Key Points
HMB is associated with rare and common variants in genes related to anemias and bleeding disorders.
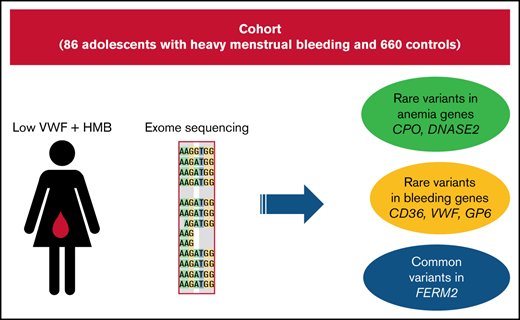
These are the first exome-sequencing results from patients with HMB, as well as their comparison with control exomes.

Abstract
Adolescents with low von Willebrand factor (VWF) levels and heavy menstrual bleeding (HMB) experience significant morbidity. There is a need to better characterize these patients genetically and improve our understanding of the pathophysiology of bleeding. We performed whole-exome sequencing on 86 postmenarchal patients diagnosed with low VWF levels (30-50 IU/dL) and HMB and compared them with 660 in-house controls. We compared the number of rare stop-gain/stop-loss and rare ClinVar “pathogenic” variants between cases and controls, as well as performed gene burden and gene-set burden analyses. We found an enrichment in cases of rare stop-gain/stop-loss variants in genes involved in bleeding disorders and an enrichment of rare ClinVar “pathogenic” variants in genes involved in anemias. The 2 most significant genes in the gene burden analysis, CFB and DNASE2, are associated with atypical hemolytic uremia and severe anemia, respectively. VWF also surpassed exome-wide significance in the gene burden analysis (P = 7.31 × 10−6). Gene-set burden analysis revealed an enrichment of rare nonsynonymous variants in cases in several hematologically relevant pathways. Further, common variants in FERMT2, a gene involved in the regulation of hemostasis and angiogenesis, surpassed genome-wide significance. We demonstrate that adolescents with HMB and low VWF have an excess of rare nonsynonymous and pathogenic variants in genes involved in bleeding disorders and anemia. Variants of variable penetrance in these genes may contribute to the spectrum of phenotypes observed in patients with HMB and could partially explain the bleeding phenotype. By identifying patients with HMB who possess these variants, we may be able to improve risk stratification and patient outcomes.
Introduction
Heavy menstrual bleeding (HMB) is a pathologic state defined as excessive or prolonged menstrual blood loss > 80 mL per menstrual cycle1-3 ; it occurs in approximately one third of adolescent females.4,5 Quality of life can be significantly affected by HMB, in the form of significant iron deficiency anemia (IDA), psychological distress, missed school, limitations on activities, and prolonged periods of required rest.6,7 The co-occurrence of bleeding disorders in this population has been shown to be higher than in the general population.4,8 von Willebrand disease (VWD) is the most common inherited bleeding disorder, with a prevalence of 0.1% (defined by low levels of von Willebrand factor [VWF] and bleeding) in the general population9 but with a prevalence of 13% in patients with HMB.10,11 More recently, low VWF levels, independent of a diagnosis of a bleeding disorder, have been shown to be a significant risk factor for HMB.12
Although there have been several genome-wide association studies (GWAS) of VWF levels,13-17 as well as GWAS of hematological parameters, such as number and volume of various cell types in blood,18-20 these do not detect associations with rare variants. One study validated 1 of the top hits from these GWAS and found that homozygosity for a SNP in TMPRSS6 was protective against IDA, especially in women with HMB.21 To date, no next-generation sequencing study has specifically looked at genetic risk factors for HMB. Although it might be expected that such a study would find an excess of rare and pathogenic variants in the VWF gene in this population of patients with low VWF, by taking an agnostic approach, other genetic patterns may emerge. Here, we describe the results from the first whole-exome sequencing of 86 adolescent patients with HMB and low VWF, as well as comparison with 660 unrelated control exomes.
Methods
Subjects and samples
A multicenter observational cohort study to delineate the phenotype and genotype of adolescent females with low VWF–associated HMB was undertaken from February of 2017 to June of 2019. Tertiary care centers in North America, with expertise in hemostasis and that provided care for adolescents with HMB and were members of the Foundation for Women and Girls with Blood Disorders, participated in the study. Institutional Review Board approval was obtained by all participating centers, and parental and/or patient consent/assent was obtained from all patients prior to study participation. Ten centers participated in this multicenter study. One hundred and thirteen adolescent females with a diagnosis of HMB and low VWF, who met the study inclusion and exclusion criteria, were enrolled. One patient was withdrawn because of the subsequent detection of type 1 VWD and factor XI deficiency. The clinical and laboratory characteristics of this cohort were published recently.12 Of the 113 patients enrolled, 86 had sufficient blood samples collected for whole-exome sequencing and formed the study population for this analysis.
Eligibility criteria
The study eligibility criteria included postmenarchal females, <21 years of age who were diagnosed with HMB, defined as a Pictorial Blood Assessment Chart score > 100 and low VWF, which was defined as having ≥2 values of VWF activity ≥ 30 and ≤ 50 IU/dL (as measured by VWF ristocetin cofactor assay [VWF:RCo] and/or VWF glycoprotein IbM assay). Patients who did not meet these criteria or who were diagnosed with other bleeding disorders were ineligible for the study. Adolescent females, seen in hematology clinics in participating centers managing patients with HMB, were screened for study eligibility, eligible patients were approached for study participation, and patients who consented to participate were enrolled in the study. Clinical phenotype data were extracted prospectively from the patients and retrospectively by reviewing the patients’ electronic medical records. Deidentified patient data from each participating center were entered into the coordinating center’s electronic database; the data were maintained as confidential with access restricted to study investigators by means of password protection. Baseline hemoglobin values, as well as both measures of von Willebrand factor antigen (VWF:Ag), can be found in supplemental Table 1.
Sequencing analysis and validation
Exome sequence data for 86 unrelated HMB probands and 660 unrelated in-house controls were generated at the McDonnell Genome Institute (Washington University in St. Louis) using IDT xGen Exome Panel V1 capture on Illumina HiSeq 4000 paired-end reads. The average age of the controls was 16, with a range of 3 to 40 years. Fifty-seven percent of controls (376/660) were female. Analysis of exome sequencing data was performed in-house using our previously described methods,22,23 with the addition of the Sentieon software package. Briefly, FASTQ formatted sequences were aligned to the hg19 human reference sequence (NCBI GRCh37) using Burrows-Wheeler Aligner (BWA).24 Mapped reads were filtered to remove duplicate reads with the same paired start sites. Median depth per sample at captured bases for HMB cases was 76× (range, 17-202); it was 71× (range, 21-121) for in-house controls. All cases and controls had >97% of captured bases covered with >10 reads. The Binary sequence Alignment/Map formatted alignments were processed using the Genome Analysis Toolkit (GATK) Haplotype Caller25,26 and genotypes jointly called together with all in-house control exome-sequenced individuals. Genotypes were filtered for read depth (>10×), genotype quality > 20, and allele balance > 0.3 and <0.7. Variants were filtered for GATK-calculated variant quality score recalibration and genotype call rate >90%. Allele frequencies were annotated using the gnomAD database.27 Variants were functionally annotated using ANNOVAR.28 To reduce the risk of population stratification, only cases and controls with principal components–confirmed European ancestry were included in all analyses. Principal components were calculated using EIGENSTRAT29 from whole-exome single-nucleotide polymorphism (SNP) data using all common (minor allele frequency [MAF] > 5%) SNPs. We did not remove any cases, because principal components confirmed self-reported race. For the purposes of all single SNP analyses, the standard cutoffs for genome-wide significance (5 × 10−8) and exome-wide significance (2.5 × 10−6) were used. We reasonably excluded HMB and low VWF from the control cohort because the estimate of the prevalence of low VWF–associated HMB in the general population is likely ∼1% given that the prevalence of low VWF is generally ∼2.5% and the prevalence of HMB is ∼35%.4,30,31
Common SNP analysis
Fisher’s exact tests were performed using PLINK to compare the MAF of common and rare variants in cases and controls. SNPs were dropped if the genotyping success rate was <90% per SNP or per individual, the Hardy-Weinberg equilibrium (P < 1 × 10−6), and MAF or missingness was <0.05.
Gene burden and gene-set burden analysis
To quantify the enrichment of rare nonsynonymous/splice-site variants and ClinVar “pathogenic” variants in HMB cases compared with controls for each gene, we compared the collapsed MAF of variants in cases and controls using a Fisher’s exact test using only filtered variants. Variants were filtered by rareness and quality: (1) max MAF in all populations < 0.01 in gnomAD exomes and genomes, (2) genome quality > 20, (3) GATK variant quality score recalibration of ‘PASS,’ (4) minimum sequencing depth of 8 reads in each participant, and (5) allele balance > 0.3 and <0.7. For statistical analysis of exome sequencing data, affected individuals were compared with in-house exome controls. For individual gene association analyses, Fisher’s exact tests were performed using PLINK to compare the collapsed MAF of rare variants in cases and controls. Student t tests were performed using R to compare the quantitative traits. For gene-set burden analyses, nonsynonymous/splice-site variants within a gene were collapsed to obtain the number of rare (<1% in gnomAD) variants per gene. The numbers of variants within groups of genes were then summed based on membership within a given gene set and used as the dependent variable in a linear regression. Gene Ontology (GO) terms were obtained from the UniProt Knowledgebase (http://www.uniprot.org/help/uniprotkb). The exome-wide P-value cutoff was used in the gene burden analyses, because that is the number of independent tests (0.05/20 000 genes). Additionally, for our disease-associated gene-set burden analyses, we created a disease-defined set of genes involved in anemia using Online Mendelian Inheritance in Man and UniProt disease descriptions by searching for the term “anemia.” For the bleeding disorder disease–defined gene set we searched the same databases for the terms “bleeding,” “platelet,” and “hemostasis.”
Results
Exome sequencing reveals excess of rare stop-gain/stop-loss mutations in bleeding disorder genes and rare pathogenic variants in anemia genes
Because rare likely pathogenic variants are necessarily enriched for causal variants, we first examined rare stop-gain/stop-loss and rare ClinVar “pathogenic” variants in our sample. Among the 86 patients, we observed 4 (4.6%) rare stop-gain or stop-loss mutations in genes associated with bleeding or hematological diseases: CD36 (p.I337Vfs*10, NM_001371081), CPO (p.R184X, NM_173077), VWF (p.E383X, NM_000552), and GP6 (p.X340R, NM_016363). Variants in these genes are known to cause platelet glycoprotein IV deficiency, hereditary coproporphyria, VWD, and bleeding disorder, platelet type 11, respectively. In controls, there was 1 rare stop-gain variant in CPO (0.15%). This difference was statistically significant (Fisher’s exact text, P = 7.6 × 10−4, odds ratio [OR], 31.8; 95% confidence interval [CI], 27.7-35.9). The patient we found with a stop-gain mutation (p.E383X) in VWF had a frequency of this allele in gnomAD of nearly 0, because only 1 instance of it was observed. This variant was previously identified in 1 patient with VWD.32 This variant is not listed in ClinVar but is likely to be the cause of the HMB in that individual and also may be contributing to any VWD phenotype that they have.
We also pooled all rare (<1% in gnomAD) ClinVar “pathogenic” variants present in cases that were in genes known to cause a dominant or recessive form of anemia or a disease with anemia as a major symptom; 9.3% of cases and 4.2% of unrelated controls harbored such a variant, and this difference was significant (Fisher’s exact test, P = .04; OR, 2.3; 95% CI, 1.1-3.5) (Table 1). We performed the same comparison in genes involved in hemostasis and platelet function. We found that 4.6% of cases and 1.4% of controls had rare ClinVar “pathogenic” variants in these genes (Fisher’s exact test, P = .05; OR, 3.5; 95% CI, 2.5-4.5). This association was driven by variants in cases in VWF (2), F2 (1) and LYST (1). All significant SNPs and genes remained significant after correcting for the presence of the D1472H variant. We compared our variants with the ISTH VWDdb (Sheffield database). None of the variants that we found are listed as “segregates” in the database, but all are listed, giving no specificity with regard to pathogenicity. Additionally, there was only 1 patient within the cohort that had a VWF:RCo/VWF:Ag ratio < 0.6 from the first set of VWF:Ag measurements, indicating that the cohort was not skewed toward lower VWF:RCo levels because of the presence of this variant. To ensure these were not false positive results, we also examined 5 “control” gene sets in cases and controls including the GO terms “regulation of lipid metabolic processes,” “breast/ovarian cancer,” “microvascular complications from diabetes,” “epilepsy,” and “autism.” None of these were significant between cases and controls (.08 < P < .99).
Common and rare single SNP analyses
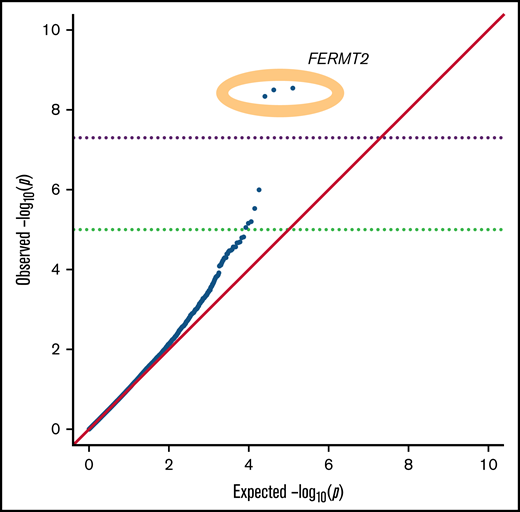
We tested for association with all common SNPs present in the exome and genome data (>5% in gnomAD). Three common SNPs in linkage disequilibrium in or downstream of FERMT2 (fermitin family member 2) achieved genome-wide significance in the common SNP analysis (P = 2.9 × 10−9; OR, 4.4; 95% CI, 3.0-7.8) (Figure 1). Although the MAF of these SNP is 10% in non-Finnish Europeans in gnomAD, we found these SNPs to be present in 24% of cases and 6% of controls. FERMT2 encodes a cytoskeletal protein that is a crucial regulator of integrin function.33 It is known to regulate hemostasis and is required for angiogenesis and blood vessel homeostasis.34 Additionally, the SNPs that we found to be associated with HMB are expression quantitative trait loci for FERMT2, according to the Genotype Tissue Expression Project (https://www.gtexportal.org/home/), further supporting their potential functional role in the phenotype.

QQ plot of the P values for common SNPs. Three SNPs in FERMT2 achieved genome-wide significance (yellow oval).
QQ plot of the P values for common SNPs. Three SNPs in FERMT2 achieved genome-wide significance (yellow oval).
We also discovered SNPs in 3 additional genes with P values reaching exome-wide significance (Figure 2). EBAG9 (estrogen receptor binding site associated antigen 9) is primarily produced by macrophages in hematopoietic tissue and has a crucial role in controlling erythropoiesis.35 TTC18 (a.k.a. CFAP70 [cilia and flagella associated protein 70]) is expressed in the cilia of glandular cells in fallopian tube, endometrium, and respiratory tract36 (http://www.proteinatlas.org), and TCN1 (transcobalamin 1) is highly expressed in granulocytes,36 which have been identified as critical determinants of uterine bleeding and tissue remodeling in a mouse menstruation model.37
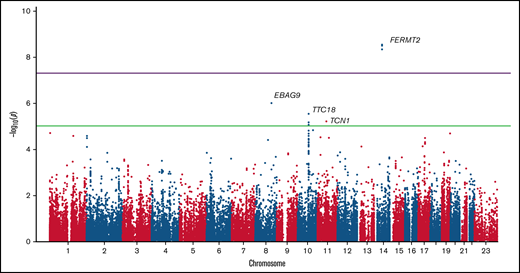
Manhattan plot of the P values for common SNPs. Three SNPs in FERMT2 achieved genome-wide significance (purple line), whereas SNPs in EBAG9, TTC18, and TCN1 achieved exome-wide significance (green line).
Manhattan plot of the P values for common SNPs. Three SNPs in FERMT2 achieved genome-wide significance (purple line), whereas SNPs in EBAG9, TTC18, and TCN1 achieved exome-wide significance (green line).
Gene burden analysis uncovers associations with anemia and bleeding disorder genes
Only coding variants that caused non-sense, splice-site, missense, or insertion/deletion mutations that were rare (defined as <1% in gnomAD) were included in the analysis. This filtering strategy was selected to enrich for mutations that are very rare and most likely to be deleterious. Using a gene burden analysis, we compared the genome-wide frequency of rare coding variants in HMB cases and controls. An increased burden of rare variants that surpassed exome-wide significance was seen in 10 genes (Table 2). The top hit in the results was CFB (complement factor B), which achieved exome-wide significance and was the only gene in this analysis to also achieve genome-wide significance (P = 3.0 × 10−8). This gene encodes for a component of the alternative pathway of complement activation. The collapsed MAF (cMAF) for all rare variants in CFB was 0.02 in cases (n = 86) and 0.0 in controls (n = 660). Rare variants in this gene have been associated with atypical hemolytic uremic syndrome,38,39 which is characterized by microangiopathic hemolytic anemia and thrombocytopenia, as well as acute renal failure.40
The second most significant hit in our results was DNASE2 (deoxyribonuclease 2, lysosomal), which achieved exome-wide significance (P = 3.5 × 10−7). The cMAF for all rare variants in DNASE2 was 0.04 in cases compared with 0.004 in controls. This gene encodes a protein that mediates the breakdown of DNA during erythropoiesis and apoptosis. Patients with complete loss of function of this gene were observed to have severe anemia and thrombocytopenia.41 Because it has been shown that patients with low VWF have an increased rate of rare variants in VWF,42 it is perhaps expected that VWF was also significant in the gene burden analysis. There were 19 cases (22%) and 47 controls (7%) with ≥1 rare nonsynonymous variant in VWF (P = 7.3 × 10−6).
Rare variants in hematologically relevant pathways collectively influence HMB risk
Although some genes achieved exome-wide significance in the gene burden analyses (Table 2), we used our previously developed pathway burden analysis framework43 that, unlike some methods,44 preserves power by using data from all genes, not only those with significant single-gene associations. With this method, variants are first collapsed at the gene level and then by GO term membership.
Exome-wide pathway burden analysis yielded a strong association between HMB and novel variants in genes within the GO terms “oxygen transporter activity,” “hemoglobin complex,” “platelet degranulation,” “positive regulation of erythrocyte differentiation,” and platelet α granule lumen” (Table 3). These terms are not mutually exclusive; in fact, some have a significant amount of overlap. Notably, these and several other top associated GO terms are highly correlated, often consisting of gene lists that are subsets of one another. For example, nearly all of the genes in the term “hemoglobin complex” are included in the term “oxygen transporter activity.”
Discussion
It is estimated that up to 30% of women experience HMB, and it accounts for two thirds of all hysterectomies.45 Many systemic disruptions of hemostasis, such as liver disease and hypothyroidism, are well-described causes of HMB,10 yet 50% of women with hysterectomies resulting from HMB do not have the underlying cause identified.46 Thus, it is clinically important to understand the underlying causes of HMB. Although it is known that bleeding disorders, such as VWD, occur more frequently in this subpopulation than in the overall population, IDA is a common, but underappreciated, complication in adolescents with HMB.2 The anemia is a direct result of the excessive blood loss during menses and occurs when iron depletion is severe enough to suppress erythropoiesis.47 Genetic causes of anemia do not necessarily imply a mechanism beyond bleeding. In many cases the underlying biological mechanism is unknown and could be a presdisposition to bleeding.
HMB is associated with rare and common variants in anemia genes
We performed several types of whole-exome analyses on 86 HMB cases compared with our in-house controls. We focused primarily on rare variant analyses, because, as a class, rare variants (defined as <5% population frequency) constitute the majority of genetic variation and are 4 times more likely to be deleterious.48 In single SNP rare variant and rare variant gene burden analyses, HMB cases had significantly more rare variants in genes known to cause different subtypes of anemias than did controls. Additionally, common variant analysis revealed an association with 1 gene that is known to contribute to anemia. That the HMB patients do not have known diagnoses of these Mendelian disorders suggests that the variants we identified could be causing less severe or incompletely penetrant forms of anemia.
First, we observed significantly more rare ClinVar “pathogenic” variants in 8 genes associated with varying types of anemias in cases than in controls. A subset of these genes causes dominant forms of anemia, suggesting that it is possible that at least a subset of patients with these rare pathogenic variants may actually have undiagnosed disease. In these patients, the anemia may precede the HMB and, thus, would be exacerbated by the blood loss. Patients with variants in the genes causing recessively inherited anemias may also have milder forms of the disease if they have 1 functional copy of the gene.
Second, the 2 most significant genes from the gene burden analysis are directly linked to anemia. As previously mentioned, rare missense mutations in CFB are associated with atypical hemolytic uremic syndrome.38,39 Biallelic mutations in DNASE2 have been associated with a loss of DNase II endonuclease activity, causing severe neonatal anemia and thrombocytopenia, among other symptoms.41 This loss of DNase II induces interferon signaling, inhibiting macrophages from destroying nuclear DNA expelled from erythroid precursor cells. Increased erythroblasts in peripheral blood are observed, suggestive of ineffective erythropoiesis.41 This phenotype is also seen in mice, because DNase II–null mice accumulate undigested DNA in the lysosomes of macrophages, activating the production of type 1 interferon and resulting in lethal perinatal anemia.49,50 Because the patients with HMB in our study did not appear to have biallelic loss of DNASE2, it is possible that having haploinsufficiency may cause a milder anemia.
Lastly, common variant analysis revealed an association with EBAG9, which induces apoptosis in normal human erythroid progenitor cells.35 EBAG9 has been detected in monocytes and macrophages. When macrophages were stimulated with lipopolysaccharide, expression of the protein increased, and cell death of erythroid progenitor cells was induced by this increased expression. It has been suggested that erythropoiesis is negatively regulated, in part, by macrophages through the production of EBAG9, contributing to the pathogenesis of anemia in patients with inflammatory disorders.51
It was recently shown that elevated hematocrit is associated with increased platelet accumulation at the site of injury in mice and human, establishing the role of red cells in normal hemostasis and further underscoring the association of anemia with increased bleeding.52
Rare and common variants in bleeding genes, including VWF, in HMB patients
We also replicate what other investigators have noted in patients with HMB: an enrichment of pathogenic variants in genes known to cause bleeding disorders.10-12 We found 4 rare stop-gain/stop-loss mutations in our cases. One of these was a rare stop-gain mutation in VWF that has not been seen in gnomAD. This mutation may be contributing to the undiagnosed VWD in that patient. Two of these mutations are in genes that are known to cause platelet deficiency (CD36) and platelet-type bleeding disorder (GP6). The last was a stop-gain mutation in CPO, a gene that causes a type of hereditary porphyria. Porphyrias are a group of inborn errors of heme biosynthesis.53 We also observed more rare ClinVar “pathogenic” variants in hemostasis and platelet function genes in cases vs controls, including 2 people with the same rare ClinVar “pathogenic” variant in VWF (P1266Q, NM_000552), 1 variant in F2, and 1 variant in LYST. The variant in F2 (c.*97G>A, NM_000506.5) has been associated with prothrombin-related thrombophilia,54 and the variant in LYST (p.R1563H, NM_000081.3) has been shown to be associated with Chediak-Higashi syndrome.55,56 Coagulation defects are 1 of the known symptoms of Chediak-Higashi syndrome.57 Although the VWF variant that we found has been observed in patients with type 2 VWD,58 the patients with this variant had a normal multimer pattern and normal plasma and platelet VWF levels. Of the 2 patients with P1266Q_NM_000552, 1 patient had an additional variant in VWF (p.P2297L_NM_000552). However, there were also 3 controls with the P1266Q variant, suggesting that it is not pathogenic on its own. VWF was also significant in the rare variant gene burden analysis, and common variant analysis showed an association with FERMT2, which is involved in the regulation of hemostasis, angiogenesis, and blood vessel homeostasis.34 This gene encodes kindlin-2, a widely distributed cytoskeletal protein that is involved in integrin activation. Its absence is embryonically lethal in mice and causes severe developmental defects in zebrafish.59 Even partial reduction of kindlin-2 in mice resulted in fewer blood vessels, whereas the vessels that form lack smooth muscle cells and are thinner and shorter than normal.34,59 Kindlin-2 is present in platelets,60 and kindlin-2–knockdown mice had prolonged bleeding and vascular occlusion times as a result of the suppression of platelet aggregation caused by the elevated expression of 2 enzymes (CD39, CD73) on endothelial cells.34 Therefore, an enrichment of variants in FERMT2 in cases is consistent with what is known about the pathophysiology of this gene and its encoded protein. Additionally, the gene-set burden analysis revealed an enrichment of rare variants in cases in several pathways relevant to hemostasis.
Limitations
Although 86 patients are sufficient for detecting an association with variants of modest effect size, we recognize that our study is underpowered to detect variants of smaller effect sizes. We evaluated the clinical phenotypes that we ascertained to associate them with genetic variants, but there was too little variation in the laboratory values to identify any associations. This is due to the fact that all patients have the HMB clinical phenotypes, regardless of any differences in the underlying reason for the HMB. In the future we hope to sequence more patients to further fine-tune the associations, as well as to replicate these associations in other similarly phenotyped data sets.
Conclusions
We present the first whole-exome sequencing results of patients with HMB, as well as their comparison with control exomes. In addition to observing an excess of rare nonsynonymous variants in genes involved in several bleeding disorders, we observed excess variants in genes causing anemias and disorders with anemia as a symptom. Although most of these patients may not have the full disorder associated with variants in that gene, they may simply have a milder version due to incomplete penetrance of the variants, as well as potential interactions with variants in other genes that are needed to cause the severe form of the disorder. Our findings need validation in larger cohorts. This work may begin to shed some light on the large proportion of patients with HMB who do not have a cause for their symptoms. Eventually, identifying these patients with HMB and with these risk variants may improve risk stratification and patient outcomes.
Acknowledgment
This work was supported by an investigator-initiated research grant (IIR[1]USA-BXLT-001980-H16-30985) from Shire US Inc., now part of Takeda (L.S.).
Authorship
Contribution: B.S., L.S., J.E.D., and P.A.K. conceived the study; B.S. and G.H. performed all genetic analyses; C.G.M. analyzed the clinical phenotype data; G.H. and C.A.G. provided control samples; L.S., P.A.K., S.H.O., A.W., S.J., M.S., A.Z., R.K., M.V.R., R.S., and J.E.D. collected samples; and B.S. and J.E.D. wrote the manuscript; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jorge Di Paola, 660 South Euclid Ave, Campus Box 8208, Washington University in St. Louis, St. Louis, MO 63110; e-mail: dipaolaj@wustl.edu; and Lakshmi Srivaths, Gulf States Hemophilia and Thrombophilia Center, University of Texas Health Science Center at Houston and McGovern School of Medicine, 6655 Travis St, Suite 400 HMC, Houston, TX 77030; e-mail: lakshmi.v.srivaths@uth.tmc.edu.