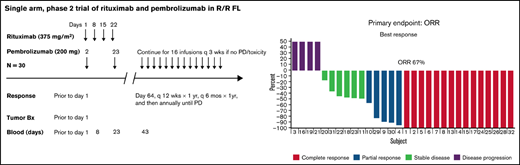
Key Points
Combination therapy with pembrolizumab and rituximab was well tolerated in patients with relapsed/refractory follicular lymphoma.
In this single-arm, phase 2 study, the overall response rate was 67%, with 50% complete response and median PFS of 12.6 months.

Abstract
PD-1 blockade enhances the function of antitumor T cells and antibody-dependent, cell-mediated cytotoxicity (ADCC) of NK cells. In a single-center, open-label, phase 2 trial, we tested the combination of pembrolizumab, an anti-PD-1 monoclonal antibody, and rituximab, an anti-CD20 monoclonal antibody that induces ADCC, in 30 patients with follicular lymphoma (FL) with rituximab-sensitive disease who had relapsed after ≥1 prior therapy. Pembrolizumab was administered at 200 mg IV every 3 weeks for up to 16 cycles, and rituximab was given at 375 mg/m2 IV weekly for 4 weeks in cycle 1 only. The most common grade 3/4 adverse events (AEs) were liver enzyme abnormalities (3%), diarrhea (3%), nausea (3%), aseptic meningitis (3%), and pancreatitis (3%). Low-grade immune-related AEs were reported in 80% of patients, including diarrhea (43%), liver enzyme abnormalities (33%), thyroid dysfunction (27%), and rash (23%). Grade 3 or 4 immune-related AEs occurred in 13% of the patients. Treatment-related AEs led to discontinuation in 6 (20%) patients. The overall response rate (primary end point) was 67%, and the complete response (CR) rate was 50%. Median progression-free survival (PFS) was 12.6 months (95% confidence interval, 8.2-27.6), the 3-year overall survival rate was 97%, and 23% of patients were in remission at a median follow-up of 35 months. The presence of a high CD8+ T-effector score at baseline in the tumor was associated with induction of a CR and improved PFS. In this single-arm, phase 2 study, the combination of pembrolizumab and rituximab demonstrates favorable efficacy and safety profile in relapsed FL. This trial is registered at www.clinicaltrials.gov as #NCT02446457.
Introduction
Follicular lymphoma (FL) is the most common indolent non-Hodgkin lymphoma worldwide and is characterized by a pattern of stable, relapsing and/or remitting disease.1 Although the tumor microenvironment maintains disease control through endogenous immune surveillance, tumor cells escape through mechanisms including anergy and exhaustion of tumor infiltrating lymphocytes by expression of inhibitory checkpoint markers including programmed cell death 1/programmed cell death ligand-1 (PD1/PD-L1), lymphocyte activation gene 3 (LAG3), and cluster of differentiation 47 (CD47).2-4 The programmed cell death protein 1 (PD-1) axis regulates the immune responses of activated T cells to infection and prevents autoimmunity.5 The PD-1 ligands PD-L1 and PD-L2 may be expressed on tumor cells and/or stromal cells, and, when bound to PD-1, inhibit T-cell activation and facilitate immune escape. PD-1 expression is markedly elevated on endogenous T cells after chronic antigenic stimulation by viral infection or tumor exposure and is associated with T-cell exhaustion. PD-1 blockade has been demonstrated in preclinical studies to restore the function of antitumor T cells and enhance the antibody-dependent, cell-mediated cytotoxicity (ADCC) effect of natural killer (NK) cells.6-12 Therefore, targeting the PD-1/PD-ligand pathway in combination with rituximab, an anti-CD20 antibody that induces ADCC, is likely to be synergistic.
Pembrolizumab is a humanized immunoglobulin G4 (IgG4) isotype antibody that targets PD-1 with demonstrable activity across a range of solid organ and lymphoid malignancies.13 PD-1 blockade with nivolumab, a humanized IgG monoclonal antibody has been shown in a small phase 1b study to have promising single-agent activity in 10 relapsed or refractory (R/R) patients with FL who have an overall response rate (ORR) of 40% and complete response (CR) rate of 10%.14 A phase 2 study of pidilizumab, a humanized IgG-1κ recombinant monoclonal antibody thought to target PD-1, in combination with rituximab demonstrated synergistic activity in patients with relapsed FL (ORR 66%, CR 52%).15 However, recent evidence suggests that inhibition of PD-1 inhibition by pidilizumab is a secondary effect, with an alternate mechanism of action through binding the deltalike 1 (DLL1) gene.16,17 Therefore, a gap exists in the literature regarding the safety and efficacy of PD-1 inhibition in combination with rituximab in the treatment of R/R FL. We reasoned that the combination of pembrolizumab and rituximab would be synergistic by activating both the innate (NK-cell) and adaptive (T-cell) systems to improve clinical activity without excess toxicity. We aimed to assess the safety and activity of pembrolizumab and rituximab and performed correlative studies in an open-label, single-center, phase 2 study in patients with relapsed FL.
Patients and methods
Patients
This was an investigator-initiated, open-label, single-center, phase 2 study of pembrolizumab+rituximab in R/R FL. The MD Anderson Institutional Review Board approved the study, which was performed in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. All patients provided written informed consent. Eligibility criteria included World Health Organization grade 1 to 3A FL18 ; relapsed or refractory disease after ≥1 prior therapy; rituximab-sensitive disease, defined as a CR or partial response (PR) lasting at least 6 months after the most recent rituximab-containing therapy; age ≥18 years; an Eastern Cooperative Oncology Group (ECOG) performance status <2; an absolute neutrophil count of at least 1.0 × 109 cells/L; a platelet count of at least 50 × 109 cells/L; and adequate hepatic, renal, cardiac, and pulmonary function. Patients were ineligible if they had a history of autoimmune disease within the past 2 years requiring systemic therapy, prior immune checkpoint inhibitor therapy, history of any malignancy within the previous 5 years, uncontrolled serious illness or comorbidities, human immunodeficiency virus infection, or active hepatitis B or C infection.
Study design
Patients received rituximab 375 mg/m2 as an IV infusion on days 1, 8, 15, and 22 of cycle 1 and 200 mg pembrolizumab IV every 3 weeks for up to 16 cycles starting on day 2 of cycle 1. Adverse events (AEs) were graded using the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. Responses were assessed according to the 2014 Lugano classification.19 Two independent radiologists reviewed any equivocal findings. Diagnostic quality positron emission tomography computerized tomography (PET-CT) with IV and oral/water contrast and bone marrow biopsy were used for initial disease assessment within 42 days before the first dose of study treatment. Th PET-CT scan was repeated to confirm CR and/or at treatment discontinuation; otherwise follow-up assessments were performed with CT scans. Response assessments occurred after week 12, every 3 months during therapy, 1 year after treatment discontinuation, and then every 6 months until the start of another anticancer treatment or documented disease progression, death, or the end of the study, whichever occurred first. Bone marrow biopsy was performed to confirm CR if bone marrow involvement was present at study enrollment.
Immunohistochemical staining for PD-L1
Immunohistochemical (IHC) tumor staining was performed by QualTek with an automated staining system for PD-L1 (22C3 PharmDx) on pretreatment tumor samples. The staining was reviewed and scored according to the percentage of malignant cells with positive staining (0% to 100%) and the intensity of staining of tumor and nonmalignant cells (0, no staining; 1+, weak/equivocal staining; 2+, moderate staining; and 3+, strong staining). Previously published criteria for categorizing cases as positive for PD-L1 expression were used.20 Tumor cells had to exhibit 2+ or 3+ membrane staining in ≥1% of malignant cells for PD-L1 to be considered positive.21 For the tumor microenvironment, ≥1% of nonmalignant cells had to exhibit positive staining for PD-L1 to be categorized as positive.
Assessment of immune signatures
Core needle biopsy specimens from involved lymph nodes obtained before treatment were collected in RNAlater fixative (Life Technologies, Carlsbad, CA) and stored at −80°C until RNA isolation. RNA was isolated by RNeasy Mini Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer’s guidance. RNA quantity and quality were established with a 2100 Bioanalyzer (Agilent, Santa Clara, CA), and RNA was profiled by using a semicustom ∼1500-gene panel on the NanoString platform.
Pretreatment peripheral blood samples were obtained, and mRNA was extracted for gene expression analysis conducted on the NanoString nCounter gene expression platform (NanoString Technologies). A 10-gene interferon-γ (IFN-γ) signature panel (IFNG, STAT1, CCR5, CXCL9, CXCL10, CXCL11, IDO1, PRF1, GZMA, and major histocompatibility complex II HLA-DRA) and an expanded immune 28-gene IFN-γ signature that encompasses genes related to cytolytic activity (granzyme A/B/K, PRF1), cytokines/chemokines for initiation of inflammation (CXCR6, CXCL9, CCL5, and CCR5), T-cell markers (CD3D, CD3E, CD2, and IL2RG), NK cell activity (NKG7 and HLA-E), antigen presentation (CIITA and HLA-DRA), and additional immunomodulatory factors (LAG3, IDO1, and SLAMF6) were tested.22
Statistical analysis
The primary end point was ORR, defined as the proportion of patients who exhibited a CR or PR. Secondary end points included CR and PR rates; progression-free survival (PFS) assessed from first treatment administration to disease progression or death from any cause, whichever occurred first; overall survival (OS) assessed from first treatment administration to death; and safety.
The null hypothesis predicted ORR in no more than 40% of patients. We expected to improve the proportion of patients achieving an overall response to at least 60% with the current regimen. The proposed sample size of 30 patients was expected to achieve a width of 0.23 for the posterior 90% credibility interval under the assumption of an overall response in 60% of patients. Patients were assessed for response if they had any postbaseline tumor assessment.
In this analysis, we evaluated the associations between various categorical patient characteristics (age, sex, stage, B symptoms, ECOG performance status, and whether tumor burden, as defined by Groupe d’Etude des Lymphomes Folliculaires [GELF] criteria for treatment, were met) with response to pembrolizumab+rituximab, as well as the duration of disease control. Summary statistics including mean, standard deviation, median, and range of continuous variables, such as age and time to response. Frequency counts and percentages for categorical variables, such as stage and response are provided. The best response rates and their 95% confidence intervals (CI) using the exact method are reported. The Kaplan-Meier method was used to estimate PFS and response duration. A log-rank test was used to compare survival between different groups. Statistical software SAS 9.4 (SAS, Cary, NC) and TIBCO Spotfire S+ 8.2 (TIBCO Software Inc, Palo Alto, CA) were used for all the analyses.
Results
Patient demographics
Thirty patients were enrolled in the trial from 1 August 2015 to 7 February 2017. The median age was 64 years (range, 43-84 years) and 57% were male (Table 1). Twenty-two (73%) patients had a Follicular Lymphoma International Prognostic Index (FLIPI) score of ≥2. Twenty-six (87%) patients had stage III or IV disease, and 15 (50%) met the GELF criteria for high tumor burden. The median number of prior lines of therapy was 1 (range, 1-4) with a median PFS after the last therapy of 27.5 months (range, 3-162 months).
The median follow-up for the study was 34.9 months (range, 8.8-48.5 months). The median duration of pembrolizumab treatment was 6.7 months (range, 2-13 months). Twenty-six patients (87%) completed at least 6 cycles; 10 (33%) patients completed 16 cycles without early discontinuation. Thirteen (43%) patients discontinued therapy because of disease progression; 6 (20%) patients discontinued secondary to AEs; 1 (3%) patient discontinued therapy early because of relocation; no patients were lost to long-term follow-up.
Safety
Treatment-related AEs occurred in all patients; most were mild to moderate, with 5 patients (17%) having grade 3 or 4 AEs (n = 1; 3% each), including liver enzyme abnormalities, diarrhea, aseptic meningitis, pancreatitis, and nausea, and vomiting (Table 2). Immune-related AEs were reported for 80% of patients; most were low grade (grade 3 or 4: n = 4, 13%). The most commonly reported immune-related AEs were diarrhea (n = 13, 43%), liver enzyme abnormalities (n = 12, 40%), oral mucositis (n = 10, 33%), thyroid dysfunction (n = 9, 30%), and rash (n = 7, 23%). Median time to onset of the first immune-related AE was 35 days (range, 1-207 days). None of the assessed patient characteristics were significantly associated with immune-related AEs. Grade 2 or higher immune-related AEs occurred in 16 patients (53%). With regard to management of immune-related AEs, 7 patients received oral corticosteroids (median of 125 days of corticosteroids; range, 40-319 days), 3 patients received topical corticosteroids only for rash, and the remaining were observed until resolution. Three patients discontinued therapy permanently and were never rechallenged. Four patients were rechallenged with pembrolizumab after a median of 28.5 days (range, 14-51 days), but permanently discontinued therapy because of the recurrence of treatment-related diarrhea (n = 3) or pneumonitis (n = 1). All were in CR at the time of discontinuation of treatment. One patient was rechallenged with pembrolizumab and completed all 16 cycles without further immune-related AEs. No treatment-related deaths were observed.
Efficacy
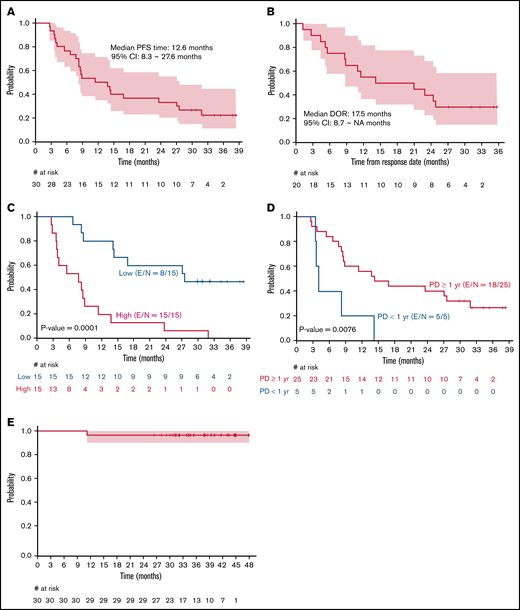
The ORR among the 30 patients was 67%, including 15 (50%) patients with CR and 5 (17%) with PR. The median time to best response was 3 months (range, 2.5-9.0). Among those who achieved a CR, 12 of 15 achieved it at their first response assessment, and 3 converted from a PR at their first response assessment to a CR by the second response assessment. A lower ORR was observed in patients with high tumor burden by GELF criteria and progression of disease within 24 months after first-line therapy (POD24) when compared with patients with low tumor burden and without POD24, respectively (53% vs 80%, P = .002; 36% vs 84%, P = .04). Twenty-two events (progression or death) had occurred at the time of this analysis. Median PFS was 12.6 months (95% CI, 8.2-27.6; Figure 1A). For patients who achieved a CR or PR (n = 20), the median duration of response was 17.5 months (95% CI, 8.7 to not reached, Figure 1B). High tumor burden by GELF (P = .0001) and PFS <1 year for a prior line of therapy (P = .008) were associated with inferior PFS (Figures 1C-D). Median OS was not reached, and 3-year OS was 97% (95% CI, 90-100; Figure 1E). Sex, stage, FLIPI, ECOG performance status, number of prior therapies, POD24 status, and occurrence of immune-related AEs was not significantly associated with inferior PFS. At last follow-up, 28 (93%) patients were alive, 7 (23%) were in remission, 23 (77%) demonstrated progression/relapse, and 2 (7%) had died of disease progression.
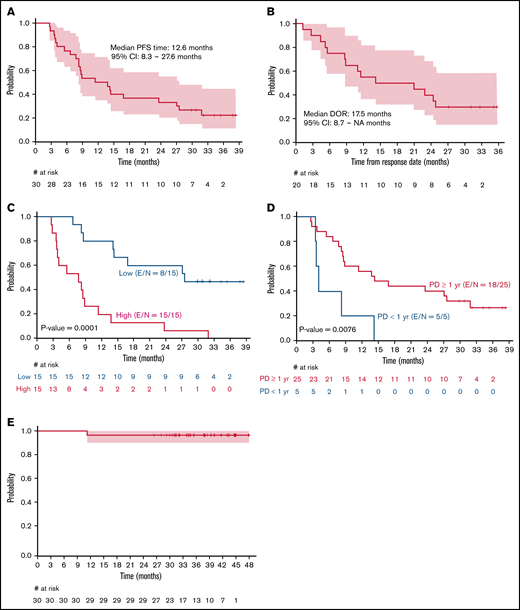
Kaplan-Meier curves. PFS (A); duration of response (B); PFS by high or low tumor burden according to GELF criteria (C); PFS by PD <1 year or PD >/=1 year from prior treatment (D); OS in patients with R/R FL receiving pembrolizumab+rituximab (E). E, number of events; N, sample size.
Kaplan-Meier curves. PFS (A); duration of response (B); PFS by high or low tumor burden according to GELF criteria (C); PFS by PD <1 year or PD >/=1 year from prior treatment (D); OS in patients with R/R FL receiving pembrolizumab+rituximab (E). E, number of events; N, sample size.
Correlative studies
Of 19 pretreatment tumor samples available for assessment, PD-L1 expression by immunohistochemistry was detectable on background histiocytes in all samples (100%) and tumor cells in 11 samples (58%). Of the tumors with detectable PD-L1 expression (n = 11), PD-L1 was present in 1% to 8% of tumor cells in 10 samples and in 20% of tumor cells in 1 sample. PD-L1 status was not a significant predictor of response (P = .71) with 7 of 11 (64%; 6 CR, 1 PR) responses observed in patients with detectable PD-L1 tumor expression and 4 of 8 (50%; 2 CR, 2 PR) responses in patients with undetectable tumor PD-L1 expression (supplemental Figure 1). PD-L1 expression of background histiocytes was not predictive of PFS (P = .68).
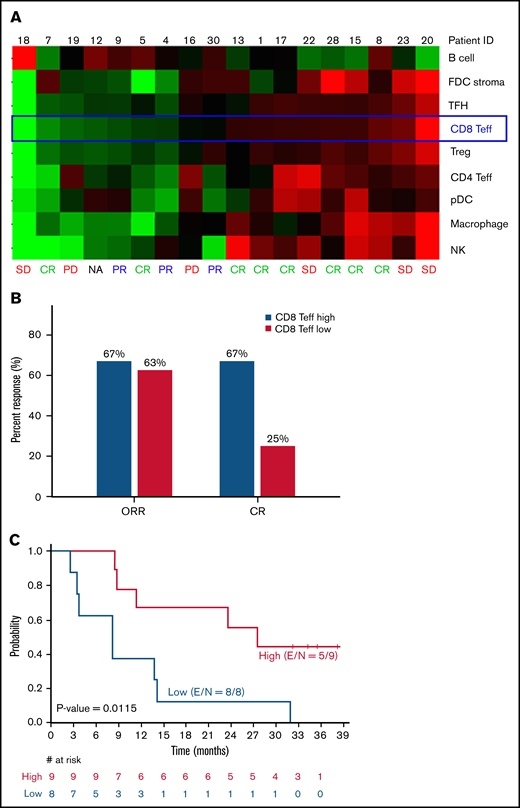
Pretreatment tumor immune cell gene signature analysis was performed on 18 tumor samples, and a significant difference between patients with high CD8+ T-effector score and patients with low CD8+ T-effector score was observed in achievement of a CR (67% vs 25%; P = .046) and PFS (27.6 months vs 8.3 months; P = .012; Figure 2). After adjustment for multiple testing at a false-discovery rate of 0.1, per the Benjamin-Hochberg procedure,23 the CD8+ T-effector signature was the only gene score that significantly associated with PFS among all immune cell signatures tested (Figure 2; supplemental Figure 2). IFN-γ–related gene signatures in pretreatment peripheral blood mononuclear cells (n = 26) showed an association with objective response using both a previously described 10-gene panel (P = .016) and a 28-gene panel (P = .023; supplemental Figure 3).22 However, this finding did not translate into a PFS benefit (P = .93; data not shown).
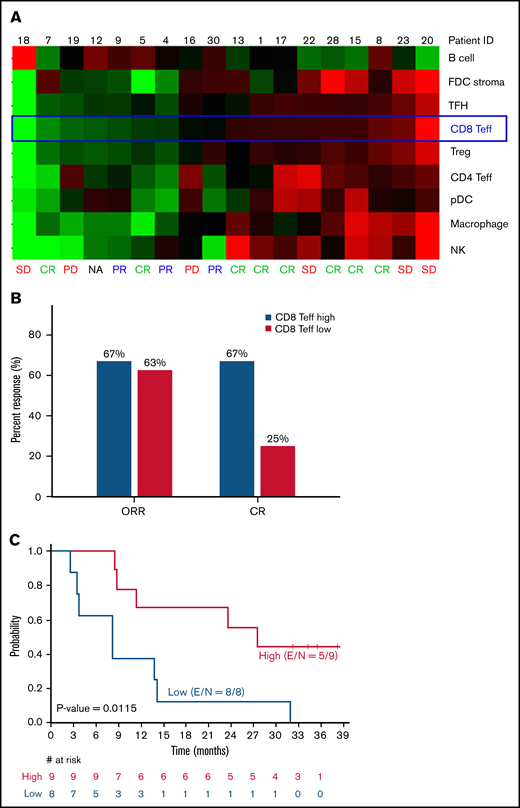
Immune profiling of pretreatment tumor samples by gene signatures. (A) Core tumor biopsy specimens (n = 18) were profiled by using the ∼1500-gene custom NanoString panel. Relative expression of signatures of 9 different immune cell subsets and tumor B cells are shown as heat map with samples ranked by CD8 Teff score. (B) ORR and CR rates stratified by CD8 Teff score. (C) PFS based on CD8 Teff score. FDC, follicular dendritic cell; NA, not assessable; PD, progressive disease; pDC, plasmacytoid dendritic cell; SD, stable disease; Teff, T-effector cell; TFH, T follicular helper cell; E, number of events; S, sample size.
Immune profiling of pretreatment tumor samples by gene signatures. (A) Core tumor biopsy specimens (n = 18) were profiled by using the ∼1500-gene custom NanoString panel. Relative expression of signatures of 9 different immune cell subsets and tumor B cells are shown as heat map with samples ranked by CD8 Teff score. (B) ORR and CR rates stratified by CD8 Teff score. (C) PFS based on CD8 Teff score. FDC, follicular dendritic cell; NA, not assessable; PD, progressive disease; pDC, plasmacytoid dendritic cell; SD, stable disease; Teff, T-effector cell; TFH, T follicular helper cell; E, number of events; S, sample size.
Discussion
The combination of pembrolizumab and rituximab is clinically active and well tolerated in patients with rituximab-sensitive R/R FL, with an objective response observed in 67% of patients with rare grade 3 or 4 treatment or autoimmune-related AEs. Although we acknowledge the potential bias with patient selection in single-arm trials, our study of pembrolizumab in combination with rituximab demonstrated superior response rates when compared with the rates in a previous phase 2 study of retreatment with single-agent rituximab (4 weekly doses, 375 mg/m2) in rituximab-sensitive patients with FL (ORR 67% vs 40%; CR 50% vs 11%, respectively).24 However, PFS was not significantly different with a reported median PFS of 17.8 months in rituximab-sensitive patients treated with rituximab monotherapy compared with 12.6 months with pembrolizumab plusrituximab. Although our trial was designed to include only rituximab-sensitive patients, to augment the ADCC effect of rituximab in combination with pembrolizumab, the synergistic effects of PD-1 inhibition may be modest and limit applicability of this combination to patients with rituximab-refractory disease. A separate phase 3 study randomized patients with rituximab-sensitive R/R FL to either lenalidomide or placebo (12 cycles) plus rituximab (4 weekly doses in cycle 1 and day 1 of cycles 2 through 5).25 Lenalidomide plusrituximab resulted in a significant improvement in PFS when compared with placebo plusrituximab (39.4 vs 14.1 months, respectively). Although the lenalidomide plus rituximab study used a different dosing schedule from that used in our study (6 vs 1 cycle of rituximab), critical review of our results raise the question of whether the combination is any better than rituximab monotherapy or whether extended dosing of rituximab may be required to prolong the duration of response when used in combination with pembrolizumab. A limitation of our single-arm, phase 2 study design with a primary end point of ORR, is that we cannot discern whether pembrolizumab contributes to the efficacy observed. A randomized study would be necessary to explore whether pembrolizumab in combination with rituximab is more effective than rituximab monotherapy. Alternatively, if the therapy had been sequenced and a response assessment performed before the addition of the second agent, this may have provided clarity on whether this combination is synergistic or additive.
Pembrolizumab has impressive single-agent activity in R/R classic Hodgkin lymphoma (cHL) with an ORR of 72% and median duration of response of 16.5 months, supporting the hypothesis that HL evades endogenous immune surveillance by tumor expression of inhibitory checkpoint markers, and this can be therapeutically targeted with checkpoint inhibitors. In the pivotal phase 2 study of single-agent pembrolizumab in R/R cHL, the safety profile was similar to that reported with our pembrolizumab plusrituximab study in R/R FL with treatment-related AEs occurring in most patients (73%, all grades) with rare grade ≥3 AEs (11%).26 The most commonly encountered grade ≥3 treatment-related AEs were similar (neutropenia, diarrhea) as were the reported immune-related AEs (thyroid dysfunction, pneumonitis). No assessed patient characteristics were predictive of immune-related AEs in our study. In the phase 2 study of pidilizumab plus rituximab in R/R FL, no immune-related or grade ≥3 AEs were reported, probably because of the alternative mechanism of pidilizumab via the DLL-1 gene pathway.15 Importantly, no unexpected toxicities were encountered; however, 7 patients (23%) discontinued therapy permanently because of immune-related AEs, higher than the rates of discontinuation of single-agent pembrolizumab in R/R cHL (6.7%).26 This may reflect the low tolerance for toxicity in patients with indolent lymphoma, given the prolonged natural history of the disease and the high response rates. With no treatment-related deaths and infrequent grade 3 or higher AEs, further exploration of anti-PD1 antibodies in combination with rituximab and other immune-activating agents such as lenalidomide are warranted (NCT02446457).
Immune checkpoint inhibitors enhance endogenous antitumor immune response; therefore, patients with preexisting antitumor immunity and baseline intratumoral lymphocyte infiltration are most likely to benefit. Pretreatment tumor immune gene profiling identified a high CD8+ T-effector signature to have the most significant association and strongly correlated with a higher CR rate and significantly longer PFS (27.6 vs 8.3 months; P = .012) when compared with tumors with low CD8+ T-effector signature (Figure 2), suggesting that PD-1/PD-L1 checkpoint blockade may effectively overcome immune evasion in immunocompetent patients with “inflamed” tumors. These findings provide the rationale for triplet combinations with the addition of immunomodulatory agents such as lenalidomide. In preclinical studies, lenalidomide has demonstrated the ability to augment antitumor responses by increasing the proportion of T-effector cells within the tumor microenvironment, repair the immune synapse between tumor cells and cytotoxic T cells, and restore impaired T-cell motility.27-31
Recent studies have identified IFN-γ as a critical driver of PD-L1 expression in tumor cells that downregulates the cytotoxic response from tumor-infiltrating T cells and upregulates key immune suppressive molecules within the tumor microenvironment.32-36 In a previous study, a 10-gene IFN-γ signature panel and an expanded immune 28-gene IFN-γ signature panel were evaluated in tumor samples to identify immunocompetent patients with T-cell–inflamed phenotypes who may benefit the most from PD-1 inhibition. This analysis demonstrated a significant association between the gene expression signature and best ORR and PFS in patients with metastatic melanoma treated with pembrolizumab.22 We performed the same analysis using the 10- and 28-gene IFN-γ signature panels on pretreatment peripheral blood samples of patients with FL and identified a correlation between gene expression signature and tumor reduction, but without a PFS benefit (supplemental Figure 3). Although these findings suggest that a peripheral blood IFN-γ gene signature may serve as a surrogate marker for inflamed tumors and identify patients who are likely to have an initial response to a PD-1 checkpoint blockade, the lack of a sustained clinical response may be a result of immunosuppressive pathways other than PD-1 within the tumor microenvironment. In support of this, the lack of association between tumor and background histiocyte expression of PD-L1 determined by IHC and clinical outcomes suggests a more complex interaction than a single checkpoint pathway. Therefore, there is a need to identify additional immunosuppressive mechanisms and develop novel agents to block other immune checkpoints and improve intratumoral trafficking of antitumor immune cells, which may act synergistically with PD-1 inhibitors. Preclinical studies have demonstrated that immunosuppressive molecules such as IDO1 and LAG3 are also overexpressed in inflamed tumors, suggesting that dual checkpoint blockade with PD-1 and LAG3 inhibition may overcome resistance, with a phase 1/2 study now recruiting.37 In patients lacking the inflamed phenotype, the strategy should be aimed at stimulating a host antitumor immune response, with further work needed to understand the distinct mechanisms that inhibit T-cell infiltration into the tumor.38
In summary, the combination of pembrolizumab and rituximab is associated with a favorable safety profile and a high objective response rate in patients with rituximab-sensitive R/R FL. However, for this approach to be practice changing, improved durability of response is desirable. Several approaches could be considered to enhance the durability, including extended dosing of rituximab instead of the single cycle of 4 weekly doses administered in this study, or combination approaches to enhance T-cell trafficking into the tumor and blocking of multiple immunosuppressive mechanisms in the tumor microenvironment. Accordingly, several phase 2 trials with triplet combination therapies are underway. These future combination studies will define the growing role of targeted agents and checkpoint inhibition within the tumor microenvironment in the treatment of hematologic malignancies and enable further development of novel chemotherapy-free approaches.
Acknowledgments
This work is supported by grants from Merck, a Leukemia and Lymphoma Society Quest for Cure grant (P-QFC-3068-14) (S.S.N.), generous philanthropic contributions to the University of Texas Anderson Moon Shots Program, and The University of Texas Anderson Cancer Center Support from National Institutes of Health, National Cancer Institute grant P30CA016672.
Funding support for this article was provided by the The University of Texas MD Anderson Cancer Center Support Grant from National Institutes of Health (P30 CA016672)
Authorship
Contribution: L.J.N., and S.S.N. designed the study; L.J.N., C.K.C., J.R.W, N.H.F., F.S., L.D., C.O., J.M., L.F., and S.S.N., participated in enrollment of patients, treatment of patients, data collection, data analysis, and data interpretation; L.J.N., C.K.C., X.C., M.C.J.M., Z.W., F.C., S.Z., M.G., R.E.D., S.S.N., contributed to correlative studies, data analysis, and data interpretation; L.F. performed the statistical analysis; X.C., M.C.J.M., Z.W., F.C., and S.S.N., obtained tumor biopsy specimens for correlative studies; and all authors read and approved the final manuscript.
Conflict-of-interest disclosure: L.J.N. has received honoraria from ADC Therapeutics, Bayer, BMS/Celgene, Epizyme, Genentech, Gilead/KITE, Janssen, MEI, Novartis, Pfizer, Takeda, and TG Therapeutics; and research funding from BMS/Celgene, Caribou Biosciences, Epizyme, Genentech, IGM Biosciences, Janssen, Merck, Novartis, Takeda, and TG Therapeutics. S.S.N. has received research support from Kite/Gilead, Cellectis, Poseida, Merck, Acerta, Karus, BMS, Unum Therapeutics, Allogene, and Precision Biosciences; has served as a consultant and an advisory board member for Kite/Gilead, Celgene, Novartis, Unum Therapeutics, Pfizer, Merck, Precision Biosciences, Cell Medica, Incyte, Allogene, Calibr, and Legend Biotech; has received royalties from Takeda Pharmaceuticals; and holds patents related to cell therapy. The remaining authors declare no competing financial interests.
The current affiliation for M.C.J. M. is BioLegend, Inc.
Correspondence: Loretta J. Nastoupil, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030-4009; e-mail: lnastoupil@mdanderson.org.