Key Points
KAT2A is required at different stages of human cord blood erythroid development with ATAC and SAGA complex specificity.
KAT2A regulates leukemia cell maintenance and identity through specific participation in ATAC and SAGA complexes.

Abstract
Epigenetic histone modifiers are key regulators of cell fate decisions in normal and malignant hematopoiesis. Their enzymatic activities are of particular significance as putative therapeutic targets in leukemia. In contrast, less is known about the contextual role in which those enzymatic activities are exercised and specifically how different macromolecular complexes configure the same enzymatic activity with distinct molecular and cellular consequences. We focus on KAT2A, a lysine acetyltransferase responsible for histone H3 lysine 9 acetylation, which we recently identified as a dependence in acute myeloid leukemia stem cells and that participates in 2 distinct macromolecular complexes: Ada two-A-containing (ATAC) and Spt-Ada-Gcn5-Acetyltransferase (SAGA). Through analysis of human cord blood hematopoietic stem cells and progenitors, and of myeloid leukemia cells, we identify unique respective contributions of the ATAC complex to regulation of biosynthetic activity in undifferentiated self-renewing cells and of the SAGA complex to stabilization or correct progression of cell type–specific programs with putative preservation of cell identity. Cell type and stage-specific dependencies on ATAC and SAGA-regulated programs explain multilevel KAT2A requirements in leukemia and in erythroid lineage specification and development. Importantly, they set a paradigm against which lineage specification and identity can be explored across developmental stem cell systems.
Introduction
KAT2A is a histone acetyltransferase required for correct mesodermal specification in the developing mouse embryo.1 It stabilizes pluripotency of mouse embryonic stem (ES) cells2 and is required for survival of neural stem and progenitor cells.3 In hematopoiesis, Kat2a has been shown to regulate proliferation and activation of T-cell subsets4 and maturation of invariant natural killer T (iNKT) cells,5 and it restricts terminal differentiation of granulocytic cells.6 We7 and others6 have not found that Kat2a play a central role in hematopoietic stem cells. In contrast, we have identified KAT2A as a requirement in acute myeloid leukemia (AML) cell lines and patient samples.8 Using a conditional knockout mouse model, we showed that Kat2a loss results in transcriptional instability of general metabolic regulation programs such as translation and ribosomal protein synthesis, leading to probabilistic loss of functional leukemia stem-like cells.7 Kat2a exerts transcriptional control through histone or nonhistone lysine acetylation, including in the examples cited within the hematopoietic system. Specifically, regulation of leukemia stem-like cells7 and control of T-cell subset activation4 are linked to histone H3 lysine 9 acetylation (H3K9ac) at gene promoters, a role that is evolutionarily conserved from the original identification of Gcn5 in yeast9 and associated with transcriptional activation.10 Granulocytic and iNKT cell maturation, on the other hand, depend on protein acetylation of key transcriptional regulators, respectively Cebpa6 and Egr2.5 More recently, Kat2a has been shown to catalyze histone succinylation, another acyl modification that, like H3K9ac, is associated with transcriptional activation.11
KAT2A exerts its activity in the context of 2 macromolecular complexes, Ada-Two-A-Containing (ATAC) and Spt-Ada-Gcn5-Acetyltransferase (SAGA) (Figure 1A). These complexes share the HAT activity by KAT2A, with a single subunit difference within the HAT module (TADA2A in ATAC and TADA2B in SAGA).12 Additionally, the 2 complexes exhibit different chromatin specificities and regulate distinct sets of genes.13 Integration of KAT2A in either complex is a requirement for its full HAT activity14 ; however, analysis of their differential contributions to KAT2A function in a given system has not been performed to date. The yeast KAT2A-containing SAGA was the first multimodular HAT complex to be isolated.10 Since then, several studies unveiled its molecular architecture: SAGA comprises 19 subunits organized in 4 functionally distinct modules15 , a structure highly conserved from yeast to human.12 In addition to the KAT2A participating HAT module, which also includes TADA2B, TADA3, and CCDC101 (SGF29), SAGA contains: (1) a H2B DUB module centered on USP22 enzymatic activity; (2) a core module comprising SPT20, which is specific to SAGA, and 5 TBP-associated factors (TAFs), 3 of which are shared with TFIID; and (3) the transcription factor interaction module, TRRAP. Two recent reports on the cryo-electron microscopy structure of the yeast SAGA complex described the interactions between the different modules with key implications for gene activation.16,17 The SAGA core module contains an octamer-like fold that facilitates TBP loading onto TATA promoters17 ; the 2 enzymatic HAT and DUB modules connect flexibly to the core,16,17 suggesting functional independence between them. The ATAC complex, in turn, is exclusive to multicellular eukaryotes.12 It was first linked to chromatin remodeling functions in Drosophila,18 where it preferentially targets histone H4.19 ATAC histone substrates in mammalian cells are less clear, although human ATAC preferentially modifies H3.20 ATAC-specific elements include DNA-binding subunit ZZZ3, HAT module component TADA2A, and YEATS2, which is required for assembly of the ATAC complex and has acetyl-reading activity.21 Moreover, ATAC comprises additional HAT activity by KAT14, which is essential for ATAC assembly and required in embryonic development.22 Additionally, both KAT2A-containing complexes have been implicated in malignant transformation.23 ATAC-YEATS2 was shown to be highly amplified in non–small cell lung cancer and required for malignant cell survival.21 SAGA complex cofactor TRRAP interacts with multiple proteins key to oncogenesis, such as c-Myc and E2F proteins.24 Expression of SAGA USP22 associates with an oncogenic signature of poor prognosis.25 However, recent studies propose a tumor suppressor function of USP22,26 including in AML,27 suggesting dependency on cell context for functional consequences and fate decisions.
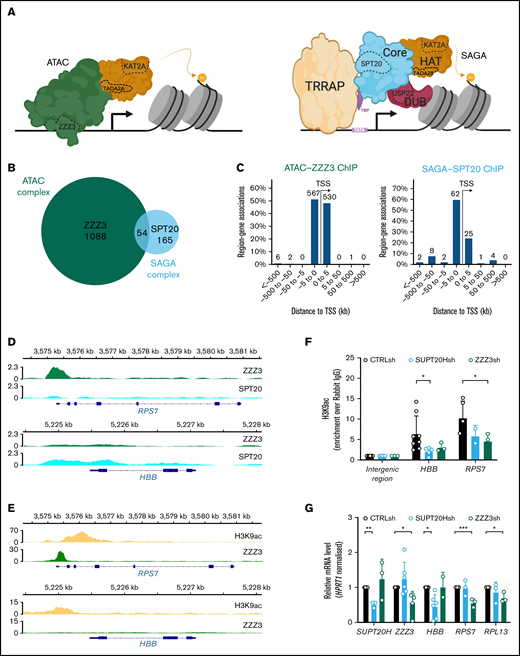
KAT2A-containing ATAC and SAGA complexes have unique targets in hematopoietic cells. (A) Schematic representation of the human ATAC (left) and SAGA (right) multiprotein complexes. SAGA (right) is organized into distinct structural and functional modules, colored similarly. The KAT2A-containing histone acetyltransferase (HAT) module is depicted in orange and partially hared with ATAC (left), with exception of TADA2B, which is replaced by TADA2A in ATAC. The histone deubiquitination (DUB) module is shown in red; the core module, which includes SPT20, in blue. TF-binding module, TRRAP, is shown in yellow. TATA-binding protein (TBP), in purple, is not part of the complex architecture, but it associates with SUPT3H to recruit SAGA to TATA box and facilitate transcription. The ATAC complex does not have a modular organization. Its main structure is shown in green and includes DNA-binding subunit ZZZ3. Subunits tested in this study are delineated with a dashed line. (B) Venn diagram of consensus ZZZ3 and SPT20 ChIP-seq binding from 2 independent experiments. (C) Genomic location of ZZZ3 (left) and SPT20 (right) ChIP-seq binding in K562 cells. Summary of consensus peaks from 2 independent ChIP-seq experiments is shown. (D) Representative ChIP-seq peak for ZZZ3 target in K562 cells (RPS7) and representative ChIP-seq peak for SPT20 target in K562 cells (HBB). (E) Publicly available ChIP-seq tracks for H3K9ac (ENCFF257CLC) and ZZZ3 (ENCFF856KCV) in K562 cells at the RPS7 and HBB loci retrieved from the ENCODE project portal (www.encodeproject.org). The RPS7 and HBB loci in panel D are represented confirming the presence of H3K9ac peaks and reproducing the selective ZZZ3 binding at RPS7 also observed in our data. (F) H3K9ac ChIP-qPCR analysis of representative SPT20 and ZZZ3 targets upon knockdown in K562 cells. N ≥ 3 independent experiments. Mean ± SEM of enrichment relative to rabbit IgG, with normalization to control intergenic region with no significant H3K9ac enrichment. Two-tailed Student t test for significance *P < .05. (G) qRT-PCR analysis of expression of ATAC and SAGA complex targets in K562 cells. N ≥ 3 independent experiments, mean ± SEM of gene expression relative to CTRLsh, normalized to HPRT1 housekeeping gene. Two-tailed Student t test for significance *P < .05, **P < .01, ***P < .001.
KAT2A-containing ATAC and SAGA complexes have unique targets in hematopoietic cells. (A) Schematic representation of the human ATAC (left) and SAGA (right) multiprotein complexes. SAGA (right) is organized into distinct structural and functional modules, colored similarly. The KAT2A-containing histone acetyltransferase (HAT) module is depicted in orange and partially hared with ATAC (left), with exception of TADA2B, which is replaced by TADA2A in ATAC. The histone deubiquitination (DUB) module is shown in red; the core module, which includes SPT20, in blue. TF-binding module, TRRAP, is shown in yellow. TATA-binding protein (TBP), in purple, is not part of the complex architecture, but it associates with SUPT3H to recruit SAGA to TATA box and facilitate transcription. The ATAC complex does not have a modular organization. Its main structure is shown in green and includes DNA-binding subunit ZZZ3. Subunits tested in this study are delineated with a dashed line. (B) Venn diagram of consensus ZZZ3 and SPT20 ChIP-seq binding from 2 independent experiments. (C) Genomic location of ZZZ3 (left) and SPT20 (right) ChIP-seq binding in K562 cells. Summary of consensus peaks from 2 independent ChIP-seq experiments is shown. (D) Representative ChIP-seq peak for ZZZ3 target in K562 cells (RPS7) and representative ChIP-seq peak for SPT20 target in K562 cells (HBB). (E) Publicly available ChIP-seq tracks for H3K9ac (ENCFF257CLC) and ZZZ3 (ENCFF856KCV) in K562 cells at the RPS7 and HBB loci retrieved from the ENCODE project portal (www.encodeproject.org). The RPS7 and HBB loci in panel D are represented confirming the presence of H3K9ac peaks and reproducing the selective ZZZ3 binding at RPS7 also observed in our data. (F) H3K9ac ChIP-qPCR analysis of representative SPT20 and ZZZ3 targets upon knockdown in K562 cells. N ≥ 3 independent experiments. Mean ± SEM of enrichment relative to rabbit IgG, with normalization to control intergenic region with no significant H3K9ac enrichment. Two-tailed Student t test for significance *P < .05. (G) qRT-PCR analysis of expression of ATAC and SAGA complex targets in K562 cells. N ≥ 3 independent experiments, mean ± SEM of gene expression relative to CTRLsh, normalized to HPRT1 housekeeping gene. Two-tailed Student t test for significance *P < .05, **P < .01, ***P < .001.
Herein, we attempted to characterize the role of KAT2A-containing ATAC and SAGA complexes in normal and leukemic blood cell function. We identified unique ATAC-specific requirements for specification or propagation of early erythroid-committed progenitors, which were distinct from the participation of the SAGA complex in progression of erythroid differentiation. In AML cells, ATAC and SAGA controlled distinct aspects of biosynthetic maintenance and identity preservation of leukemia cells, thus suggesting a dichotomy between ATAC- and SAGA-centered KAT2A functions that has implications for normal and malignant developmental decisions.
Methods
Cell lines
K562, MOLM13, and Kasumi-1 lines (kind gift from Brian Huntly, Cambridge, UK) and KG1a (kind gift from Joanna Baxter, Cambridge Blood and Stem Cell Biobank) were maintained in RPMI supplemented with 20% fetal bovine serum (FBS; Invitrogen), 1% penicillin/streptomycin/amphotericin (P/S/A; Invitrogen) and 2 mM L-Gln. HEK 293T cells were grown in Dulbecco’s modified Eagle medium supplemented with 10% FBS, P/S/A, and L-Gln as above. Cultures were kept at 37°C and 5% CO2.
Human CB CD34+ cells and AML patient samples
Cord blood (CB) samples and AML patient samples were obtained with informed consent under local ethical approval, research ethics committee 07MRE05-44 (Cambridge) and ethics committee code number 94/2016/O/Tess (Bologna). Mononuclear cells (MNC) were isolated by Ficoll-Paque density gradient (StemCell Technologies) centrifugation. CB MNC were enriched for CD34+ cells using the RosetteSep Human CB CD34 Pre-Enrichment Cocktail (Stem Cell Technologies) as per manufacturer’s instructions.
Lentiviral packaging and transduction
Viral constructs containing control short hairpin RNA (shRNA) (CTRLsh, noneukaryotic gene targeting) or shRNA targeting KAT2A (KAT2Ash), SGF29 (CCDC101sh), SUPT20H (SUPT20Hsh and SUPT20sh2) USP22 (USP22sh), ZZZ3 (ZZZ3sh), TADA2A (TADA2Ash and TADA2Ash2), and TADA2B (TADA2Bsh) (supplemental Table 1) were packaged in HEK 293T cells as previously described,28 using Turbofect (Thermo) or trans-IT (Mirus) as lipofection reagents. Cell lines were transduced overnight with 1-2 T75 packaging flask-equivalents/106 cells and washed the following day as described.28 Green florescent protein-positive (GFP+) cells were sorted and used in downstream assays 4 days later. CD34+ CB cells were prestimulated in serum-free medium (hematopoietic stem cell [HSC] expansion medium XF, Miltenyi Biotec) with stem cell factor (SCF), thrombopoietin (TPO), and Flt3L (respectively 200, 20, and 20 ng/mL) for up to 24 hours and transduced overnight using 2 flask-equivalents/2-3*105 cells. Cells were washed the following day and cultured in half the cytokine concentration for an additional 2 to 3 days prior to sorting.
Colony-forming cell assays
Sorted CB cells were seeded in methyl-cellulose-based semisolid medium (StemMACS HSC-CFU media complete with erythropoietin [EPO]; Miltenyi Biotec) for assessment of multilineage colony-forming activity. Sorted cells were counted and resuspended in Iscove modified Dulbecco medium 10% FBS supplemented with P/S/A and 2 mM L-Gln prior to addition to the semisolid medium, which was plated in duplicate onto 35 mm dishes with 200 to 300 cells/dish. In the case of colony-forming cell (CFC) assays performed in the presence of the KAT2A inhibitor MB-3 (Abcam), this was added to the methylcellulose in a final concentration of 200 μM and dispersed by vortexing prior to addition of CB cells as described.8 Colonies were scored by microscopy at 10 to 12 days at the point of full colony hemoglobinization.
Mixed lineage differentiation cultures
CB CD34+ cells were cultured for up to 7 days in serum-free liquid culture (StemSpan, Stem Cell Technologies) in the presence of SCF, Flt3-L, IL-3, IL-6 (1x CC100; Stem Cell Technologies), and EPO 3 U/μL (R&D). Transduced CD34+/HSC were seeded as GFP+ cells immediately after sorting. In other experiments, enriched CD34+ cells were resuspended in serum-free medium and cytokines in the presence of MB-3 (Abcam) 100 μM. Fresh cytokines, and, where appropriate, inhibitor and vehicle DMSO, were added every 2 to 3 days. At different time points, cells were tested by flow cytometry for the presence of CD34, CD13, and CD71/CD235a surface markers.
Erythroid differentiation and induction cultures
Transduced HSC were tested in erythroid differentiation conditions in serum-free liquid culture (HSC expansion medium xeno-free; Miltenyi Biotec) in the presence of hydrocortisone (10−5M), SCF (100 ng/mL), TPO (10 ng/mL), and EPO (3 U/mL). Cultures were followed up for 9 days with daily live and dead cell counts and regular flow cytometry analysis for CD34, CD71, and CD235a markers (supplemental Table 4). K562 cells were induced to the erythroid lineage in the presence of 1.5% DMSO as described.29 Cells were monitored for differentiation markers (CD71 and CD235a) and/or activation of erythroid-affiliated molecular programs during a 6-day culture period.
AML-MS5 coculture
MS5 murine bone marrow stromal cells were grown in Iscove modified Dulbecco medium supplemented with 10% FBS, 1% P/S/A, and 2 mM L-Gln. Cells were subcultured twice a week and passage 2 was used to carry out experiments in 48-well plates. AML cells were thawed in PBS 2% FBS, 1% P/S/A, and prestimulated with H5100 (Stem Cell Technologies) supplemented with IL-3 20 ng/mL, G-CSF 20 ng/mL, and TPO 20 ng/mL (3GT), 1% N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, and 1% P/S/A for ∼4 hours. Cells were then transduced with the desired lentiviral constructs in transduction media: H5100 with SCF (100 ng/mL), Flt-3 (100 ng/mL), IL-3 (60 ng/mL), TPO (10 ng/mL), 1% N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, and 1% P/S/A. Cells were washed twice in PBS 2% FBS and 1% P/S/A, once in H5100 1% P/S/A, and seeded onto MS5 stroma. Cultures were kept at 37°C and 5% CO2 for up to 3 weeks, with twice-weekly demipopulation and fresh addition of H5100-3GT. For KAT2A inhibition experiments, AML MNC were seeded onto MS-5 stroma in H5100-3GT in the presence of 100 μM of MB-3 (Abcam) or DMSO (0.1%) and demipopulated as above with fresh addition of MB-3. Cells were stained for flow cytometry analysis once a week (supplemental Table 4).
Chromatin immunoprecipitation
For chromatin immunoprecipitation (ChIP)-sequencing (ChIP-seq), chromatin was prepared in duplicate from K562 cells (9 sonication cycles, 30”ON/30”OFF) and immunoprecipitated using anti-SPT20 and anti-ZZZ3 sera prepared in the Tora Laboratory.13 All procedures, including library preparation and sequencing, were performed as described.7 For ChIP-quantitative polymerase chain reaction (qPCR), K562 cells were immunoprecipitated with anti-H3K9ac antibody or rabbit immunoglobulin G (IgG) (supplemental Table 2); eluted DNA was diluted and quantified by SYBR green qPCR using 2 μL DNA per triplicate reaction (primers in supplemental Table 3). Peak enrichments relative to rabbit IgG were determined using the 2ΔΔCt method with a reference intergenic region or KRT5.
Statistical analysis
Statistical analysis was performed in GraphPad Prism 7 software (GraphPad Software). Data are reported as mean ± standard error of the mean (SEM). Significance calculated by 2-tailed Student t test at P < .05 (see individual figure legends for details).
Results
KAT2A-containing ATAC and SAGA complexes have unique targets in hematopoietic cells
In order to identify unique ATAC and SAGA targets and characterize complex-specific roles of KAT2A in the blood system, we performed ChIP followed by next-generation sequencing (ChIP-seq) of ZZZ3 (ATAC-specific) and SPT20 (SAGA-specific) subunits (Figure 1A) in human K562 cells. K562 are a chronic myelogenous leukemia cell line with multilineage potential: cells display aspects of molecular differentiation into erythroid, megakaryocytic, and macrophage identities under defined cytokine conditions29 ; in steady state, K562 cells represent immature leukemia blasts with self-renewal properties. K562 cells have been previously used as a model for understanding molecular mechanisms of normal, particularly erythroid, and malignant blood specification.30-33 We made use of specific sera against human ZZZ3 and SPT20 and performed ChIP-seq experiments in duplicate in self-renewing K562 cells. As in previous reports,13 we found limited overlap between ATAC and SAGA targets (Figure 1B; supplemental File 1). Bound peaks were preferentially found in the vicinity of the transcriptional start site (Figure 1C) and were robustly enriched for ZZZ3 and SPT20 experimental targets, respectively (supplemental Figure 1A-B), as cataloged in the ENCODE database.34 ZZZ3 peaks were more proximal than SPT20’s, in contrast with previously described enhancer association of ATAC complexes in lymphoblast and HeLa cells.13 Our results are nevertheless consistent with a distinct ZZZ3 ChIP-seq experiment in human non–small cell lung cancer cells, which revealed strong enrichment of ZZZ3 peaks in regions ±1 kb of the transcriptional start site.21 This suggests that ATAC-complex enhancer region occupancy may be cell type or context-dependent. Interrogation of functional associations of ZZZ3 and SPT20 peaks highlighted complex-specific biologies (supplemental Figure 1C-D). These are reflected in distinct gene ontology categories associated with complex-specific peaks, which encompass RNA and ribosomal metabolism in the case of ATAC (Figure 1D; supplemental Figure 1C) and transcriptional activity for SAGA (supplemental Figure 1D). Moreover, SAGA specifically binds red blood cell–associated genes (Figure 1D), which may indicate a unique role in erythroid differentiation or identity. Inspection of publicly available K562 ChIP-seq datasets in the ENCODE database confirmed that SPT20 and ZZZ3-bound regions coincided with H3K9ac peaks, a hallmark of KAT2A enzymatic activity (Figure 1E).9 By making use of lentiviral-delivered shRNA targeting SPT20 or ZZZ3, we verified that selected SAGA and ATAC ChIP-seq target loci did indeed reduce promoter H3K9ac (Figure 1F) upon SUPT20H and ZZZ3 knockdown (Figure 1G; supplemental Figure 1E), respectively. The same target loci had reduced gene expression upon SUPT20H or ZZZ3 loss, suggesting that the chromatin binding had activating consequences for gene expression in a complex-specific manner (Figure 1G). Observed gene expression changes could be partially recapitulated upon KAT2A gene expression knockdown (supplemental Figure 1F-G), particularly in respect of ZZZ3-bound elements, which could reflect differential dependence and/or redundancy of histone acetyltransferase activity in either complex. Indeed, K562 cells were more clearly dependent on ZZZ3 than on SPT20 for propagation in culture (supplemental Figure 1H). Loss of KAT2A had an intermediate effect (supplemental Figure 1H), in line with independent activities of each complex in hematopoietic cell maintenance and/or identity, which might be balanced by the action of KAT2A.
We have recently established a role for KAT2A7,8 in maintenance of AML cells, which could be akin to our observations in K562 cells. In contrast, we and others failed to identify a specific requirement for Kat2a in normal mouse hematopoiesis,6,7 which contrasts with SAGA SPT20 chromatin binding of erythroid lineage loci. We therefore sought to systematically dissect SAGA and ATAC-mediated KAT2A contributions to normal and myeloid leukemic hematopoiesis in primary and cultured human cells.
ATAC is selectively required for erythroid specification from CB HSC
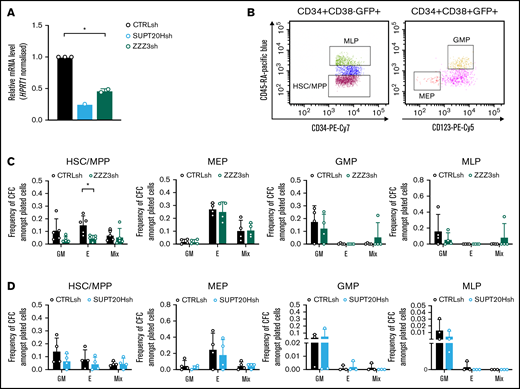
We started by inspecting putative differential functional contributions of SAGA and ATAC complexes to normal human hematopoiesis by transducing CD34+ CB cells with lentiviral-delivered ZZZ3 or SUPT20H shRNAs (Figure 2A). We flow-sorted transduced (GFP+) CD34+ cells as stem and multipotent progenitor cells (HSC) or as lineage-restricted myelo-lymphoid (MLP), megakaryocytic-erythroid (MEP), and granulocytic-monocytic progenitors (GMP) (Figure 2B) and assessed their cell differentiation and proliferation potential in CFC progenitor assays (supplemental Figure 2A). We did not observe differences in the stem and progenitor proportions of transduced cells with either construct (supplemental Figure 2B-C). In contrast, CFC assay output revealed a unique defect in erythroid specification from HSC upon ZZZ3 loss (Figure 2C), with no changes to generation of mixed-lineage or GM colonies. Colony formation from downstream lineage-restricted progenitors was not affected, suggesting a unique requirement for ZZZ3 in early erythroid commitment and a dispensable role for the ATAC component posterythroid commitment, as well as in the myelo-monocytic lineages. SUPT20H expression knockdown, on the other hand, did not result in changes in colony-forming efficiency from either HSC or lineage-restricted progenitors (Figure 2D). However, knockdown of the deubiquitinase component of the SAGA complex, USP22 (supplemental Figure 2D-E), resulted in loss of erythroid colony formation from committed MEP (supplemental Figure 2F), which did not significantly affect precommitment HSC and multipotent progenitors (supplemental Figure 2F). In agreement, proportions of CD34+ progenitors were unaffected (supplemental Figure 2G). Taken together, the data support contrasting roles for the ATAC and SAGA complexes in normal human blood progenitor biology, with early erythroid lineage specification dependent on ZZZ3. SAGA elements, in turn, participate in the development of the erythroid lineage postcommitment as suggested by gene regulation in K562 cells, although their requirement may be less absolute.
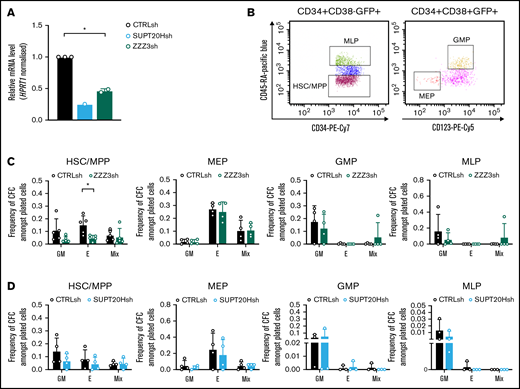
ATAC is selectively required for erythroid specification from CB HSC. (A) qRT-PCR validation of SUPT20H and ZZZ3 knockdown in human CB HSC. Representative experiment for SUPT20H; mean ± SEM of 2 individual experiments for ZZZ3; gene expression relative to CTRLsh, normalized to HPRT1 housekeeping gene. Paired 2-tailed Student t test for significance *P < .05. (B) Representative sorting plot for transduced HSC and progenitor cells from CB. (C) Frequency of CFC efficiency in the HSC/multipotent progenitor (MPP), MEP, GMP, and MLP compartments transduced with ZZZ3sh. Mean ± SEM of 5 individual CB samples (4 for GMP, MLP). Two-tailed paired Student t test for significance; *P < .05. (D) Frequency of CFC efficiency in the HSC/MPP, MEP, GMP, and MLP compartments transduced with SUPT20Hsh. Mean ± SEM of 4 individual CB samples. Two-tailed paired Student t test for significance; no significant differences.
ATAC is selectively required for erythroid specification from CB HSC. (A) qRT-PCR validation of SUPT20H and ZZZ3 knockdown in human CB HSC. Representative experiment for SUPT20H; mean ± SEM of 2 individual experiments for ZZZ3; gene expression relative to CTRLsh, normalized to HPRT1 housekeeping gene. Paired 2-tailed Student t test for significance *P < .05. (B) Representative sorting plot for transduced HSC and progenitor cells from CB. (C) Frequency of CFC efficiency in the HSC/multipotent progenitor (MPP), MEP, GMP, and MLP compartments transduced with ZZZ3sh. Mean ± SEM of 5 individual CB samples (4 for GMP, MLP). Two-tailed paired Student t test for significance; *P < .05. (D) Frequency of CFC efficiency in the HSC/MPP, MEP, GMP, and MLP compartments transduced with SUPT20Hsh. Mean ± SEM of 4 individual CB samples. Two-tailed paired Student t test for significance; no significant differences.
KAT2A regulates human CB erythroid progenitor specification and survival
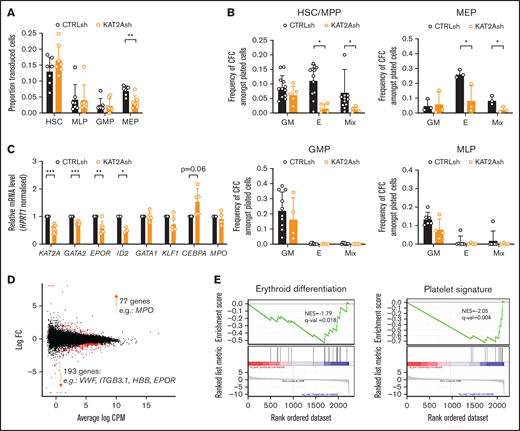
Given the differential role of ATAC and SAGA elements in the erythroid lineage, we asked if KAT2A itself was required in human CB progenitor specification or differentiation. To reiterate, chemical inhibition of KAT2A activity at the 100 μM dose effective in AML cell lines had not resulted in an overall reduction in colony formation from CB CD34+ cells,8 and we and others had not found a requirement for Kat2a in mouse bone marrow hematopoiesis.1,2 Nevertheless, reinspection of Kat2a expression in bone marrow subpopulations of our conditional knockout model7 revealed that gene expression ablation in MEP was less extensive than in myelo-monocytic cells (n = 4; mean ± standard deviation: 0.39 ± 0.20 in MEP vs 0.13 ± 0.08 in GMP, 2-tailed Student t test; P = .054), compatible with selective preservation of unexcised Kat2a allele-carrying MEP cells and a possible partial requirement for Kat2a expression. In agreement, transduction of CB CD34+ cells with KAT2A shRNA resulted in a significant and specific proportional decrease in KAT2A-depleted MEP (Figure 3A), suggesting a defect in specification of CB progenitors committed to the erythroid lineage. Inspection of progenitor activity of CB CD34+ cells in CFC assays upon KAT2A knockdown (supplemental Figure 3A) or chemical inhibition (supplemental Figure 3B) detected a reduction in erythroid colony formation. The same reduction was observed upon depletion of the Tudor domain protein SGF29 (gene CCDC101) (supplemental Figure 3C-D), a KAT2A partner common to SAGA and ATAC complexes. Equally, culture of KAT2A knockdown (supplemental Figure 3E) or MB-3 inhibited (supplemental Figure 3F) CD34+ cells in mixed-lineage differentiation conditions resulted in relative loss of differentiated erythroid cells, compatible with hindered specification of the red blood cell lineage. Dissection of HSC and progenitor CD34+ compartments revealed that loss of KAT2A affected erythroid lineage output from both HSC and MEP (Figure 3B) without changes to myelo-monocytic lineage output (Figure 3B), thus capturing both ATAC and SAGA-associated effects. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) transcriptional analysis of KAT2A knockdown HSC shows downregulation of early erythroid regulatory genes, namely GATA2 and EPOR, (Figure 3C), a finding compatible with the global reduction of erythroid and megakaryocytic programs apparent upon RNA-seq analysis (Figure 3D-E; supplemental File 2). Analysis of effects of KAT2A knockdown on cell cycle and apoptosis is suggestive of putative effects on cell survival peri-erythroid commitment (supplemental Figure 3G-H), with specific increases in apoptotic HSC and MEP, which do not extend to other lineages. Taken together, the data support a function for KAT2A in specification and/or survival of erythroid-megakaryocytic progenitors.
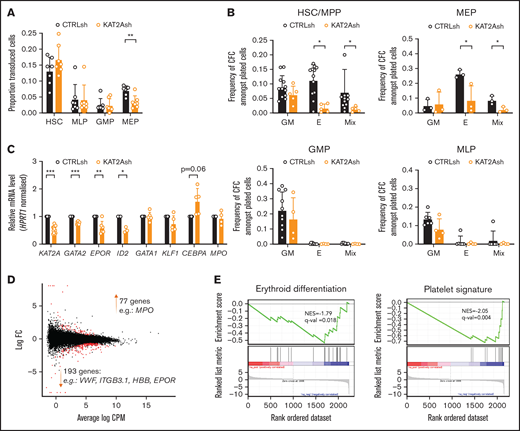
KAT2A regulates human CB erythroid progenitor specification and survival. (A) Proportion of KAT2Ash-transduced CB HSC and progenitors. Mean ± SEM of >7 individual sorting experiments. Two-tailed paired Student t test for significance; **P < .01. (B) Frequency of CFC efficiency in the HSC/MPP, MEP (top), GMP, and MLP (bottom) compartments transduced with KAT2Ash. Mean ± SEM of 7 individual CB samples. Two-tailed paired Student t test for significance; *P < .05. (C) qRT-PCR analysis of expression of erythroid-associated genes in KAT2Ash-transduced HSC from individual CB samples. N ≥ 3 independent experiments, mean ± SEM of gene expression relative to CTRLsh, normalized to HPRT1 housekeeping gene. Two-tailed paired Student t test for significance *P < .05, **P < .01, ***P < .001. (D) MA plot of RNA-seq gene expression analysis of differentially-expressed genes (red) in CTRLsh vs KAT2Ash transduced HSC/MPP cells. (E) Gene set enrichment analysis plot for erythroid differentiation (right) and platelet signature (left) in the RNA-seq data in panel D. Gene signature identifiers are ADDYA erythroid differentiation by HEMIN and GNATENKO platelet signature, respectively, as obtained from the UC San Diego and BROAD Institute Molecular Signatures Database (MSigDB).
KAT2A regulates human CB erythroid progenitor specification and survival. (A) Proportion of KAT2Ash-transduced CB HSC and progenitors. Mean ± SEM of >7 individual sorting experiments. Two-tailed paired Student t test for significance; **P < .01. (B) Frequency of CFC efficiency in the HSC/MPP, MEP (top), GMP, and MLP (bottom) compartments transduced with KAT2Ash. Mean ± SEM of 7 individual CB samples. Two-tailed paired Student t test for significance; *P < .05. (C) qRT-PCR analysis of expression of erythroid-associated genes in KAT2Ash-transduced HSC from individual CB samples. N ≥ 3 independent experiments, mean ± SEM of gene expression relative to CTRLsh, normalized to HPRT1 housekeeping gene. Two-tailed paired Student t test for significance *P < .05, **P < .01, ***P < .001. (D) MA plot of RNA-seq gene expression analysis of differentially-expressed genes (red) in CTRLsh vs KAT2Ash transduced HSC/MPP cells. (E) Gene set enrichment analysis plot for erythroid differentiation (right) and platelet signature (left) in the RNA-seq data in panel D. Gene signature identifiers are ADDYA erythroid differentiation by HEMIN and GNATENKO platelet signature, respectively, as obtained from the UC San Diego and BROAD Institute Molecular Signatures Database (MSigDB).
SAGA facilitates progression of erythroid differentiation
Colony-forming progenitor assays of CB CD34+ cells associated with loss of KAT2A-containing ATAC complexes with impairment of early erythroid and megakaryocytic specification (Figure 2C). The data also suggested that KAT2A-containing SAGA complexes may play a role in erythroid differentiation postcommitment (supplemental Figure 2F). However, CFC assays are end-point assays that do not permit a detailed dissection of mechanisms of differentiation, and we attempted to overcome this limitation using a liquid culture system. Differentiation of total CB CD34+ cells in the presence of hydrocortisone, SCF, TPO, and EPO resulted in progressive loss of CD34 and accumulation of glycophorin A (CD235a)-positive cells, denoting effective erythroid differentiation (Figure 4A), which was not qualitatively impaired upon KAT2A knockdown (Figure 4B). However, erythroid differentiation cultures initiated from KAT2A-depleted HSC were quantitatively impaired, being severely restricted in their expansion (Figure 4C). Reduced KAT2A knockdown cell growth was accompanied by a general trend toward increased cell death (supplemental Figure 4A), compatible with the observed KAT2A role in HSC and MEP cell survival. To circumvent the limiting cell numbers in transduced primary CB cultures, we inspected erythroid lineage progression and associated molecular programs in K562 cells. Exposure of K562 cells to 1.5% DMSO resulted in accumulation of CD235a+ cells and progressive loss of the earlier CD71 transferrin receptor (supplemental Figure 4B) and was accompanied by loss of GATA2 and upregulation of EPOR and TAL1, denoting molecular erythroid lineage progression29 (supplemental Figure 4C). DMSO-induced erythroid cultures initiated by KAT2A knockdown cells followed a similar molecular progression (Figure 4D). In contrast, SUPT20H depletion significantly perturbed induction of erythroid programs in K562 cells, which failed to downregulate GATA2, and variably upregulated EPOR and TAL1, suggestive of a defect in lineage progression (Figure 4D). We confirmed the findings with a second SUPT20H shRNA (SUPT20Hsh2) (supplemental Figure 4D-F). Accordingly, inspection of the expression pattern of SAGA-specific elements during in vitro maturation of committed erythroid progenitors from human CB35 showed that several elements associate with late differentiation (supplemental Figure 4G), including members of the core, the SAGA-specific HAT subunit TADA2B, and the H2B deubiquitinase USP22 (supplemental File 3), which specifically affected erythroid colony formation from MEP in human CB. In contrast, inspection of the detailed single-cell profiling of erythroid development by Tusi et al36 captured Kat2a and Zzz3 enrichment, but no elements of the SAGA complex, at the transition of multipotent progenitors to the erythroid and megakaryocytic lineages (supplemental Figure 4H; supplemental File 4), compatible with an early specification role of ATAC that does not extend into terminal erythroid differentiation. Indeed, loss of ZZZ3 allowed downregulation of GATA2 and upregulation of TAL1 during K562 erythroid induction (Figure 4D). However, EPOR was not consistently upregulated (Figure 4D), a trend confirmed upon knockdown of a second ATAC element, TADA2A (supplemental Figure 4E), which resulted in minimal EPOR upregulation, which was significantly lower than that observed for CTRLsh (1.52 ± 0.13 TADA2Ash vs 3.19 ± 0.49 CTRLsh fold change relative to undifferentiated cells; P = .0214). Inspection of the K562 ZZZ3 ChIP-seq data showed binding of the EPOR locus in one of the 2 replicate samples (supplemental File 1; supplemental Figure 4I), and we checked if ZZZ3 knockdown resulted in loss of H3K9ac at the EPOR promoter. Indeed, we observed that ZZZ3, but not SUPT20H loss, decreased EPOR H3K9 acetylation (Figure 4E), suggesting that KAT2A requirement for early erythroid specification may be mediated by ATAC control of EPOR expression and downstream signaling. However, we could not detect consistent downregulation of EPOR in steady-state K562 cells upon ZZZ3 or TADA2A knockdown (supplemental Figure 4J), suggesting that regulation of EPOR by ATAC may be context-dependent and not an absolute requirement for KAT2A-mediated regulation of early erythroid specification or survival.
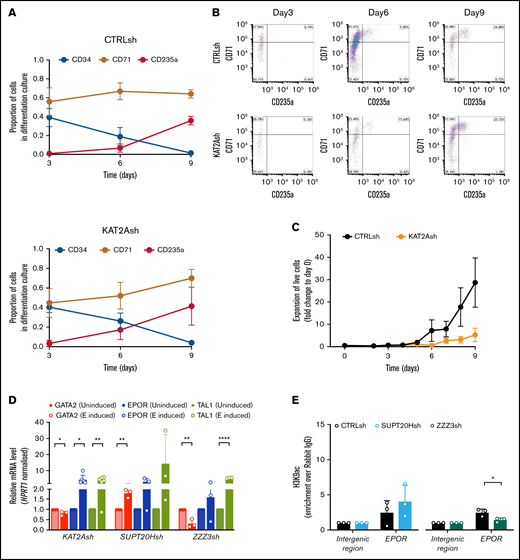
KAT2A SAGA and ATAC complexes differentially affect erythroid lineage progression. (A) Erythroid differentiation of HSC/MPP sorted from CD34+ cells of individual CB samples transduced with CTRLsh (top) and KAT2Ash (bottom) in liquid cultures. CD34 marks HSC and progenitors; CD71 and CD235a mark early and late differentiated erythroid cells. Data summarize mean ± SEM of 4 independent differentiation experiments. (B) Flow cytometry quantitative analysis of CTRLsh and KAT2Ash HSC in erythroid differentiation cultures in panel A. Representative plots. (C) Expansion of CTRLsh and KAT2Ash HSC in erythroid differentiation cultures in panel A. (D) qRT-PCR analysis of erythroid gene expression progression in K562 cells transduced with KAT2Ash, SUPT20Hsh, or ZZZ3sh and treated with 1.5% DMSO for erythroid molecular induction. Mean ± SD of n >3 independent experiments; data are represented relative to day 0 normalized to HPRT1 housekeeping gene. Two-tailed paired Student t test for significance *P < .05, **P < .01, ****P < .0001. CTRLsh-transduced cells for the same experiment shown in supplemental Figure 4C. (E) H3K9ac ChIP-qPCR analysis of EPOR locus in K562 cells upon SUPT20H and ZZZ3 knockdown. N ≥ 3 independent experiments. Mean ± SEM of enrichment relative to rabbit IgG, with normalization to control intergenic region with no significant H3K9ac enrichment. Two-tailed Student t test for significance *P < .05.
KAT2A SAGA and ATAC complexes differentially affect erythroid lineage progression. (A) Erythroid differentiation of HSC/MPP sorted from CD34+ cells of individual CB samples transduced with CTRLsh (top) and KAT2Ash (bottom) in liquid cultures. CD34 marks HSC and progenitors; CD71 and CD235a mark early and late differentiated erythroid cells. Data summarize mean ± SEM of 4 independent differentiation experiments. (B) Flow cytometry quantitative analysis of CTRLsh and KAT2Ash HSC in erythroid differentiation cultures in panel A. Representative plots. (C) Expansion of CTRLsh and KAT2Ash HSC in erythroid differentiation cultures in panel A. (D) qRT-PCR analysis of erythroid gene expression progression in K562 cells transduced with KAT2Ash, SUPT20Hsh, or ZZZ3sh and treated with 1.5% DMSO for erythroid molecular induction. Mean ± SD of n >3 independent experiments; data are represented relative to day 0 normalized to HPRT1 housekeeping gene. Two-tailed paired Student t test for significance *P < .05, **P < .01, ****P < .0001. CTRLsh-transduced cells for the same experiment shown in supplemental Figure 4C. (E) H3K9ac ChIP-qPCR analysis of EPOR locus in K562 cells upon SUPT20H and ZZZ3 knockdown. N ≥ 3 independent experiments. Mean ± SEM of enrichment relative to rabbit IgG, with normalization to control intergenic region with no significant H3K9ac enrichment. Two-tailed Student t test for significance *P < .05.
In conclusion, our analysis of normal erythroid lineage progression indicates that KAT2A plays stage-specific roles, which differentially align with each of the 2 complexes it integrates. On the one hand, KAT2A regulates specification and survival of erythroid progenitors through participation in the ATAC complex, which controls biosynthetic activity, and to some extent, EPOR expression. On the other hand, KAT2A fine-tunes progression of erythroid lineage programs through participation in SAGA, which is only required in fully-committed progenitors and may play a contributory rather than an absolute role. Notably, SUPT20H regulation of erythroid molecular progression in K562 cells exceeds the contribution of KAT2A itself. It is possible that SAGA complexes also use the ortholog histone acetyltransferase KAT2B in this context, which can compensate for KAT2A loss. Increased KAT2B expression in progressed erythroid differentiation, accompanying upregulation of SAGA elements (supplemental File 3), is compatible with this view.
KAT2A complexes uniquely maintain proliferation and identity of MOLM13 AML cells
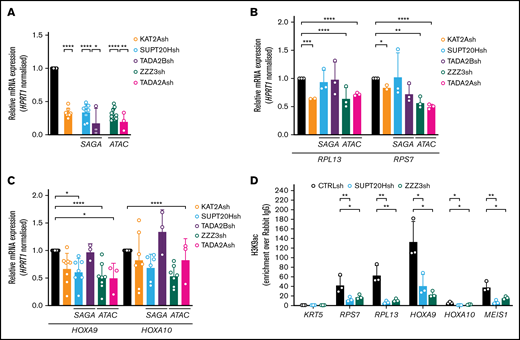
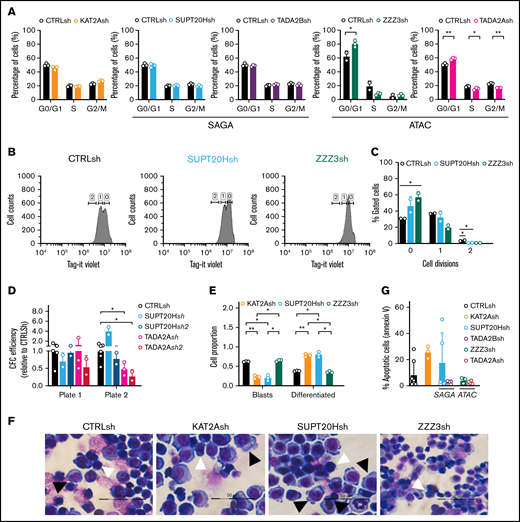
Having demonstrated that KAT2A-containing ATAC and SAGA complexes differentially sustain early lineage establishment and differentiation of erythroid progenitor cells from human CB, we asked whether they also made specific contributions to AML biology. We started by investigating the model AML cell line MOLM13, which we had previously shown to be dependent on KAT2A expression and activity.8 We knocked down expression of ATAC elements ZZZ3 and TADA2A and of SAGA components SUPT20H and TADA2B (Figure 5A), all of which, like KAT2A depletion, impacted expansion of MOLM13 cultures (supplemental Figure 5A-B). At a molecular level, depletion of ATAC, but not SAGA elements, impacted expression of ribosomal protein genes (Figure 5B), in line with a pervasive control of protein biosynthetic activity by the ATAC complex, which we observed in K562 cells and others also reported in lung cancer cell lines depleted of ZZZ3 and YEATS2 expression.21,37 Analysis of expression of self-renewal signature genes HOXA9 and HOXA10 showed a more extensive association with ATAC elements (Figure 5C). Interestingly, despite the selective impact of ATAC elements on gene expression, loss of both SUPT20H and ZZZ3 resulted in depletion of H3K9ac in the respective ribosomal protein and HOX gene promoters (Figure 5D), suggesting that locus regulation may be more complex than the promoter binding observed in K562 cells. Loss of KAT2A itself affects ribosomal protein gene expression but not HOXA genes (Figure 5B-C). This recapitulates our MLL-AF9 Kat2a knockout leukemia mouse model7 in which progressive depletion of leukemia stem-like cells does not seem dependent on the classical Hoxa signature. Despite some ambiguity at a molecular level, ATAC and SAGA complexes do make a distinct cellular impact on the biology of MOLM-13 cells. Loss of ATAC ZZZ3 and TADA2A arrest cell cycle progression in G0/G1 (Figure 6A; supplemental Figure 5C). The proliferation defect is reflected by a reduced number of ZZZ3 knockdown cells entering cell division (Figure 6B-C) over a 3-day culture period (supplemental Figure 5D) as captured by divisional tracking (supplemental Figure 5E). We asked if the proliferation defect consequent to ATAC loss affected MOLM13 cell self-renewal as measured by in vitro replating of CFC assays. Indeed, although neither ATAC (TADA2A) nor SAGA (SUPT20H) knockdowns impacted initial colony formation, ATAC alone affected replating of colony assays (Figure 6D; supplemental Figure 5F), suggesting an effect on self-renewal. In contrast, loss of SAGA SUPT20H resulted in extensive differentiation of MOLM13 cells (Figure 6E-F; supplemental Figure 5G), suggesting a role in preservation of cell identity, which is not unlike SAGA contribution to the development of human erythroid cells. Despite a trend toward enhanced apoptosis of KAT2A knockdown cells (Figure 6G; supplemental Figure 5H), which could be an end-point consequence of enhanced differentiation (Figure 6E-F; supplemental Figure 5G), we did not observe a consistent apoptotic response to knockdown of SAGA (SUPT20H and TADA2B) or indeed ATAC (ZZZ3 and TADA2A) elements (Figure 6G; supplemental Figure 5H), suggesting that control of cell survival is not central to KAT2A complex-mediated maintenance of MOLM13 AML cells.
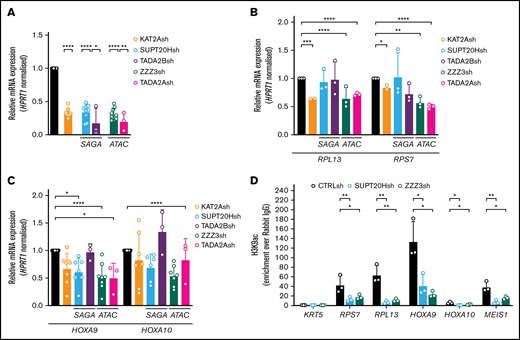
ATAC and SAGA differentially impact histone acetylation and gene expression in MOLM13 AML cells. (A) qRT-PCR validation of KAT2A, SUPT20H, TADA2B (SAGA-specific subunits), ZZZ3, and TADA2A (ATAC-specific subunits) knockdown in MOLM-13 cells. N ≥3 independent experiments, mean ± SEM of gene expression relative to CTRLsh, normalized to HPRT1 housekeeping gene. Two-tailed Student t test for significance *P < .05, **P < .01, ****P < .0001. (B) qRT-PCR analysis of ribosomal protein gene expression in MOLM-13 cells transduced with KAT2Ash, SAGA-specific SUPT20Hsh and TADA2Bsh, and ATAC-specific ZZZ3sh and TADA2Ash. N ≥3 independent experiments, mean ± SEM of gene expression relative to CTRLsh, normalized to HPRT1 housekeeping gene. Two-tailed Student t test for significance *P < .05, **P < .01, ***P < .001, ****P < .0001. (C) qRT-PCR analysis of self-renewal gene signature in MOLM-13 transduced with KAT2Ash, SAGA-specific SUPT20Hsh and TADA2Bsh, and ATAC-specific ZZZ3sh and TADA2Ash. N = 2 biological replicates, each run as 2 or 3 technical repeats; mean ± SEM of relative gene expression relative to CTRLsh, normalized to HPRT1 housekeeping gene. Two-tailed nested Student t test for significance *P < .05, ****P < .0001. (D) H3K9ac ChIP-qPCR analysis of ribosomal protein genes and self-renewal genes in MOLM-13 cells upon knockdown of SAGA and ATAC elements SUPT20H and ZZZ3, respectively. N ≥ 3 independent experiments. Mean ± SEM of enrichment relative to rabbit IgG, with normalization to control region in KRT5 locus with no significant H3K9ac enrichment. Two-tailed Student t test for significance *P < .05, **P < .01.
ATAC and SAGA differentially impact histone acetylation and gene expression in MOLM13 AML cells. (A) qRT-PCR validation of KAT2A, SUPT20H, TADA2B (SAGA-specific subunits), ZZZ3, and TADA2A (ATAC-specific subunits) knockdown in MOLM-13 cells. N ≥3 independent experiments, mean ± SEM of gene expression relative to CTRLsh, normalized to HPRT1 housekeeping gene. Two-tailed Student t test for significance *P < .05, **P < .01, ****P < .0001. (B) qRT-PCR analysis of ribosomal protein gene expression in MOLM-13 cells transduced with KAT2Ash, SAGA-specific SUPT20Hsh and TADA2Bsh, and ATAC-specific ZZZ3sh and TADA2Ash. N ≥3 independent experiments, mean ± SEM of gene expression relative to CTRLsh, normalized to HPRT1 housekeeping gene. Two-tailed Student t test for significance *P < .05, **P < .01, ***P < .001, ****P < .0001. (C) qRT-PCR analysis of self-renewal gene signature in MOLM-13 transduced with KAT2Ash, SAGA-specific SUPT20Hsh and TADA2Bsh, and ATAC-specific ZZZ3sh and TADA2Ash. N = 2 biological replicates, each run as 2 or 3 technical repeats; mean ± SEM of relative gene expression relative to CTRLsh, normalized to HPRT1 housekeeping gene. Two-tailed nested Student t test for significance *P < .05, ****P < .0001. (D) H3K9ac ChIP-qPCR analysis of ribosomal protein genes and self-renewal genes in MOLM-13 cells upon knockdown of SAGA and ATAC elements SUPT20H and ZZZ3, respectively. N ≥ 3 independent experiments. Mean ± SEM of enrichment relative to rabbit IgG, with normalization to control region in KRT5 locus with no significant H3K9ac enrichment. Two-tailed Student t test for significance *P < .05, **P < .01.
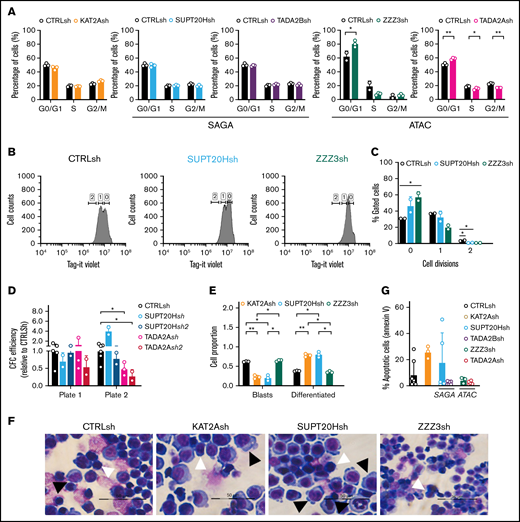
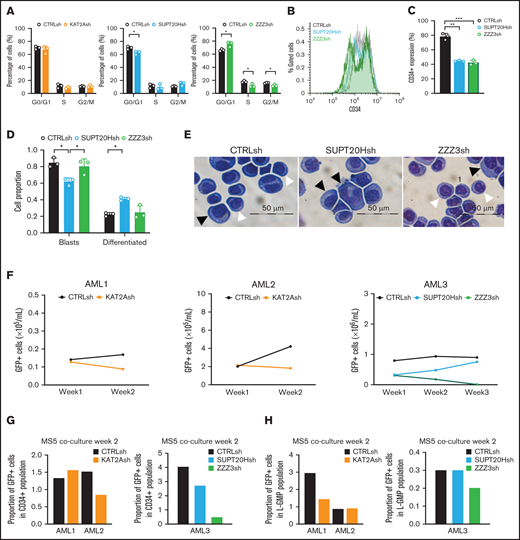
ATAC maintains self-propagation and SAGA blocks differentiation of MOLM13 AML cells. (A) Quantification of flow cytometry analysis of cell cycle in MOLM-13 cells transduced with KAT2Ash, SAGA-specific SUPT20Hsh and TADA2Bsh, and ATAC-specific ZZZ3sh and TADA2Ash. Corresponding representative plots in supplemental Figure 5C. Mean ± SEM of 3 independent experiments. Two-tailed Student t test for significance *P < .05, **P < .01. (B) Representative flow cytometry plots of divisional tracking of MOLM-13 cells transduced with CTRLsh, SUPT20Hsh, and ZZZ3sh and loaded with the Tag-IT violet dye (Biolegend) after 3 days of culture. Regions 0, 1, and 2 represent the number of cell divisions relative to initial loading control (also see supplemental Figure 5D-E). (C) Quantification of results in panel B representing the distribution of MOLM-13 cells transduced with CTRLsh, SUPT20Hsh, and ZZZ3sh undergone 0-2 cell divisions after 3 days in culture. Mean ± SEM of 3 independent experiments. Two-tailed Student t test for significance *P < .05. (D) Colony replating of MOLM-13 cells transduced with SUPT20Hsh1, SUPT20Hsh2 (SAGA), TADA2Ash1, and TADA2Ash2 (ATAC) as a measure of in vitro self-renewal. Mean ± SEM of 2-3 independent experiments. Two-tailed Student t test for significance *P < .05. (E) Quantification of blast-like and differentiated cells in MOLM-13 cultures transduced with CTRLsh, KAT2Ash, SUPT20Hsh, and ZZZ3sh. Scoring of 3 randomly selected fields of >100 cells; Two-tailed Student t test for significance; *P < .05, **P < .01. (F) Representative photographs of MOLM-13 cytospins. White arrow heads denote blast-like cells; black arrow heads denote differentiated cells. Bar represents 50 μm. (G) Quantification of Annexin V+ apoptotic cells in MOLM13 cultures analyzed by flow cytometry (representative plots in supplemental Figure 4F). Mean ± SEM of n > 3 independent experiments. Two-tailed Student t test for significance.
ATAC maintains self-propagation and SAGA blocks differentiation of MOLM13 AML cells. (A) Quantification of flow cytometry analysis of cell cycle in MOLM-13 cells transduced with KAT2Ash, SAGA-specific SUPT20Hsh and TADA2Bsh, and ATAC-specific ZZZ3sh and TADA2Ash. Corresponding representative plots in supplemental Figure 5C. Mean ± SEM of 3 independent experiments. Two-tailed Student t test for significance *P < .05, **P < .01. (B) Representative flow cytometry plots of divisional tracking of MOLM-13 cells transduced with CTRLsh, SUPT20Hsh, and ZZZ3sh and loaded with the Tag-IT violet dye (Biolegend) after 3 days of culture. Regions 0, 1, and 2 represent the number of cell divisions relative to initial loading control (also see supplemental Figure 5D-E). (C) Quantification of results in panel B representing the distribution of MOLM-13 cells transduced with CTRLsh, SUPT20Hsh, and ZZZ3sh undergone 0-2 cell divisions after 3 days in culture. Mean ± SEM of 3 independent experiments. Two-tailed Student t test for significance *P < .05. (D) Colony replating of MOLM-13 cells transduced with SUPT20Hsh1, SUPT20Hsh2 (SAGA), TADA2Ash1, and TADA2Ash2 (ATAC) as a measure of in vitro self-renewal. Mean ± SEM of 2-3 independent experiments. Two-tailed Student t test for significance *P < .05. (E) Quantification of blast-like and differentiated cells in MOLM-13 cultures transduced with CTRLsh, KAT2Ash, SUPT20Hsh, and ZZZ3sh. Scoring of 3 randomly selected fields of >100 cells; Two-tailed Student t test for significance; *P < .05, **P < .01. (F) Representative photographs of MOLM-13 cytospins. White arrow heads denote blast-like cells; black arrow heads denote differentiated cells. Bar represents 50 μm. (G) Quantification of Annexin V+ apoptotic cells in MOLM13 cultures analyzed by flow cytometry (representative plots in supplemental Figure 4F). Mean ± SEM of n > 3 independent experiments. Two-tailed Student t test for significance.
Overall, the data support distinct roles for ATAC and SAGA in the model AML cell line MOLM13. ATAC controls biosynthetic activity and impacts self-renewal of MOLM13 cells through molecular control of HOXA genes and regulation of cell division. SAGA, on the other hand, preserves cell identity and impedes differentiation. The dichotomy of effects captures different aspects of KAT2A regulation of MOLM13 cells and MLL-AF9–driven leukemia7 and broadly aligns with differential participation of KAT2A complexes in normal hematopoiesis.
KAT2A-containing SAGA and ATAC complexes regulate cultured and primary CD34+ AML cells
Finally, we sought to extend our analysis of AML cells to include CD34+ lines not originally covered by the screen that established KAT2A as a critical regulator of AML cells.8 Similarly, we investigated primary CD34+ AML patient samples for which stem-like compartments have been more clearly established.38 We initially focused on the CD34+ AML cell lines KG1a and Kasumi1, both of which represent minimally differentiated AML French-American-British M1 and M2, respectively, and carry distinct molecular abnormalities (KG1a, FGFR1OP2-FGFR1 fusion; Kasumi1, RUNX1-RUNX1T1 fusion) representative of human disease.39,40 In both models, loss of ATAC, but not SAGA elements, restricted cell cycle progression (Figure 7A; supplemental Figure 6A). Similar to what had been observed in MOLM13 cells, none of the cell lines displayed increased apoptosis in response to loss of KAT2A complexes (supplemental Figure 6B-C). We investigated differentiation consequences of loss of ATAC or SAGA components in KG1a cells and observed downregulation of the CD34 marker by flow cytometry upon ZZZ3 and SUPT20H loss (Figure 7B-C). However, only SUPT20H knockdown resulted in morphological differentiation along the monocytic lineage (Figure 7D-E), supporting the notion that the undifferentiated state of AML cells is sustained through SAGA activity. Finally, we attempted to transduce primary CD34+ AML patient blasts (supplemental Table 5) with our lentiviral vectors delivering shRNAs against KAT2A, SUPT20H, or ZZZ3. We could observe transduced GFP+ cells in <50% of the samples transduced and kept the cells in culture for 2-3 weeks in the presence of MS5 stroma as described,41 following up their expansion as well as the preservation of CD34+ and candidate phenotypic leukemia stem-like (L-)GMP (supplemental Figure 7A). Downregulation of KAT2A and ZZZ3, but in the sample analyzed, not of SUPT20H, resulted in reduced expansion of transduced AML cells (Figure 7F), which specifically affected CD34+ (Figure 7G) and/or L-GMP (Figure 7H), suggesting a loss of self-renewal potential. Chemical inhibition of KAT2A activity using MB-38 had a similar effect on the preservation of CD34+ cells (supplemental Figure 7B), which impacted the proportion of L-GMP (supplemental Figure 7C).
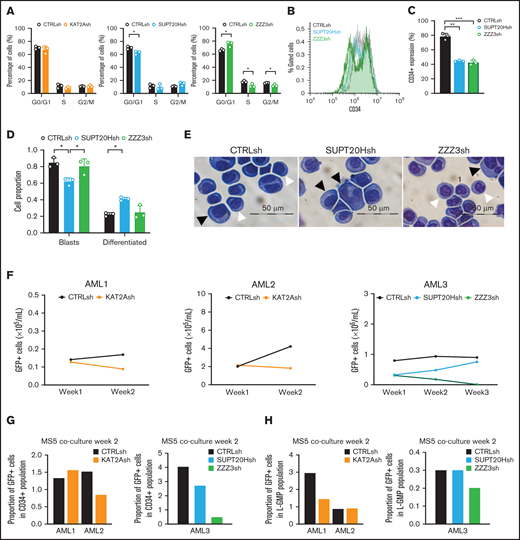
KAT2A complex activity maintains propagation of undifferentiated cultured and primary CD34+AML cells. (A) Quantification of flow cytometry analysis of cell cycle in Kasumi-1 cells transduced with KAT2Ash, SUPT20Hsh, and ZZZ3sh. Mean ± SEM of 3 independent experiments. Two-tailed Student t test for significance *P < .05. (B) Representative flow cytometry overlay plot of analysis of undifferentiated marker CD34 in KG1a cells transduced with CTRLsh, SUPT20Hsh, and ZZZ3sh. (C) Quantification of CD34+cells in transduced KG1a cells in panel B. N = 3 independent experiments. Two-tailed Student t test for significance **P < .01. (D) Quantification of blast-like and differentiated cells in KG1a cultures transduced with CTRLsh, SUPT20Hsh, and ZZZ3sh. Scoring of 3 randomly selected fields of >100 cells; Two-tailed Student t test for significance; *P < .05. (E) Representative photographs of KG1a cytospins. White arrow heads denote blast-like cells; black arrow heads denote differentiated cells. Bar represents 50 μm. (F) Growth of human primary AML cells (CD34+samples; details in supplemental File 5) transduced with CTRLsh, KAT2Ash, SUPT20Hsh, or ZZZ3sh and maintained in the MS5 coculture system for 2-3 weeks. (G) Percentage of GFP+ CD34+ cells in AML samples in panel F analyzed at week 2 of the MS5 coculture. Analysis gates are presented in supplemental Figure 6A. Data are normalized to global GFP level to correct for unequal GFP transduction levels. (H) Percentage of GFP+ GMP-like (L-GMP) cells at week 2 of the MS5 coculture system in AML samples in panel F. Analysis gates in supplemental Figure 7A. GFP transduction correction as in panel G.
KAT2A complex activity maintains propagation of undifferentiated cultured and primary CD34+AML cells. (A) Quantification of flow cytometry analysis of cell cycle in Kasumi-1 cells transduced with KAT2Ash, SUPT20Hsh, and ZZZ3sh. Mean ± SEM of 3 independent experiments. Two-tailed Student t test for significance *P < .05. (B) Representative flow cytometry overlay plot of analysis of undifferentiated marker CD34 in KG1a cells transduced with CTRLsh, SUPT20Hsh, and ZZZ3sh. (C) Quantification of CD34+cells in transduced KG1a cells in panel B. N = 3 independent experiments. Two-tailed Student t test for significance **P < .01. (D) Quantification of blast-like and differentiated cells in KG1a cultures transduced with CTRLsh, SUPT20Hsh, and ZZZ3sh. Scoring of 3 randomly selected fields of >100 cells; Two-tailed Student t test for significance; *P < .05. (E) Representative photographs of KG1a cytospins. White arrow heads denote blast-like cells; black arrow heads denote differentiated cells. Bar represents 50 μm. (F) Growth of human primary AML cells (CD34+samples; details in supplemental File 5) transduced with CTRLsh, KAT2Ash, SUPT20Hsh, or ZZZ3sh and maintained in the MS5 coculture system for 2-3 weeks. (G) Percentage of GFP+ CD34+ cells in AML samples in panel F analyzed at week 2 of the MS5 coculture. Analysis gates are presented in supplemental Figure 6A. Data are normalized to global GFP level to correct for unequal GFP transduction levels. (H) Percentage of GFP+ GMP-like (L-GMP) cells at week 2 of the MS5 coculture system in AML samples in panel F. Analysis gates in supplemental Figure 7A. GFP transduction correction as in panel G.
Thus, the data suggest that KAT2A-containing complexes, particularly ATAC, play a role in the maintenance of candidate leukemia self-renewing CD34+ cells, with SAGA putatively contributing to the AML differentiation block.
Discussion
In this study, we dissected the roles of KAT2A in normal and malignant human hematopoiesis through analysis of the macromolecular complexes ATAC and SAGA, in which KAT2A exerts its HAT activity. We have aligned the roles of ATAC with maintenance of biosynthetic molecular activity, namely control of ribosomal protein and translation-associated genes. SAGA, on the other hand, participates in activation or maintenance of molecular programs underlying the characteristics of individual cell types, a property we equate with preservation of cell identity.
In the context of normal hematopoiesis, ATAC control of ribosomal protein genes selectively affects early stages of erythropoiesis, a lineage previously shown to be uniquely dependent on ribosomal assembly and rates of protein synthesis42,43 as captured by the selective erythroid defects of congenital ribosomopathies such as Diamond-Blackfan Anemia.44 Selective loss of individual ribosomal proteins45 or of global ribosomal protein regulators such as BMI146 have been shown to associate with proliferation and survival of early erythroid committed cells, with minimal changes to terminal differentiation. In the case of ATAC, the early nature of the defect may be potentiated by deregulation of the EPOR locus, a candidate instructor of erythroid lineage commitment.47 However, the transcriptional consequences of reduced EPOR promoter acetylation upon ATAC loss are inconsistent, suggesting that it may contribute to rather than drive the erythroid defect. In the context of leukemia, ribosomal protein abundance and translational activity have been shown to sustain AML self-renewal.42 We found that ribosomal protein genes are selectively affected by loss of ATAC elements, which may nevertheless control other self-renewal associated genes, namely the underlying HOXA signature associated with KMT2A/MLL rearrangements.
In contrast with the universal targeting of ribosomal protein genes by ATAC,21,37 SAGA may not regulate a constant set of genes but instead sustain the expression of the unique transcriptomes of individual cell types. This could in part be achieved through control of other transcriptional regulators as suggested by the functional ChIP enrichments in our data. In the context of normal hematopoiesis, SAGA may exert its putative identity control by fine-tuning normal progression of erythroid programs, a role for which its requirement may be contributory rather than absolute. The fact that SAGA, as indeed ATAC, requirements seem restricted to the erythroid lineage may reflect the distinctive nature of erythroid/megakaryocytic commitment and differentiation, which segregate from the stem cell root upstream of myeloid and lymphoid lineages48,49 with de novo establishment of transcription programs.50 In the context of malignant hematopoiesis, SAGA participates in maintenance of the AML differentiation block, irrespective of the exact stage of cell differentiation, compatible with a general control of cell identity through preservation of existing transcriptional programs.51 Indeed, like KAT2A itself, we previously captured several SAGA elements (SUPT20H, TRRAP, TAF5L, TAF6L, and TAF12) as candidate genetic vulnerabilities in AML cell lines in a Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) drop-out screen.8 In contrast, we failed to capture ATAC components in the same screen,8 which nevertheless identified ribosomal proteins as universal vulnerabilities. One possible explanation is that cultured AML cell lines may be permissive to mild or moderate reduction in ribosomal assembly and protein synthesis. Our observed milder consequences to cell expansion of ATAC element loss are compatible with this view, as the screen readout8 was dependent on acute consequences to proliferation or survival. Also, the specific ATAC-mediated defect of in vitro self-renewal of CFCs is progressively displayed, becoming apparent upon colony replating but not affecting initial colony formation. Nevertheless, ATAC biosynthetic regulatory activities are required for supporting proliferation and, at least in some contexts self-renewal, of cells dependent on SAGA-mediated regulation through putative stabilization of their unique transcriptional programs. The resulting KAT2A activity can be described as preservation of the cellular status quo with minimal deviation toward alternative fate choices.
Preservation of the cellular status quo as the suggested operating mode of KAT2A-containing complexes is compatible with the role of KAT2A in sustaining rather than initiating gene transcription,52 as well as in stabilizing promoter activity and minimizing variability in transcriptional output as recently described by us in leukemia7 and ES cells.2 Control of transcriptional variability by KAT2A-like acetyltransferases is well established in yeast GCN5,53 which integrates a SAGA-like complex that was shown to regulate the global transcriptome.54 Our7 and other5 observations in mammalian cells is that Kat2a only acetylates a subset of promoters, which are circumscribed into limited numbers of SAGA-specific and ATAC-specific targets (this paper and Krebs et al13 ). Although precise identification of KAT2A complex targets may be limited by the tools available, the number of promoters dependent on KAT2A for H3K9ac is restricted7 and functionally aligns with the categories we captured for ATAC and SAGA binding, suggesting a more restricted use in mammalian species as compared with yeast. Furthermore, our data suggest that ATAC and SAGA control of gene transcription may be more specific than control of promoter H3K9 acetylation, indicating that specificity of transcriptional regulation by either complex may rely on unique effects of their additional enzymatic activities, namely H2B deubiquitination by USP22 and H4 acetylation by KAT14.55 Accordingly, our inspection of USP22 requirements in erythropoiesis suggest that they align with the stage specificity of SAGA but may exceed the effects of loss of SUPT20H and more directly resemble postcommitment contributions of KAT2A. Future studies investigating single and combined requirements of ATAC and SAGA enzymatic subunits in locus and cellular regulation will enhance our understanding of the transcriptional control of cell state and fate decisions, including in leukemia and the normal blood system.
In respect of control of transcriptional variability, it is somewhat surprising that Kat2a-dependent promoters that respond to Kat2a KO with loss of H3K9ac and increased transcriptional variability are enriched in ATAC-dependent translation-associated genes7 and indeed enriched for ZZZ3 bound targets (ENCODE ChIP-seq significance tool; P = 1.94e−4). Although both complexes regulate H3K9ac, control of transcriptional variability is conserved from yeast, which only contains a SAGA-like complex. One possibility is that ATAC and SAGA have evolved to nucleate distinct molecular functions of KAT2A. A more trivial explanation is that SAGA being preferentially recruited to cell type–specific promoters, comparison with ChIP-seq targets in different cell types may fail to detect enrichment despite a similar molecular regulation. Future analysis of transcriptional variability in response to parallel ATAC and SAGA-specific depletions in the same cell type should clarify this discrepancy. Additional studies will also be needed to understand the discrepancies between the identified requirements for KAT2A in human hematopoiesis and the broadly unperturbed hematopoietic development in the mouse. As we pointed out, detailed analysis of our own conditional Kat2a knockout model7 suggests a retention of MEP carrying unexcised Kat2a alleles, compatible with a requirement in erythroid specification. Also, we reported reduced mixed colony formation from HSC in Kat2a KO cells, which could portray a defect in early erythroid specification. We did not observe defective colony formation from MEP but cannot exclude these may have been masked by persistence of unexcised cells. On the other hand, a distinct conditional knockout model6 also failed to identify gross defects in hematopoiesis, although detailed analysis of lineage specification was not performed. To date, no other knockouts of ATAC and SAGA subunits have been investigated in the hematopoietic system, at the exception of Usp22, which did not affect stem and progenitor compartments or normal myeloid differentiation27 but had a surprising tumor suppressor effect in Kras-driven transformation. However, a recent screen of deubiquitinating enzymes required for 4-week bone marrow engraftment that identified Usp15 as a regulator of HSC activity in vitro and in vivo56 also identified Usp2227 as a possible hit, reenforcing the notion that additional analysis of ATAC and SAGA-mediated roles in mouse hematopoiesis is necessary.
In conclusion, we have identified unique requirements for ATAC and SAGA complexes in normal and leukemic hematopoiesis that are suggestive of respective pervasive roles in maintenance of biosynthetic activity and cell type–specific programs through complex-coordinated control of histone modifications and transcriptional stability. Individual cell types may be differentially dependent on the activity of either complex as suggested by a relatively stronger dependence on ATAC at early stages of erythropoiesis. Indeed, this may pose an opportunity for targeting of KAT2A activity through the SAGA complex in leukemia. In support of the ATAC/SAGA functional dichotomy, a paper contemporary with this report identifies distinct targets and roles of ATAC and SAGA elements in mouse ES cells.57 Similar to our findings, it associates ATAC with regulation of ribosomal protein genes and a pervasive control of pluripotency; SAGA, on the other hand, specifically regulates naïve pluripotency genes, akin to a role in cell identity. However, the authors find that SAGA and ATAC functional roles in mouse ES cells are independent of HAT activity,57 which is distinct from our observations in the human hematopoietic system and, clearly, in AML. Detailed understanding of distinct chromatin regulatory strategies of SAGA and ATAC complexes for gene transcription and their usage in different cells will clarify the discrepancy. This knowledge can be explored for stability or perturbation of cell fate in regeneration and disease.
Acknowledgments
The authors thank the Cell Phenotyping Hub NIHR BRC, the University of Cambridge Department of Pathology Flow Sorting Facility, and Yanping Guo at the UCL Cancer Institute Flow Cytometry Translational Technology Platform for their expert support in cell sorting.
This work was funded by a Rosetrees Trust Studentship to L.A. (M650) and a Kay Kendall Leukaemia Fund Intermediate Fellowship (KKL888) to C.P. Work in the C.P. laboratory was also funded by a Leuka John Goldman Fellowship for Future Science (2017) and a Wellcome Trust/University of Cambridge ISSF Grant to C.P. S.G. was funded by a Lady Tata Memorial Trust Studentship, a Trinity Henry Barlow Trust Studentship, and by the Cambridge Trust. This study was also supported by an NIH RO1 grant (1R01GM131626-01) to L.T., by Agence Nationale de la Recherche (ANR) Program grants, AAPG2019 PICen to L.T., ANR PRCI AAPG2019 EpiCAST to L.T., grant ANR-10-LABX-0030-INRT, and a French State fund managed by the ANR under the frame program Investissements d’Avenir ANR-10IDEX-0002-02 to Institut de Genetique et de Biologie Moleculaire et Cellulaire (IGBMC). Samples were provided by the Cambridge Blood and Stem Cell Biobank, which is supported by the Cambridge NIHR BRC Wellcome Trust MRC Stem Cell Institute and the Cambridge Experimental Cancer Medicine Centre, UK.
Authorship
Contribution: C.P. conceived the study; L.A. and C.P. designed the study; L.A., E.F., S.W., A.F.D., G.G., S.K., M.M.-S., H.D., A. Chandru, R.A., R.S., and S.G. collected and assembled data; G.G., D.F., A. Curti, E.S., B.J.P.H., and L.T. contributed critical reagents; L.A., E.F., S.W., R.K., and C.P. analyzed data; L.A. and C.P. interpreted data; L.A. and C.P. wrote the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Cristina Pina, College of Health, Medicine and Life Sciences, Division of Biosciences, Brunel University London, Uxbridge UB8 3PH, United Kingdom; e-mail: cristina.pina@brunel.ac.uk.