged DNMT1 depletion in addition to increased efficacy.49 A retrospective analysis of patients treated using this dosing strategy across a spectrum of myeloid malignancies demonstrated a response rate of 43% with median overall survival of 31 months in those achieving a response.50 As previously discussed in this article, similar dosing regimens of oral decitabine with THU have been investigated in sickle cell disease and demonstrated DNMT1 depletion.39
There are prospective studies investigating alternative dosing regimens of oral HMAs across a spectrum of myeloid malignancies. CC-486 was originally investigated in myeloid malignancies with a dosing schedule of once daily for the first 7 days of a 28-day cycle. While reduction in DNA methylation, oral bioavailability, and clinical efficacy were noted, there was interest in studying an extended dosing schedule with the intent of sustained DNA demethylation.37 This schedule was investigated in patients with IPSS low/intermediate-1 risk MDS and demonstrated an ORR of 38% with grade 3/4 toxicities in 43% to 48% of patients and sustained reduction in DNA methylation for the duration of the cycle.51 Wei and colleagues demonstrated a survival benefit with use of an extended dosing schedule of CC-486 as a maintenance therapy in patients with AML who achieve a CR and do not go onto transplant. This led to FDA approval of CC-486 for patients with AML achieving a CR/complete remission with incomplete blood count recovery (CRi) after intensive induction chemotherapy who are unable to complete intensive curative therapy.52 CC-486 was also found to be tolerable and efficacious as post-allo-HCT maintenance in MDS and AML; a randomized controlled trial is currently ongoing to establish the efficacy of CC-486 in this setting (NCT04173533).53 In addition to the extended dosing schedule of CC-486 being investigated, a prospective study evaluating low-dose C-DEC in lower-risk MDS is ongoing. As the field of oral HMA therapy continues to evolve, prospective evaluation of alternative dosing regimens designed to maximize the epigenetic effect of oral HMAs in high-risk myeloid neoplasms merits additional consideration.
There is also great interest in the evaluation of oral HMA-based combination therapies. Current prospective trials include a basket trial investigating ASTX727 combination therapies in MDS/MPN overlap syndromes and ASTX727 combined with the anti-apoptosis protein inhibitor ASTX660. Given the recent data demonstrating a survival benefit in older patients with AML treated with venetoclax and an HMA when compared with HMA monotherapy,54 early-phase trials evaluating venetoclax with C-DEC are on the horizon as well. Prospective trials of triplet therapies with the backbone of parenteral HMAs and venetoclax are ongoing, and as we continue to gain information about the efficacy and tolerability of these approaches, one can envision a role for the investigation of oral HMAs in this setting as well. Current prospective trials include the oral HMA-venetoclax backbone in combination with IDH inhibitors; evaluation of combinations with FLT3 inhibitors, anti-CD47 antibodies, and mutant TP53-targeted therapies should be considered as well.
Other oral HMA/cedazuridine molecules are also on the horizon. Studies in a PDX model of AML have demonstrated equivalent pharmacokinetics in mice treated with parenteral azacitidine and those treated with oral azacitidine-cedazuridine. Furthermore, the combination of venetoclax with oral azacitidine-cedazuridine demonstrated significant antileukemia efficacy in the PDX models, emulating what is seen in the clinic with parenteral HMA therapy with venetoclax.55 Oral azacitidine-cedazuridine is currently being prospectively investigated as the compound ASTX030.
In summary, since the FDA approvals of parenteral azacitidine and decitabine, HMAs have been the most significant advance made for the treatment of higher-risk MDS. Given the emphasis on measures to improve quality of life for patients and the extended period of time that patients are typically treated with HMAs, the development of oral HMAs with equivalent bioavailability to parenteral options is predicted to be of major importance and to lend itself to combinatorial therapy with promising novel agents in myeloid malignancies in the near future.
Authorship
Contribution: A.A.P., K.C., C.S., and O.O. conceived and wrote the article.
Conflict-of-interest disclosure: O.O. has served on advisory boards convened by AbbVie, BMS, Celgene, Impact Biomedicines, Novartis, and Taiho and has received research funding (paid to institution) from AbbVie, Agios, Astex, AstraZeneca, Celgene, CTI, Gilead, Incyte, Janssen, Kartos, NS Pharma, and Oncotherapy Sciences. The remaining authors declare no competing financial interests.
Correspondence: Olatoyosi Odenike, Section of Hematology/Oncology, Department of Medicine, The University of Chicago Medicine, 5841 S. Maryland Ave, MC 2115, Chicago, IL 60637, e-mail: todenike@medicine.bsd.uchicago.edu.