

Key Points
Non-RAR gene rearrangements have been associated with patients with AML resembling APL but the underlying pathogenesis is unclear.
NUP98-JADE2 perturbs wild-type JADE2 and retinoic acid signaling thereby contributing to an APL-like phenotype.
Abstract
Acute promyelocytic leukemia (APL) is a specific subtype of acute myeloid leukemia (AML) characterized by block of differentiation at the promyelocytic stage and the presence of PML-RARA fusion. In rare instances, RARA is fused with other partners in variant APL. More infrequently, non-RARA genes are rearranged in AML patients resembling APL. However, the underlying disease pathogenesis in these atypical cases is largely unknown. Here, we report the identification and characterization of a NUP98- JADE2 fusion in a pediatric AML patient showing APL-like morphology and immunophenotype. Mechanistically, we showed that NUP98-JADE2 could impair all-trans retinoic acid (ATRA)-mediated transcriptional control and myeloid differentiation. Intriguingly, NUP98-JADE2 was found to alter the subcellular distribution of wild-type JADE2, whose down-regulation similarly led to attenuated ATRA-induced responses and myeloid activation, suggesting that NUP98-JADE2 may mediate JADE2 inhibition. To our knowledge, this is the first report of a NUP98-non-RAR rearrangement identified in an AML patient mimicking APL. Our findings suggest JADE2 as a novel myeloid player involved in retinoic acid-induced differentiation. Despite lacking a rearranged RARA, our findings implicate that altered retinoic acid signaling by JADE2 disruption may underlie the APL-like features in our case, corroborating the importance of this signaling in APL pathogenesis.
Introduction
Acute promyelocytic leukemia (APL) is featured by arrested promyelocytic differentiation and the PML-RARA fusion.1 In variant APL, RARA is fused with other partners, which dictate disease biology and treatment sensitivity.2 Rarely, genomic rearrangements involving non-RARA genes have also been reported in patients with acute myeloid leukemia (AML) mimicking APL.2-4 Although these findings suggest that alternative pathways may mediate the promyelocytic differentiation block, the underlying mechanisms have remained obscure. The nucleoporin 98 (NUP98) gene encoding a component of the nuclear pore complex is rearranged in various hematological malignancies associated with poor outcomes.5,6 Here, we identify and characterize a novel NUP98-JADE2 fusion in a pediatric patient with AML with morphological and immunophenotypic features resembling APL. Our findings provided mechanistic insights into how a non-RARA rearrangement contributes to the development of an APL-like phenotype.
Case description
The patient was a 15-month-old girl who presented with fever and gum bleeding. Physical examination showed no pallor, jaundice, petechiae, bruises, or lymphadenopathy. Complete blood count revealed a hemoglobin level of 10.3 g/dL, platelet count of 227 × 109/L, white blood cell count of 5.8 × 109/L, and neutrophil count of 0.6 × 109/L with 19% circulating blasts. Prothrombin time and activated partial thromboplastin time were 13.9 seconds (reference, 9.5-12 seconds) and 42.7 seconds (reference, 28.2-37.4 seconds), respectively. d-dimer levels were 4982 ng/mL (reference, <500 ng/mL). Bone marrow examination showed 61% of medium- to large-sized blasts with hypergranular cytoplasm (Figure 1A). The blasts were positive for CD13, CD15, CD33, CD117, and myeloperoxidase but negative for CD34, HLA-DR, CD14, CD41, glycophorin A, and lymphoid markers (CD10/CD19/CD20/CD22/CD79a/CD2/CD3/CD5/CD7/TdT) by flow cytometry. Nearly all the blasts showed Sudan Black B positivity. A diagnosis of APL was suspected and all-trans retinoic acid (ATRA; 25 mg/m2 per day) was initiated while awaiting confirmatory results.

Identification of the NUP98-JADE2 fusion. (A) Left, May-Grünwald-Giemsa staining of leukemic blasts in the diagnostic bone marrow (BM). The blasts were medium to large size with fine nuclear chromatin and nucleoli showing hypergranular cytoplasm. Occasional cells with 1 to 2 Auer rods (arrow) were seen but no faggot cell was detected. Right, Sudan Black B-stained positive blasts. Original magnification ×1000. (B) A karyotype performed on the diagnostic BM revealed 46,XX,t(5;11)(q31;p15). (C) Upper, a schematic diagram showing NUP98, JADE2, and NUP98-JADE2 generated by ProteinPaint. The phenylalanine-glycine (FG)/glycine-leucine-phenylalanine-glycine (GLFG) repeats and Gle2-binding-sequence (GLEBS) domain in the amino-terminal portion of NUP98 are retained in the fusion protein. Lower, RT-PCR analysis of NUP98-JADE2 (NJ) and JADE2-NUP98 (JN) fusions in the diagnostic BM. Whole BM cells were used for the analysis. Amplification of GAPDH served as the control. Expected products are indicated by arrowheads. Sanger sequencing of the NJ PCR product is also shown. (D) Immunofluorescence analysis of NUP98-JADE2 and wild-type JADE2. HeLa cells were transfected with pCMV-HA-NUP98-JADE2 or pCMV-HA-JADE2, and the tagged proteins were detected as described in supplemental Methods. DAPI was used for nuclear staining. Original magnification ×1000. (E) Interaction between NUP98-JADE2 and wild-type JADE2. 293T cells were transfected with the indicated Myc- and HA-tagged expression vectors. Myc-tagged proteins were immunoprecipitated and samples were analyzed by immunoblotting for NUP98-JADE2 (NJ) and JADE2 (J2) detection. Similar results were obtained when HA-tagged proteins were immunoprecipitated before immunoblotting (data not shown). (F) Altered subcellular distribution of wild-type JADE2 in the presence of NUP98-JADE2. Upper, HeLa cells were cotransfected with both pCMV-HA-NUP98-JADE2 and pCMV-Myc-JADE2, and the tagged proteins were detected as mentioned previously. Original magnification ×1000. Lower, HeLa cells were transfected with the indicated Myc- and HA-tagged expression vectors. Protein lysates were analyzed by immunoblotting. No apparent change in JADE2 expression was noted when NUP98-JADE2 was coexpressed.
Identification of the NUP98-JADE2 fusion. (A) Left, May-Grünwald-Giemsa staining of leukemic blasts in the diagnostic bone marrow (BM). The blasts were medium to large size with fine nuclear chromatin and nucleoli showing hypergranular cytoplasm. Occasional cells with 1 to 2 Auer rods (arrow) were seen but no faggot cell was detected. Right, Sudan Black B-stained positive blasts. Original magnification ×1000. (B) A karyotype performed on the diagnostic BM revealed 46,XX,t(5;11)(q31;p15). (C) Upper, a schematic diagram showing NUP98, JADE2, and NUP98-JADE2 generated by ProteinPaint. The phenylalanine-glycine (FG)/glycine-leucine-phenylalanine-glycine (GLFG) repeats and Gle2-binding-sequence (GLEBS) domain in the amino-terminal portion of NUP98 are retained in the fusion protein. Lower, RT-PCR analysis of NUP98-JADE2 (NJ) and JADE2-NUP98 (JN) fusions in the diagnostic BM. Whole BM cells were used for the analysis. Amplification of GAPDH served as the control. Expected products are indicated by arrowheads. Sanger sequencing of the NJ PCR product is also shown. (D) Immunofluorescence analysis of NUP98-JADE2 and wild-type JADE2. HeLa cells were transfected with pCMV-HA-NUP98-JADE2 or pCMV-HA-JADE2, and the tagged proteins were detected as described in supplemental Methods. DAPI was used for nuclear staining. Original magnification ×1000. (E) Interaction between NUP98-JADE2 and wild-type JADE2. 293T cells were transfected with the indicated Myc- and HA-tagged expression vectors. Myc-tagged proteins were immunoprecipitated and samples were analyzed by immunoblotting for NUP98-JADE2 (NJ) and JADE2 (J2) detection. Similar results were obtained when HA-tagged proteins were immunoprecipitated before immunoblotting (data not shown). (F) Altered subcellular distribution of wild-type JADE2 in the presence of NUP98-JADE2. Upper, HeLa cells were cotransfected with both pCMV-HA-NUP98-JADE2 and pCMV-Myc-JADE2, and the tagged proteins were detected as mentioned previously. Original magnification ×1000. Lower, HeLa cells were transfected with the indicated Myc- and HA-tagged expression vectors. Protein lysates were analyzed by immunoblotting. No apparent change in JADE2 expression was noted when NUP98-JADE2 was coexpressed.
Reverse transcription-polymerase chain reaction (RT-PCR) and fluorescence in situ hybridization failed to detect PML-RARA or variant RARA fusions. Consistently, cytogenetic studies revealed 46,XX,t(5;11)(q31;p15)[20] (Figure 1B), indicating the absence of RARA translocations. No somatic mutation/copy number change in 141 myeloid-related genes (supplemental Table 1) including FLT3, NPM1, WT1, CEBPA, KIT, and K/NRAS was detected by targeted next-generation sequencing. In view of the PML-RARA negativity, ATRA was stopped after administration for 6 days and a shortened prothrombin time (12.9 seconds) and activated partial thromboplastin time (36.8 seconds) was noted. A diagnosis of AML not otherwise specified was made and the patient was treated with the Nordic Society of Paediatric Haematology and Oncology-AML 2004 protocol.7 Complete remission was achieved after the first induction course. However, the patient developed cardiac failure after the sixth course of high-dose cytarabine and etoposide and died 9 months after the initial diagnosis.
Methods
Detailed experimental procedures are provided in supplemental Methods. The study was approved by the institutional review board of the Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee. Informed consent has been obtained from the parents of the patient.
Results and discussion
RNA-sequencing revealed an in-frame NUP98-JADE2 fusion in which exon 13 of NUP98 was fused to exon 2 of JADE2, which encodes a plant homeodomain zinc finger protein (Figure 1C). The full-length fusion transcript is predicted to encode a 1304-amino acid protein containing the amino-terminal portion of NUP98 plus the entire JADE2. The reciprocal JADE2-NUP98 fusion was undetected. NUP98-JADE2 exhibited a speckled nuclear pattern in transfected HeLa cells (Figure 1D). On the contrary, wild-type JADE2 was uniformly and predominantly expressed in the nucleus. Coimmunoprecipitation studies in 293T cells showed that NUP98-JADE2 could heterodimerize with wild-type JADE2, suggesting that the fusion protein may perturb JADE2-associated pathways (Figure 1E). Accordingly, we found altered subcellular distribution of wild-type JADE2 with a speckled appearance overlapping with that of NUP98-JADE2 when the 2 proteins were overexpressed in HeLa cells (Figure 1F).
Deregulated RARA transcriptional control is central to APL pathogenesis.1 The association of NUP98-JADE2 with APL-like features prompted us to examine the transcriptional properties of NUP98-JADE2 using a retinoic acid-response element (RARE)-luciferase reporter. Without ATRA, NUP98-JADE2 exerted no apparent effect on luciferase activity, whereas wild-type JADE2 mildly activated the reporter (Figure 2A). Intriguingly, NUP98-JADE2 dose-dependently inhibited ATRA-mediated transcriptional activation (Figure 2B; supplemental Figure 1). In contrast, no significant effect on luciferase induction was noted when wild-type JADE2 was overexpressed (Figure 2B). Consistent with these findings, overexpression of NUP98-JADE2 in NB4 and U937 cells diminished ATRA-induced CD11b expression, suggesting impaired myeloid differentiation (Figure 2C; supplemental Figure 2). Because NUP98-JADE2 altered wild-type JADE2 distribution and thus potentially affected its functions, we investigated the role of the wild-type protein. Interestingly, knockdown of JADE2 by small-interfering RNAs (siRNAs) in NB4 promyelocytic leukemia cells could similarly attenuate ATRA-induced expression of CD11b and CCAAT/enhancer binding protein ε (CEBPE) (Figure 2D), a marker of terminal granulopoiesis.8 Further transcriptome analysis of JADE2 knocked-down NB4 cells identified 89 genes whose expression was consistently altered (56 downregulated/33 upregulated) by ≥1.3-fold with 2 different siRNAs in 2 independent experiments (Figure 2E; supplemental Table 2). Pathway analysis revealed that the downregulated genes were enriched in inflammatory response, myeloid activation, and cytokine production (Figure 2F). In contrast, no enriched pathway (adjusted P < .05) was identified from the upregulated genes. By analyzing the expression profiles of the downregulated genes in purified myeloid cells, 65% of the genes were found to exhibit increased expression (≥twofold) during granulocytic maturation (Figure 2G). Knockdown of JADE2 also repressed RARE-mediated gene transcription (supplemental Figure 3).
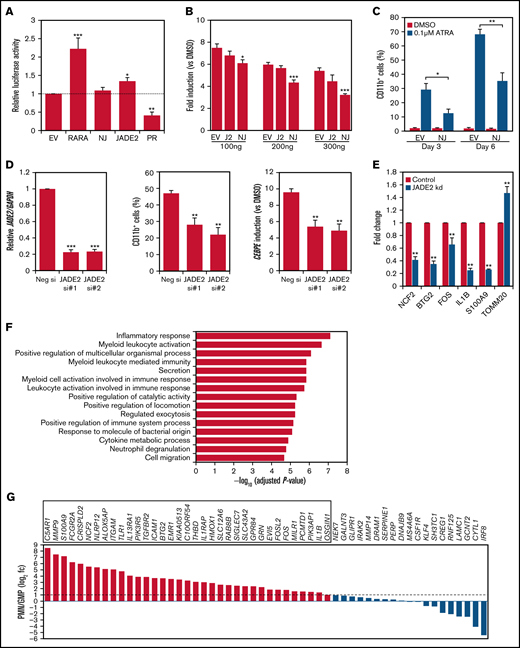
Molecular characterization of NUP98-JADE2 and JADE2 in ATRA-mediated responses and myeloid differentiation. (A) HeLa cells were cotransfected with RARE reporter and the indicated pCMV-HA plasmids. EV, empty vector; NJ, NUP98-JADE2; PR, PML-RARA. Luciferase activities were determined as described in supplemental Methods. (B) HeLa cells were cotransfected with RARE reporter and an increasing pCMV-HA-NUP98-JADE2 (NJ), pCMV-HA-JADE2 (J2), or the empty pCMV-HA (EV), and then treated with ATRA or DMSO for 6 hours before analysis. Results are presented as fold induction vs DMSO treatment. (A-B) *P < .05, **P < .01, and ***P < .001 vs EV, respectively by 1-way analysis of variance followed by Dunnett test. (C) NB4 cells transduced with NUP98-JADE2 (NJ) or control (EV) lentiviruses were treated with ATRA or DMSO. CD11b on GFP-positive cells was measured by flow cytometry. *P < .05 and **P < .01, respectively by paired t test. (D) Left, confirmation of JADE2 knockdown in NB4 cells after 24 hours of siRNA transfection by quantitative RT-PCR. After the transfection, cells were treated with 0.1 µM of ATRA or DMSO for 3 days. CD11b was measured by flow cytometry (middle) and CEBPE by quantitative RT-PCR and normalized to GAPDH (right). **P < .01 and ***P < .001 vs the negative siRNA, respectively by 1-way analysis of variance followed by Dunnett test. (E) Quantitative RT-PCR to validate selected JADE2 target genes in NB4 cells after 48 hours of siRNA transfection. **P < .01 vs negative siRNA by the Mann-Whitney U test. (A-E) Results are expressed as mean plus standard error from 3 independent experiments. (F) Pathway analysis of the 56 downregulated genes by ConsensusPathDB.15 The top 15 enriched Gene Ontology Biological Processes are shown. (G) Expression of the downregulated genes in purified polymorphonuclear cells (PMN) and granulocyte-monocyte progenitors (GMP). Data (from GSE42519) were available for 54 of the downregulated genes. Genes that are upregulated by ≥twofold (log2 fc≥1) in PMN are boxed.
Molecular characterization of NUP98-JADE2 and JADE2 in ATRA-mediated responses and myeloid differentiation. (A) HeLa cells were cotransfected with RARE reporter and the indicated pCMV-HA plasmids. EV, empty vector; NJ, NUP98-JADE2; PR, PML-RARA. Luciferase activities were determined as described in supplemental Methods. (B) HeLa cells were cotransfected with RARE reporter and an increasing pCMV-HA-NUP98-JADE2 (NJ), pCMV-HA-JADE2 (J2), or the empty pCMV-HA (EV), and then treated with ATRA or DMSO for 6 hours before analysis. Results are presented as fold induction vs DMSO treatment. (A-B) *P < .05, **P < .01, and ***P < .001 vs EV, respectively by 1-way analysis of variance followed by Dunnett test. (C) NB4 cells transduced with NUP98-JADE2 (NJ) or control (EV) lentiviruses were treated with ATRA or DMSO. CD11b on GFP-positive cells was measured by flow cytometry. *P < .05 and **P < .01, respectively by paired t test. (D) Left, confirmation of JADE2 knockdown in NB4 cells after 24 hours of siRNA transfection by quantitative RT-PCR. After the transfection, cells were treated with 0.1 µM of ATRA or DMSO for 3 days. CD11b was measured by flow cytometry (middle) and CEBPE by quantitative RT-PCR and normalized to GAPDH (right). **P < .01 and ***P < .001 vs the negative siRNA, respectively by 1-way analysis of variance followed by Dunnett test. (E) Quantitative RT-PCR to validate selected JADE2 target genes in NB4 cells after 48 hours of siRNA transfection. **P < .01 vs negative siRNA by the Mann-Whitney U test. (A-E) Results are expressed as mean plus standard error from 3 independent experiments. (F) Pathway analysis of the 56 downregulated genes by ConsensusPathDB.15 The top 15 enriched Gene Ontology Biological Processes are shown. (G) Expression of the downregulated genes in purified polymorphonuclear cells (PMN) and granulocyte-monocyte progenitors (GMP). Data (from GSE42519) were available for 54 of the downregulated genes. Genes that are upregulated by ≥twofold (log2 fc≥1) in PMN are boxed.
In this study, we characterized a NUP98-JADE2 fusion in a pediatric patient with AML resembling APL and uncovered a role of JADE2 in myeloid differentiation. JADE2 is located at chromosome 5q31, a region frequently deleted in AML and myelodysplastic syndromes. The encoded protein has been shown to be a component of the histone acetyltransferase HBO1 complexes and a regulator of the histone demethylase LSD1/KDM1A.9,10 NUP98-JADE2 is similar to other NUP98 fusions in multiple aspects including a preserved NUP98 amino-terminal portion, speckled nuclear distribution, aberrant HOXA and CDK6 expression (supplemental Figure 4), a histone recognition domain (the plant homeodomain) in the partner, and few cooperating mutations.5,11,12 Concerning the latter, common AML-associated changes were absent in our case, implying that the fusion protein may be sufficient for leukemic transformation. On the other hand, NUP98-JADE2 is distinct from other NUP98 fusions in its association with an APL-like phenotype that is not observed with other non-RAR partners. However, the therapeutic efficacy of ATRA against NUP98-JADE2 is uncertain.
Localization to the correct cellular compartment is critical for proper functioning of a protein. The NUP98-JADE2-induced alteration of the subcellular distribution of JADE2 suggests that the fusion protein may act as a dominant-negative mutant to interfere with the wild-type protein. Concordantly, we observed comparable inhibitory effects of NUP98-JADE2 overexpression and JADE2 knockdown on ATRA-induced responses. On the other hand, how JADE2 perturbation affects ATRA-mediated transcriptional control remains incompletely understood. Because JADE2 is apparently a histone modulator, it is possible that the altered transcriptional response may involve epigenetic deregulation. Interestingly, JADE2 has been shown to promote degradation of LSD1,10 which has been implicated as a negative modulator of ATRA response.13 Thus, NUP98-JADE2 may impede ATRA response through LSD1 stabilization. A previous study has reported a presumed NUP98-JADE2 fusion in a juvenile myelomonocytic leukemia patient.14 However, the fusion breakpoints in both NUP98 and JADE2 were different and with concurrent NRAS/EZH2/KAT6A mutations, which were absent in our case.
In conclusion, we have identified a NUP98-JADE2 fusion conferring an APL-like phenotype in an AML patient and revealed a link between impaired retinoic acid signaling and the observed phenotype. Further studies characterizing the leukemogenic properties of NUP98-JADE2 shall shed new lights on APL pathogenesis.
Acknowledgments
The authors thank the Core Utilities of Cancer Genomics and Pathobiology (The Chinese University of Hong Kong) for providing the facilities and assistance in support of this research.
The work was partially supported by a grant from the General Research Fund program sponsored by the Research Grants Council in Hong Kong (CUHK M14108719).
Authorship
Contribution: C.-K.C. designed and performed research and wrote the manuscript; H.-Y.C., Y.-L.Y., and T.S.K.W. performed research and analyzed data; A.W.K.L. and C.-K.L. managed the patient, collected clinical data, and advised on revision of the manuscript; K.T., N.P.H.C., and J.S.C. collected clinical samples and analyzed clinicopathological data; and M.H.L.N. designed and coordinated research and advised on revision of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Margaret H. L. Ng, Blood Cancer Cytogenetics and Genomics Laboratory, Department of Anatomical and Cellular Pathology, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, NT, Hong Kong SAR, China; e-mail: margaretng@cuhk.edu.hk.


