

Key Points
FT-4202, an oral investigational agent, increases the hemoglobin S O2 affinity to reduce sickling and improve RBC membrane deformability.
Two-week administration of FT-4202 improves RBC survival and Hb levels in SCA mice.
Abstract
Sickle cell anemia (SCA) results from an abnormal sickle hemoglobin (HbS). HbS polymerizes upon deoxygenation, resulting in red blood cell (RBC) sickling and membrane damage that cause vaso-occlusions and hemolysis. Sickle RBCs contain less adenosine triphosphate and more 2,3-diphosphoglycerate than normal RBCs, which allosterically reduces hemoglobin (Hb) oxygen (O2) affinity (ie, increases the partial pressure of oxygen at which hemoglobin is 50% saturated with oxygen [P50]), potentiating HbS polymerization. Herein, we tested the effect of investigational agent FT-4202, an RBC pyruvate kinase (PKR) activator, on RBC sickling and membrane damage by administering it to Berkeley SCA mice. Two-week oral FT-4202 administration was well tolerated, decreasing HbS P50 to levels similar to HbA and demonstrating beneficial biological effects. In FT-4202–treated animals, there was reduced sickling in vivo, demonstrated by fewer irreversibly sickled cells, and improved RBC deformability, assessed at varying shear stress. Controlled deoxygenation followed by reoxygenation of RBCs obtained from the blood of FT-4202–treated mice showed a shift in the point of sickling to a lower partial pressure of oxygen (pO2). This led to a nearly 30% increase in RBC survival and a 1.7g/dL increase in Hb level in the FT-4202–treated SCA mice. Overall, our results in SCA mice suggest that FT-4202 might be a potentially useful oral antisickling agent that warrants investigation in patients with SCA.
Introduction
Sickle cell anemia (SCA) is caused by a mutated HBB gene, resulting in the production of sickle hemoglobin (HbS). Upon deoxygenation, HbS polymerizes into long fibers, causing red blood cell (RBC) sickling and membrane damage, resulting in vaso-occlusions, hemolysis, and end-organ damage. Additionally, sickle RBCs have elevated 2,3-diphosphoglycerate (2,3-DPG) levels compared with normal RBCs,1-3 effects we also observe in an ongoing clinical study of FT-4202 in healthy subjects and patients with SCA.3,4 This occurs secondary to the anemia, hypoxia, and excessive adenosine signaling that is seen in SCA.5-8 2,3-DPG allosterically reduces hemoglobin (Hb) oxygen (O2) affinity (ie, it increases the partial pressure of oxygen at which hemoglobin is 50% saturated with oxygen [P50]) by binding to the Hb tetramer and stabilizing its T (tense)/deoxygenated form, which further promotes HbS polymerization.1,9 More elevated levels of 2,3-DPG correlate with higher sickling,10-15 although this was not observed in the early studies.16,17
Inhibiting HbS polymerization and reduction of sickling can occur via several mechanisms. Voxelotor (GBT440) modulates Hb O2 affinity by covalently binding α-globin and stabilizing oxyhemoglobin.18,19 Increasing the activity of 2,3-DPG phosphatase decreases 2,3-DPG and reduces HbS polymerization in sickle RBCs in vitro.2 A different approach of reducing 2,3-DPG in RBCs is by increasing RBC pyruvate kinase (PKR) activity, increasing substrate utilization through the glycolytic cycle, which also generates adenosine triphosphate (ATP) (supplemental Figure 1).20,21 Mitapivat is a PKR agonist that was shown to improve the hemolytic anemia associated with pyruvate kinase deficiency.20-22
FT-4202 is an activator of PKR that decreases 2,3-DPG levels,3,4,23,24 and hence would increase the HbS O2 affinity to reduce sickling. Moreover, activation of PKR by FT-4202 would also increase glycolytic ATP production, the energy currency of the cell that increases RBC membrane deformability by improving RBC hydration.22,25-27 Here, we determined the tolerability of daily oral FT-4202 in vivo, and its effects on sickle RBC 2,3-DPG levels, sickling, membrane deformability, and survival in an SCA mouse model.
Methods
Berkeley SCA [Tg(Hu-miniLCRα1GγAγδβS)Hba0/0 Hbb0/0]28 (BERK) mice were either fed FT-4202 or control chow for 2 weeks in 4 cohorts. Health status, weight, and chow consumption were determined 3 times per week. Three cohorts were injected with sulfo-NHS-biotin 1 week into treatment, and RBC survival was assessed over the next week with serial bleeds while on treatment. One cohort was only bled before and after 2 weeks of treatment to obtain complete blood and reticulocyte counts. Mice were exsanguinated after 2 weeks of treatment. Blood was processed for (a) RBC levels of 2,3-DPG and ATP, (b) irreversibly sickled RBCs (ISCs) by ImageJ analysis, (c) kinetics of experimentally induced sickling and RBC membrane deformability via Lorrca Oxygenscan (RR Mechatronics, Zwaag, The Netherlands), (d) P50 by Hemox Analyzer (TCS Scientific Corp, New Hope, PA), and (e) plasma levels of FT-4202 by liquid chromatography with tandem mass spectrometry, bilirubin, and lactate dehydrogenase (LDH) by colorimetry. Animal experiments were performed using institutional animal care and use committee–approved protocols. Detailed methodology is available in supplemental Methods.
Results and discussion
BERK mice fed FT-4202 chow (FT-4202 BERK) consumed a similar amount of food, and had similar weights and survival, compared with BERK mice fed control chow (control BERK) throughout the 2-week period, showing that oral FT-4202 was well tolerated by FT-4202 BERK mice (supplemental Figure 2A-C). After 2 weeks of FT-4202 administration, plasma FT-4202 levels were 7702 ± 796 ng/mL in FT-4202 BERK mice. Levels of 2,3-DPG were significantly decreased and ATP levels were significantly increased in FT-4202 BERK vs control BERK mice (Figure 1A-B), which were associated with a significantly lower P50 in FT-4202 BERK compared with control BERK mice (Figure 1C-D; supplemental Figure 3). Indeed, P50 in FT-4202 BERK mice was similar to P50 of normal RBCs.19,29 Although the oral dose and the exposure of FT-4202 in BERK mice was higher than in the ongoing human trial,3,4,30 it was well tolerated and the pharmacodynamic effect on 2,3-DPG levels and P50 was similar.
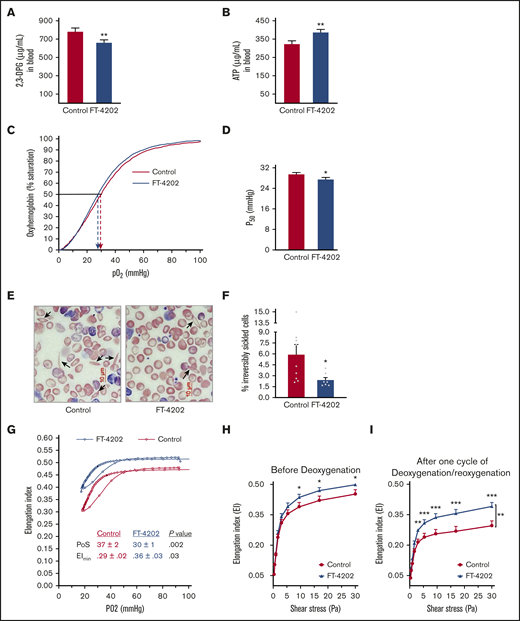
Oral administration of FT-4202 improves RBC parameters in the BERK mouse model. 2,3-DPG (A) and ATP (B) concentration in blood of BERK mice fed with control or FT-4202 chow for 2 weeks. (A-B) n = 17 mice per group. (C) P50 curve/oxygen equilibrium curve (OEC) showing the HbS P50 level in the blood of BERK assessed after 2 weeks of FT-4202 vs control chow administration. The oxygen dissociation curves are plotted using the average values of partial pressure of oxygen (pO2) and the percentage of oxygen saturation at each point of 4 control BERK mice (red line) and 4 FT-4202 BERK mice (blue line). Dashed arrows mark the P50 values for each group. (D) P50 levels. (C-D) n = 4 mice per group. (E) Representative peripheral blood smears showing ISCs (black arrow) stained with modified Wright's stain; scale bar, 10 µM. (F) Irreversibly sickled cells (ISCs) quantification in control and FT-4202 BERK mice. Images of the blood smears were taken using a Nikon Eclipse Ti inverted microscope, and 10 diffesrent high magnification fields per mouse were quantified for ISCs using ImageJ analysis software. ISCs were identified as elongated RBCs with a longitudinal-to-transverse diameter ratio of >2.0. The percentage of ISCs was calculated as the number of ISCs normalized to the total number of red blood cells; n = 9 control BERK mice; n = 7 FT-4202 BERK mice. (A-F) *P < .05; **P < .01; unpaired Student t test. (G-I) Assessment of RBC sickling kinetics and membrane deformability in control BERK vs FT-4202 BERK mice after 2 weeks of treatment, using Lorrca Oxygenscan. (G) RBCs from FT-4202 and control BERK mice were subjected to controlled deoxygenation from ambient pO2 (normoxia) to a pO2 of <15 mm Hg followed by reoxygenation to normoxia. At fixed shear stress of 30 Pa, RBC deformability (EI) was measured across an O2 gradient from normoxia to hypoxia (G). The highest (maximum) EI is seen at ambient pO2 (EImax). As pO2 decreases, HbS polymerization ensues, making the RBCs stiff/rigid, and reducing their EI. The point of sickling (PoS) is the pO2 when the EI drops to 95% of the EImax. A further precipitous decline in EI occurs during the rapid HbS polymerization phase, resulting in extremely rigid RBCs with a minimum EI (EImin) at pO2 <20 mm Hg. RBCs are then reoxygenated back and EI was measured. The graph represents an average (Avg) EI (n = 13 control BERK mice, shown in red; n = 12 FT-4202 BERK, shown in blue). PoS is denoted by the black filled circles in each group. (H-I) RBC membrane deformability (EI) measured under normoxia, across a gradient of shear stress between 0.3 and 30 Pa. Because HbS polymerization does not occur under normoxia, this assay measures membrane damage reflected in reduced deformability. Deformability before deoxygenation (H) and after 1 cycle of deoxygenation/reoxygenation (I) on the Lorcca Oxygenscan. Here, in both instances, measurements of EI under normoxia ensured assessment of membrane damage without the confounding effect of HbS polymerization/sickling, but assessed the membrane damage before and after 1 cycle of experimentally induced sickling. Comparison at the different individual shear-stress levels were made using unpaired Student t tests: *P < .05; **P < .01; ***P < .001. Comparisons at overall shear stress of the entire deformability curve were made using 2-way analysis of variance (ANOVA): **P < .01. Curves in panel H were plotted using mean plus or minus standard error of the mean (SEM), EI of n = 14 control BERK mice (depicted in red) and n = 14 FT-4202 BERK mice (depicted in blue).
Oral administration of FT-4202 improves RBC parameters in the BERK mouse model. 2,3-DPG (A) and ATP (B) concentration in blood of BERK mice fed with control or FT-4202 chow for 2 weeks. (A-B) n = 17 mice per group. (C) P50 curve/oxygen equilibrium curve (OEC) showing the HbS P50 level in the blood of BERK assessed after 2 weeks of FT-4202 vs control chow administration. The oxygen dissociation curves are plotted using the average values of partial pressure of oxygen (pO2) and the percentage of oxygen saturation at each point of 4 control BERK mice (red line) and 4 FT-4202 BERK mice (blue line). Dashed arrows mark the P50 values for each group. (D) P50 levels. (C-D) n = 4 mice per group. (E) Representative peripheral blood smears showing ISCs (black arrow) stained with modified Wright's stain; scale bar, 10 µM. (F) Irreversibly sickled cells (ISCs) quantification in control and FT-4202 BERK mice. Images of the blood smears were taken using a Nikon Eclipse Ti inverted microscope, and 10 diffesrent high magnification fields per mouse were quantified for ISCs using ImageJ analysis software. ISCs were identified as elongated RBCs with a longitudinal-to-transverse diameter ratio of >2.0. The percentage of ISCs was calculated as the number of ISCs normalized to the total number of red blood cells; n = 9 control BERK mice; n = 7 FT-4202 BERK mice. (A-F) *P < .05; **P < .01; unpaired Student t test. (G-I) Assessment of RBC sickling kinetics and membrane deformability in control BERK vs FT-4202 BERK mice after 2 weeks of treatment, using Lorrca Oxygenscan. (G) RBCs from FT-4202 and control BERK mice were subjected to controlled deoxygenation from ambient pO2 (normoxia) to a pO2 of <15 mm Hg followed by reoxygenation to normoxia. At fixed shear stress of 30 Pa, RBC deformability (EI) was measured across an O2 gradient from normoxia to hypoxia (G). The highest (maximum) EI is seen at ambient pO2 (EImax). As pO2 decreases, HbS polymerization ensues, making the RBCs stiff/rigid, and reducing their EI. The point of sickling (PoS) is the pO2 when the EI drops to 95% of the EImax. A further precipitous decline in EI occurs during the rapid HbS polymerization phase, resulting in extremely rigid RBCs with a minimum EI (EImin) at pO2 <20 mm Hg. RBCs are then reoxygenated back and EI was measured. The graph represents an average (Avg) EI (n = 13 control BERK mice, shown in red; n = 12 FT-4202 BERK, shown in blue). PoS is denoted by the black filled circles in each group. (H-I) RBC membrane deformability (EI) measured under normoxia, across a gradient of shear stress between 0.3 and 30 Pa. Because HbS polymerization does not occur under normoxia, this assay measures membrane damage reflected in reduced deformability. Deformability before deoxygenation (H) and after 1 cycle of deoxygenation/reoxygenation (I) on the Lorcca Oxygenscan. Here, in both instances, measurements of EI under normoxia ensured assessment of membrane damage without the confounding effect of HbS polymerization/sickling, but assessed the membrane damage before and after 1 cycle of experimentally induced sickling. Comparison at the different individual shear-stress levels were made using unpaired Student t tests: *P < .05; **P < .01; ***P < .001. Comparisons at overall shear stress of the entire deformability curve were made using 2-way analysis of variance (ANOVA): **P < .01. Curves in panel H were plotted using mean plus or minus standard error of the mean (SEM), EI of n = 14 control BERK mice (depicted in red) and n = 14 FT-4202 BERK mice (depicted in blue).
Repeated cycles of HbS polymerization in microvasculature and its depolymerization in lungs damage RBC membranes, which results in membrane loss and ISCs.31 Indeed, blood smears showed significantly reduced numbers of ISCs in the FT-4202 BERK compared with control BERK mice (Figure 1E-F). To demonstrate the in vivo effect of FT-4202 on HbS polymerization and RBC membrane health, we assessed the sickling kinetics and membrane deformability after 2 weeks of treatment in BERK mice using Lorrca Oxygenscan.32 FT-4202 BERK mice showed an improved Oxygenscan curve when compared with control BERK mice (Figure 1G): the point of sickling (PoS) in the FT-4202 BERK mice occurred at a much lower pO2 of 30 mm Hg compared with 37 mm Hg in controls (P < .002). The minimum elongation index (EImin) was also significantly improved in FT-4202 BERK than in control BERK mice (P < .03), whereas the maximum EI (EImax) showed an improved trend in FT-4202 BERK (0.52 ± 0.01) vs control BERK mice (0.47 ± 0.02; P = .07). In contrast, in normal mice, there was no change in EI with deoxygenation/reoxygenation, as is expected (supplemental Figure 4). Taken together, RBCs from FT-4202 BERK mice had a reduced propensity to sickle. Next, we assessed RBC membrane deformability across a gradient of shear stress under normoxia: FT-4202 BERK mice had improved EI at shear stress >3 to 9 Pa vs controls (P < .03; Figure 1H) and this effect was more pronounced after 1 cycle of deoxygenation/reoxygenation (P < .01-.001; Figure 1I).
Sickling-induced membrane damage increases hemolysis, reducing RBC lifespan in mice and humans with SCA.28,33 Indeed, hemolysis was significantly reduced in FT-4202 BERK mice, reflected in reduced bilirubin (P < .0001) and LDH levels (P < .0002) (Figure 2A-B). BERK mice have an extremely short RBC lifespan coupled with high RBC turnover as indicated by ∼50% reticulocytes.33 NHS-biotin tracing revealed a 28.5% increase in the RBC half-life to 1.8 vs 1.4 days in the FT-4202 BERK vs control BERK mice, respectively (P < .004; Figure 2C). In fact, RBC half-life inversely correlated with the percentage of ISCs in BERK FT-4202 mice (supplemental Figure 6). Reduced hemolysis and increased RBC survival was accompanied by increased RBC counts (P < .002), Hb levels (P < .0003), and hematocrit fractions (P < .003; Figure 2D-F) in FT-4202 BERK mice at 2 weeks. However, the reticulocyte counts were only minimally reduced (Figure 2G), which may be a mouse erythropoiesis-specific phenomenon, due to the inherently high reticulocytes (50%) in BERK mice. It is conceivable that the increased HbS O2 affinity results in a compensatory increase in RBC production.34 Notably, FT-4202 administration increased HbS O2 affinity to levels well within the physiological range, similar to that of HbA,19,29 and hence should not cause tissue hypoxia. Furthermore, the improved Hb should not increase vaso-occlusions as RBCs have a reduced propensity to sickle and have reduced hemolysis. Thus, the increased Hb in FT-4202 BERK mice is consistent with improved RBC survival (Figure 2C) even with mildly increased HbS O2 affinity (Figure 1C-D; supplemental Figure 3). The platelet, mean corpuscular volume, mean corpuscular Hb concentration, and total white blood cell counts did not change with FT-4202 administration, although neutrophil counts were significantly lower in the FT-4202 group, suggesting reduced inflammation (supplemental Figure 5).
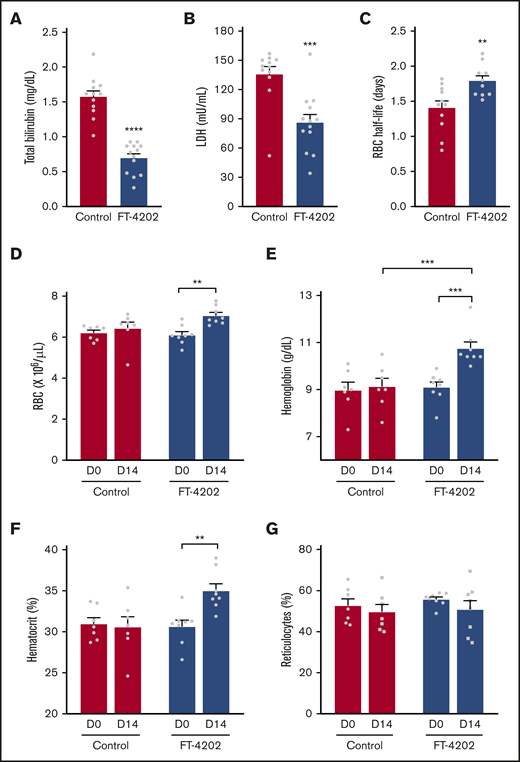
Oral administration of FT-4202 increases Hb and improves RBC parameters in BERK mice following FT-4202 administration. Total bilirubin (A) and LDH (B) concentrations in plasma collected from control and FT-4202 BERK mice after 2 weeks of placebo vs FT-4202 administration. Each symbol represents an individual animal: n = 12 control BERK mice and n = 13 FT-4202 BERK mice. (C) Half-life of RBCs in FT-4202 and control BERK mice, as determined by in vivo biotin labeling. Each symbol represents the RBC half-life of an individual mouse: n = 10 control BERK mice and n = 7 FT-4202 BERK mice. (D-G) Peripheral blood analysis at day 0 (D0) and after 2 weeks of placebo vs FT-4202 treatment (D14) showing RBC counts (D), Hb (E), hematocrit (F), and percentage of reticulocytes (G) in FT-4202 BERK as compared with control BERK mice. (D-G) n = 7 control BERK and n = 8 FT-4202 BERK. Comparison of day 0 vs day 14 was made using the paired Student t test and comparison between control and FT-4202 groups was made using the unpaired Student t test: **P < .01, ***P < .001, ****P < .0001.
Oral administration of FT-4202 increases Hb and improves RBC parameters in BERK mice following FT-4202 administration. Total bilirubin (A) and LDH (B) concentrations in plasma collected from control and FT-4202 BERK mice after 2 weeks of placebo vs FT-4202 administration. Each symbol represents an individual animal: n = 12 control BERK mice and n = 13 FT-4202 BERK mice. (C) Half-life of RBCs in FT-4202 and control BERK mice, as determined by in vivo biotin labeling. Each symbol represents the RBC half-life of an individual mouse: n = 10 control BERK mice and n = 7 FT-4202 BERK mice. (D-G) Peripheral blood analysis at day 0 (D0) and after 2 weeks of placebo vs FT-4202 treatment (D14) showing RBC counts (D), Hb (E), hematocrit (F), and percentage of reticulocytes (G) in FT-4202 BERK as compared with control BERK mice. (D-G) n = 7 control BERK and n = 8 FT-4202 BERK. Comparison of day 0 vs day 14 was made using the paired Student t test and comparison between control and FT-4202 groups was made using the unpaired Student t test: **P < .01, ***P < .001, ****P < .0001.
Other PKR activators are in clinical trials to increase Hb levels in patients with pyruvate kinase deficiency.20-22 Our studies show that FT-4202 has potential multimodal effects of reducing 2,3-DPG and increasing ATP levels in SCA RBCs. In SCA mice, these biochemical effects translated to significant biological benefits with increased HbS O2 affinity, including reduced ISCs, increased Hb levels and RBC survival, improved membrane deformability, and reduced PoS. In addition, we show that oral FT-4202 was well tolerated by SCA mice. A parallel FT-4202 human phase 1 study in healthy subjects and patients with sickle cell disease is ongoing (NCT03815695), and the preliminary data thus far are consistent with the mouse studies.3,4
For original data, please contact punam.malik@cchmc.org.
Acknowledgments
The authors thank Jeff Bailey and Victoria Summey (Comprehensive Mouse and Cancer Core) for assistance with mouse procedures, Scarlett Ripberger and the Erythrocyte Diagnostic Laboratory at CCHMC for their help with P50 measurements, Theodosia Kalfa, Katie Seu, and Rose Fessler for technical assistance with Lorrca Oxygenscan, Alexander G. Miethke for help with hemolysis assays, and Sydney Felker and Oluwabukola Gbotosho for help with blood sample processing. The authors also thank the Research Flow Cytometry Core at CCHMC for their services.
This work was supported by Forma Therapeutics, Inc.
Authorship
Contribution: A.S. supervised and performed experiments and analyzed and interpreted the data; M.C. optimized and performed the sickling kinetics and deformability experiments; K.W. performed all mouse experimental treatments and procedures and analyzed data; J.K. performed P50 measurements; A.M. performed hemolysis assays; K.F. analyzed ISC images and supervised pharmacokinetic/pharmacodynamic analysis; A.D., K.F., and S.G. provided intellectual input on study design; P.M. designed and supervised the entire study; A.S. and P.M. wrote the manuscript; and all authors edited the manuscript.
Conflict-of-interest disclosure: A.D. is a shareholder in Forma Therapeutics, Inc. K.F. is a current employee of, and a shareholder in, Forma Therapeutics, Inc. S.G. is a current employee of, and a shareholder in, Forma Therapeutics, Inc, and is a shareholder in AstraZeneca. P.M. holds patents with, and receives royalties from, Aruvant Sciences and CSL Behring, and has provided consultancy services to Aruvant Sciences and Forma Therapeutics, Inc. The remaining authors declare no competing financial interests.
Correspondence: Punam Malik, Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, ML 7013, 3333 Burnet Ave, Cincinnati, OH 45229; e-mail: punam.malik@cchmc.org.


