Key Points
Platelet reactivity via the PAR-1 thrombin receptor is mediated by GRK5.
Platelet GRK5 is associated with thrombus formation in humans and mice.

Abstract
The interindividual variation in the functional response of platelets to activation by agonists is heritable. Genome-wide association studies (GWASs) of quantitative measures of platelet function have identified fewer than 20 distinctly associated variants, some with unknown mechanisms. Here, we report GWASs of pathway-specific functional responses to agonism by adenosine 5′-diphosphate, a glycoprotein VI–specific collagen mimetic, and thrombin receptor-agonist peptides, each specific to 1 of the G protein–coupled receptors PAR-1 and PAR-4, in subsets of 1562 individuals. We identified an association (P = 2.75 × 10−40) between a common intronic variant, rs10886430, in the G protein–coupled receptor kinase 5 gene (GRK5) and the sensitivity of platelets to activate through PAR-1. The variant resides in a megakaryocyte-specific enhancer that is bound by the transcription factors GATA1 and MEIS1. The minor allele (G) is associated with fewer GRK5 transcripts in platelets and the greater sensitivity of platelets to activate through PAR-1. We show that thrombin-mediated activation of human platelets causes binding of GRK5 to PAR-1 and that deletion of the mouse homolog Grk5 enhances thrombin-induced platelet activation sensitivity and increases platelet accumulation at the site of vascular injury. This corroborates evidence that the human G allele of rs10886430 is associated with a greater risk for cardiovascular disease. In summary, by combining the results of pathway-specific GWASs and expression quantitative trait locus studies in humans with the results from platelet function studies in Grk5−/− mice, we obtain evidence that GRK5 regulates the human platelet response to thrombin via the PAR-1 pathway.
Introduction
Platelets are essential to prevent bleeding. By continuously surveying vessel walls and activating in response to damage, they initiate the formation of thrombi to close any breaches. If hemostasis is well balanced, thrombus formation is followed by thrombolysis and vessel wall repair. Unfortunately, platelet activation may initiate inappropriate thrombus formation in individuals with vascular disease, leading to vessel occlusion and ischemia. Consequently, platelet inhibitors are standard treatment for the secondary prevention and treatment of ischemic cardiovascular diseases.
Measures of the activation sensitivity of platelets, henceforth “reactivity phenotypes,” have heritable interindividual components of variation.1,2 However, this heritable variation is less well studied than that of resting platelet phenotypes, such as platelet count (PLT#) and mean platelet volume (MPV). Genome-wide association studies (GWASs) of platelet phenotypes measured by complete blood counts (CBC) in more than half a million participants identified >1800 distinctly associated genetic variants, explaining much of the heritable variation in the population with European ancestry.3,4 In contrast, studies designed to identify variants associated with platelet reactivity by agonists have each relied on <4000 participants and, in aggregate, identified <20 distinctly associated alleles.2,5 , -9 The principal reason for relatively low sample sizes is the intricate nature of platelet reactivity phenotyping. The activation of platelets in response to vascular damage is caused by the binding of a variety of agonists to cell surface receptors, in particular, the receptors for collagen and thrombin, which are important initiators of activation, and the receptors for adenosine 5′-diphosphate (ADP) and thromboxane A2, which are important for amplification. To quantify reactivity in vitro, these receptors must be stimulated and the platelet response measured by light transmission aggregometry (LTA) or by flow cytometry of surface markers of activation.
Here, we present the results of GWASs of platelet reactivity phenotypes in subsets of 1562 healthy adults who were enrolled in the Cambridge Platelet Function Cohort (PFC). Using flow cytometry, we measured responses to agonism by ADP, by the GPVI-specific ligand collagen-related peptide (CRP-XL), and by thrombin receptor-agonist peptides, each specific to PAR-1 or PAR-4. Analyses of these phenotypes replicated several known pathway-specific associations. Additionally, we identified an association, on chromosome 10q26, between a variant in a platelet expression quantitative trait locus (eQTL) for G protein–coupled receptor kinase 5 gene (GRK5) and reactivity to thrombin, replicating a recently published association in which reactivity was measured by LTA.10 By analyzing reactivity to PAR-1– and PAR-4–specific peptides, we provide compelling evidence that the association signal in GRK5 is mediated through the PAR-1 receptor. Furthermore, by deleting the mouse homolog Grk5, we show that mouse platelets without Grk5 exhibit greater ex vivo platelet reactivity to thrombin, but not to other agonists. Grk5−/− mice showed greater platelet accumulation upon laser-induced vascular injury and increased thrombin-induced pulmonary thromboembolism compared with wild-type (WT) controls. Collectively, the results of our genetic and functional studies in humans and mice provide robust evidence that G protein-coupled receptor kinase 5 (GRK5) encoded by GRK5, is a key regulator of platelet activation by thrombin via the G protein–coupled receptor (GPCR) PAR-1.
Methods
Detailed descriptions of the materials and methods used are provided in supplemental Information.
Human study participants
Donors registered with National Health Service Blood and Transplant (NHSBT) were invited to join the National Institute for Health Research BioResource, a cohort of volunteers who have consented to be recalled for studies by genotype. Subsequently, these NIHR BioResource members were invited to participate in the Cambridge PFC. Blood for the protein binding experiments was provided by healthy donors at Thomas Jefferson University. Written informed consent was obtained from participants prior to blood collection. Information about the study participants and the permissions of the institutional review board and research ethics committee are provided in supplemental Information.
Platelet function association study
DNA extracted from EDTA-treated blood from Cambridge PFC participants was genotyped using the UK Biobank Axiom Array (Thermo Fisher, Santa Clara, CA). A description of the quality control and imputation methods is included in supplemental Information.
Platelet activation was measured by whole blood flow cytometry assays, as previously described11 (supplemental Information). Platelets were agonized with ADP (Sigma-Aldrich), CRP-XL (crosslinked monomeric sequence GCO[GPO]10GCOG; Department of Biochemistry, University of Cambridge), PAR-1, PAR-4,12 and TRAP-6 (Tocris) agonist peptides. The binding of a PE-labeled anti–P-selectin monoclonal antibody (CD62P; NHSBT, Bristol, UK) and FITC-labeled polyclonal anti-fibrinogen (Agilent Dako) was quantified using Beckman Coulter flow cytometers. Individual-level summaries of platelet reactivity were derived by logit transforming the percentage of cells with a signal greater than a threshold determined from background, measured in corresponding aliquots of unagonized blood.
PLT# and MPV were measured from EDTA-treated blood using Beckman Coulter and Sysmex Complete Blood Count analyzers. We regressed the variation explained by sex, age, PLT#, MPV, and reagent batch from the individual-level summaries of reactivity (supplemental Information).
With the exception of 1 subset of 450 individuals for whom the activation response to agonism by ADP was measured in duplicate only by fibrinogen binding, each activation response was measured 4 times: twice using P-selectin expression and twice using fibrinogen binding. Outliers and discordant repeated measurements were discarded, and the average of the repeats was taken. The effect of PLT#, MPV, sample batch, sex, and age were regressed out of each average. Outliers and samples for which the P-selectin and fibrinogen binding averages were discordant were discarded. The P-selectin expression and fibrinogen binding averages correlated strongly (supplemental Figure 1); therefore, to increase the power to detect novel genetic associations, we performed a GWAS for each agonist using the score vector corresponding to the first principal component of the averaged P-selectin and averaged fibrinogen measurements as the phenotype, after further adjustment for PLT#, MPV, sample batch, sex, age, and time elapsed between the start of the study and the time of measurement. The GWAS tests were performed using linear mixed models to control for relatedness. For response to agonism by ADP, separate GWASs were performed in the subset of individuals for whom reactivity was measured by fibrinogen binding only, as well as in the subset for whom reactivity was measured by fibrinogen binding and P-selectin expression. The evidence for association was combined using inverse variance weighted meta-analysis after scaling the statistics from the fibrinogen binding GWAS to place them on the same scale of measurement as the statistics from the other GWAS.
For replication, platelet activation was measured in an independent cohort of donors using a plate-based aggregation end point assay, as described.13 The aggregation of platelets in response to increasing doses of TRAP-6 was measured using absorbance, and curves were generated to calculate the 50% effective concentration values.
A detailed description of the genetic association analysis is provided in supplemental Information.
GRK5 and PAR-1 immunoprecipitation
Washed human platelets were activated using thrombin and then lysed (supplemental Information). Proteins were precipitated with an anti–PAR-1 (ATAP-2) antibody or inert control immunoglobulin and probed for GRK5 before reprobing with an anti–PAR-1 antibody.
Mouse models
All mouse protocols were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania or Thomas Jefferson University.
Mice protocols are described in supplemental Information. Grk5-knockout mice were generated on a C57BL/6 background using CRISPR-Cas9 genome editing14 ; fetal liver chimeras were generated using transplantation from WT control or Grk5−/− mice.
Agonist-mediated platelet activation in vitro
Mouse platelet activation experiments were performed as described previously using flow cytometry and LTA15 (supplemental Information). For flow cytometry, platelets were stimulated with PAR-4 agonist peptide (PAR-4–AP), ADP, thromboxane A2 mimetic (U46619), or convulxin in diluted heparinized blood. Binding of fluorochrome-conjugated antibodies against CD41, P-selectin (CD62P; BD Bioscience), and activated integrin was used to measure platelet activation with a BD LSR II Flow Cytometer (BD Biosciences). For LTA assays, platelets were collected from platelet-rich plasma, and counts were adjusted to 2 × 108 per milliliter. Aggregation was observed in a PAP-8E Platelet Aggregation Profiler (Bio/Data Corporation). Single end point luminescence assays were used to quantify agonist-induced platelet ATP release, as described.15
Vascular injury and pulmonary thromboembolism models
Thrombus formation upon vascular injury was measured in the cremaster muscle microcirculation of 8- to 12-week-old male mice, as described.16 Pulmonary thromboembolism assay was performed as previously described with some modifications.17 Additional details about each method are provided in supplemental Information. Data sets were compared using the Student t test or the Mann-Whitney U test. P ≤. 05 was considered statistically significant.
Results
GWASs for platelet activation
We measured platelet reactivity to agonists in subsets of 1562 healthy participants in the Cambridge PFC using pathway-specific ex vivo stimulation. We agonized the platelet GPCR signaling pathways, activating the receptors P2Y1 and P2Y12 with ADP and PAR-1 and PAR-4 (the thrombin receptors) with receptor-specific peptides (PAR-1 and PAR-4). Additionally, we agonized the GPVI-FcεRI/ITAM signaling pathway with CRP-XL. We quantified platelet reactivity at the individual level by summarizing cell level measurements of surface P-selectin expression and bound fibrinogen.
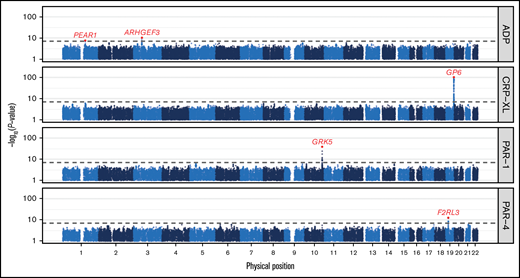
GWAS identified 5 genome-wide significant association signals (P < 5 × 10−8; Figure 1; Table 1). These association signals recapitulate previous studies, including those by us based on analyses of subsets of the present data set. They include associations with reactivity to ADP in PEAR1 (rs12566888) and ARHGEF3 (rs7624918) and associations with reactivity to CRP-XL in GP6 (rs1613662).2,9,11 The analyses of reactivity to the 2 thrombin receptor agonists relied on entirely new data and identified 2 associations. First, we replicated an association in F2RL3 (rs773902), the gene encoding the PAR-4 receptor, identified with reactivity to PAR-4 agonism measured by LTA.18 Second, we identified a newly discovered association in GRK5 (rs10886430, chr10:121010256A>G, GRCh37) with reactivity to PAR-1 agonism.10
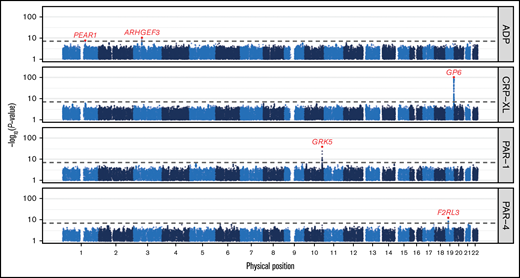
Genome-wide associations with platelet reactivity. Manhattan plot showing the P values of tests for association between genetic variants and each of 4 phenotypes measuring the reactivity of platelets to agonism by ADP, CRP-XL, PAR-1, and PAR-4 in the Cambridge PFC. Each dot corresponds to a genetic variant in the Haplotype Reference Consortium r1.1 reference panel. The position on the x-axis indicates the physical position of the variant; the position on the y-axis indicates the -log10P value of a Wald test for association from a linear mixed-model (on a log scale). Only variants with an imputation INFO score > 0.6 and a P value < .1 are shown. The horizontal dashed line corresponds to the genome-wide significance threshold (5 × 10−8). The red dots correspond to the variants showing the strongest evidence for association in those loci containing significantly associated variants. The red gene names indicate the gene causally mediating each of these associations.
Genome-wide associations with platelet reactivity. Manhattan plot showing the P values of tests for association between genetic variants and each of 4 phenotypes measuring the reactivity of platelets to agonism by ADP, CRP-XL, PAR-1, and PAR-4 in the Cambridge PFC. Each dot corresponds to a genetic variant in the Haplotype Reference Consortium r1.1 reference panel. The position on the x-axis indicates the physical position of the variant; the position on the y-axis indicates the -log10P value of a Wald test for association from a linear mixed-model (on a log scale). Only variants with an imputation INFO score > 0.6 and a P value < .1 are shown. The horizontal dashed line corresponds to the genome-wide significance threshold (5 × 10−8). The red dots correspond to the variants showing the strongest evidence for association in those loci containing significantly associated variants. The red gene names indicate the gene causally mediating each of these associations.
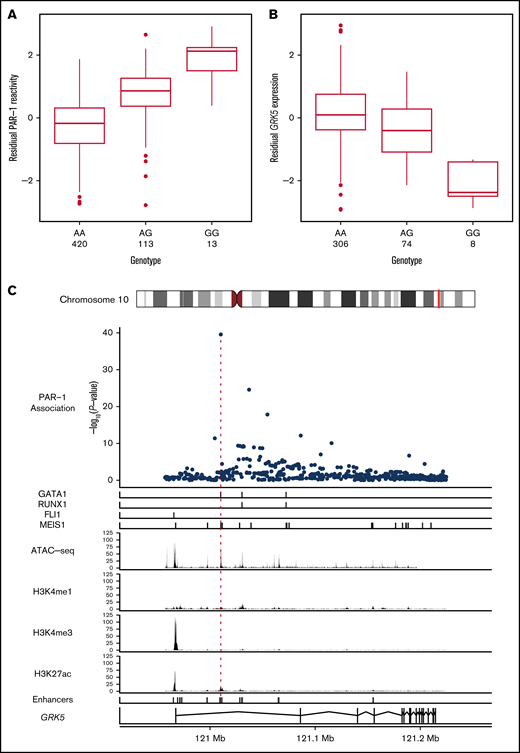
The PAR-1 association signal in the GRK5 locus was explained completely by conditioning on the imputed minor allele (G) count of rs10886430, the variant exhibiting the strongest evidence for association with platelet reactivity (n = 546; β = 1.09 standard deviations [SDs]; standard error [SE], 8.21 × 10−2; P = 2.75 × 10−40; Figure 2A; Table 1). Our analyses showed that rs10886430 is not associated with platelet activation by ADP or CRP-XL. However, an association was identified with reactivity to the PAR-4–specific peptide, although with a smaller estimated effect size (β = 0.44 SD; SE, 0.092; P = 2.20 × 10−6) (supplemental Figure 3).
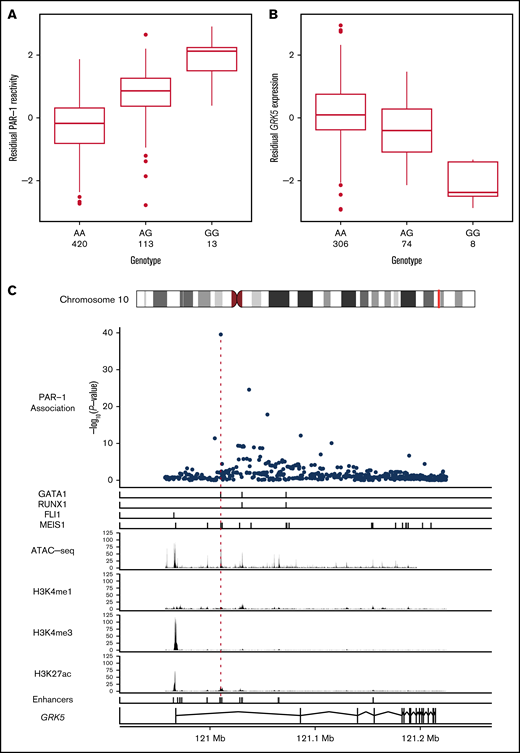
Increased platelet reactivity via PAR-1 at rs10886430 is due to decreased expression of GRK5. Box plots showing the relationships between the genotype of rs10886430 and the reactivity of platelets to PAR-1 (after adjustment for covariates) in 546 participants in the Cambridge PFC (A) and platelet expression of GRK5 measured using probe ID:3190239 of the Illumina HumanHT-12 v4.0 Expression BeadChip microarray in 388 donors (B), after adjustment for technical variation. The thick horizontal bars indicate the median of each conditional distribution and the lower and upper hinges indicate the 25th and 75th percentiles, respectively, of each distribution. The whiskers extend no further than 1.5 interquartile ranges from the hinges. (C) The genetic association between PAR-1 reactivity and rs10886430 in intron 1 of GRK5 colocalizes with the binding of the transcription factors GATA-1 and MEIS1 in human MKs and an enhancer specific to the human MK blood cell lineage. Top to bottom: the strength of evidence for the association between genetic variants in the GRK5 locus and PAR-1 reactivity, measured by −log10(P value); the binding sites of the transcription factors GATA-1, RUNX1, FLI1, and MEIS1 in human MKs; ATAC-seq read depth indicating regions of open chromatin in human MKs; ChIP-seq read depth measuring the histone modifications H3K4me1, HSK4me3, and H3K27ac in human MKs; human MK enhancer sites inferred from the ATAC-seq and ChIP-seq data; and a model of the GRK5 gene with exons indicated by rectangles.
Increased platelet reactivity via PAR-1 at rs10886430 is due to decreased expression of GRK5. Box plots showing the relationships between the genotype of rs10886430 and the reactivity of platelets to PAR-1 (after adjustment for covariates) in 546 participants in the Cambridge PFC (A) and platelet expression of GRK5 measured using probe ID:3190239 of the Illumina HumanHT-12 v4.0 Expression BeadChip microarray in 388 donors (B), after adjustment for technical variation. The thick horizontal bars indicate the median of each conditional distribution and the lower and upper hinges indicate the 25th and 75th percentiles, respectively, of each distribution. The whiskers extend no further than 1.5 interquartile ranges from the hinges. (C) The genetic association between PAR-1 reactivity and rs10886430 in intron 1 of GRK5 colocalizes with the binding of the transcription factors GATA-1 and MEIS1 in human MKs and an enhancer specific to the human MK blood cell lineage. Top to bottom: the strength of evidence for the association between genetic variants in the GRK5 locus and PAR-1 reactivity, measured by −log10(P value); the binding sites of the transcription factors GATA-1, RUNX1, FLI1, and MEIS1 in human MKs; ATAC-seq read depth indicating regions of open chromatin in human MKs; ChIP-seq read depth measuring the histone modifications H3K4me1, HSK4me3, and H3K27ac in human MKs; human MK enhancer sites inferred from the ATAC-seq and ChIP-seq data; and a model of the GRK5 gene with exons indicated by rectangles.
We replicated the association with rs10886430, in the same study participants, by replacing the PAR-1 agonizing peptide with TRAP-6, another PAR-1 receptor–specific agonist peptide (β = 1.18 SD; SE, 0.0801; P = 8.91 × 10−49). Additionally, we performed an out-of-sample replication experiment using the TRAP-6 agonist and a microplate-based platelet aggregation assay,13 which provided evidence for an association with the same direction of effect (n = 253; β = −0.103 SD; SE, 0.042; P = .014; supplemental Figure 4).
Genomic characterization of PAR-1 platelet activation association at GRK5
To explore the molecular mechanism underlying the platelet reactivity association at GRK5, we interrogated eQTL data for platelets aggregated from 2 independent studies (supplemental Information). The minor allele (G) of rs10886430, corresponding to greater platelet reactivity, was associated with a lesser amount of GRK5 messenger RNA in platelets (n = 388; = −0.50 SD; SE, 0.0473; P = 4.79 × 10−23; Figure 2B).
Inspection of our previously published erythroblast and megakaryocyte (MK) assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) data showed that rs10886430 lies in an MK-specific nucleosome-depleted element.9 Chromatin immunoprecipitation and sequencing (ChIP-seq) of MKs showed that the element has the H3K27Ac profile characteristic of an enhancer and identified strong evidence for binding by GATA1 and MEIS1, 2 of the key MK lineage transcription factors.9 However, we did not see any evidence for binding by the MK factors FLI1, GATA2, RUNX1, and TAL119,20 (Figure 2C). A difference in affinity between the 2 alleles of rs10886430 to bind to GATA1 and/or MEIS1 is the most likely explanation for the GRK5 eQTL. This hypothesis is supported by Rodriguez et al, who showed that the transcriptional activity of constructs containing the enhancer region is reduced when the G allele of rs10886430 is introduced into K562 cell and HUVEC lines.10
Together, these findings indicate that the molecular mechanism explaining the association between rs10886430 and platelet reactivity to agonism through PAR-1 is mediated by the allele-specific differences in the expression of GRK5 messenger RNA.
Binding of GRK5 to PAR-1 during platelet activation
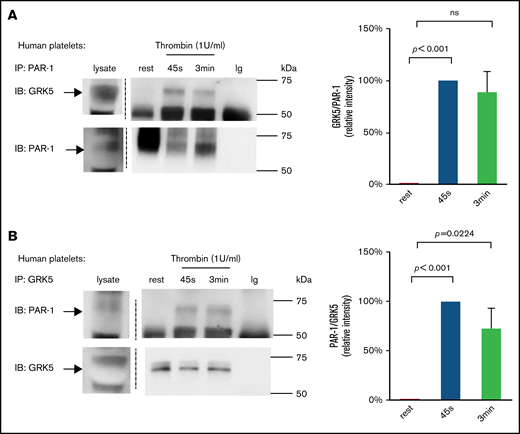
The regulation of PAR-1 signaling by GRK5 suggests that GRK5 binds to PAR-1 in response to thrombin stimulation. To evaluate this interaction, we precipitated these proteins in washed human platelets and then probed them using antibodies against GRK5 or PAR-1 (Figure 3). During platelet activation using thrombin (1 U/mL), immunoprecipitation of PAR-1 coprecipitated GRK5 (Figure 3A); conversely, immunoprecipitation of GRK5 pulled down PAR-1 (Figure 3B). Furthermore, when platelets were activated with a lower concentration of thrombin (0.1 U/mL), immunoprecipitation of GRK5 also pulled down PAR-1 (supplemental Figure 5). When we applied the same experimental procedure to resting platelets, we detected little or no binding of GRK5 to PAR-1. GRK5 and GRK6 are 72% homologous at the amino acid level21,22 ; therefore, we excluded the possibility that our results were a consequence of cross-reactivity between anti-GRK5 and GRK6 (supplemental Figure 6).
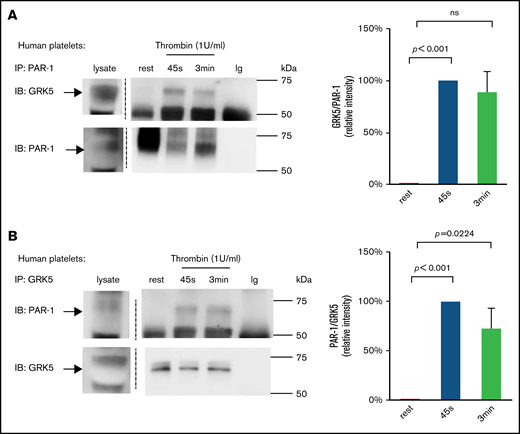
Activation of platelets with thrombin increases GRK5 and PAR-1 binding in human platelets. (A) Resting human platelets or thrombin (1 U/mL)-activated platelets were prepared and lysed. Proteins were precipitated with an anti–PAR-1 (ATAP-2) antibody or control immunoglobulin (Ig) and then probed with anti-GRK5 antibody and reprobed with an anti–PAR-1 antibody (n = 6). (B) Lysates from resting or activated platelets were precipitated with an anti-GRK5 antibody or Ig and probed with anti–PAR-1 before reprobing with anti-GRK5 (n = 3). IB, immunoblotting; IP, immunoprecipitation; ns, not significant.
Activation of platelets with thrombin increases GRK5 and PAR-1 binding in human platelets. (A) Resting human platelets or thrombin (1 U/mL)-activated platelets were prepared and lysed. Proteins were precipitated with an anti–PAR-1 (ATAP-2) antibody or control immunoglobulin (Ig) and then probed with anti-GRK5 antibody and reprobed with an anti–PAR-1 antibody (n = 6). (B) Lysates from resting or activated platelets were precipitated with an anti-GRK5 antibody or Ig and probed with anti–PAR-1 before reprobing with anti-GRK5 (n = 3). IB, immunoblotting; IP, immunoprecipitation; ns, not significant.
Generation and characterization of Grk5−/− mice
We studied the consequences of Grk5 inhibition on thrombin-mediated activation signaling using Grk5-knockout mice. Grk5−/− mice were generated by introducing a premature stop codon using CRISPR-Cas9, resulting in the loss of Grk5 expression (supplemental Figure 7). Grk5−/− mice that were generated by crossing were born in the expected Mendelian inheritance ratios and were viable and grossly normal in appearance. The average values of CBC-measured hematological variables did not differ significantly between knockout mice and their WT littermates (supplemental Table 1). Furthermore, deletion of Grk5 did not affect the expression of Grk2 and Grk6 proteins in mouse brain (supplemental Figure 7D).
Grk5−/− platelets display increased agonist-mediated platelet activation ex vivo
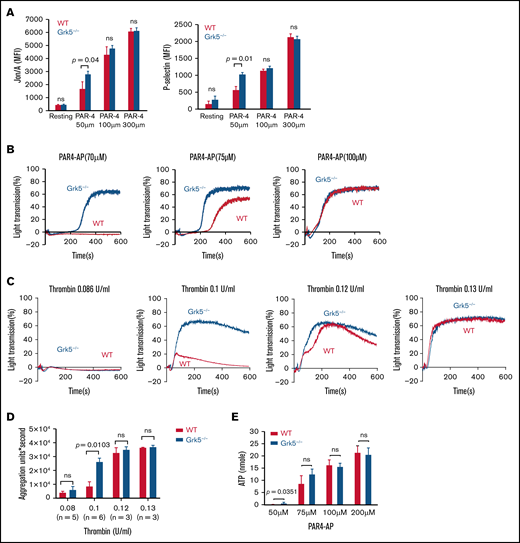
We compared the reactivity of platelets from Grk5−/− mice with those from WT mice, using flow cytometry to measure surface-bound fibrinogen and anti–P-selectin as markers of αIIbβ3 activation and α-granule secretion respectively.15,23 We agonized platelets using a mouse Par-4 thrombin receptor agonist peptide (AYPGKF), because Par-4 is the mouse homolog of human PAR-1. Mean integrin activation (P = .04) and P-selectin expression (P = .01) were greater in platelets generated by Grk5−/− than by WT mice in response to a low-dose of the Par-4 agonist peptide. However, there was no significant difference when high doses of Par-4 agonist peptide were used (Figure 4A). There was no evidence for differences in activation response to agonism by ADP, by the thromboxane A2 analog U46619, or by convulxin, which is an alternative ligand to CRP-XL for GpVI (supplemental Figure 8). Phenotyping of reactivity to the Par-4 agonist using LTA corroborated the flow cytometry data (Figure 4B). Furthermore, when platelets were incubated with thrombin, an increased response was also observed in Grk5−/− platelets (Figure 4C-D). LTA-measured responses to other agonists indicated a slight initial increase in Grk5−/− platelet response to ADP, but there was no difference in the response to U46619 or convulxin (supplemental Figure 9).
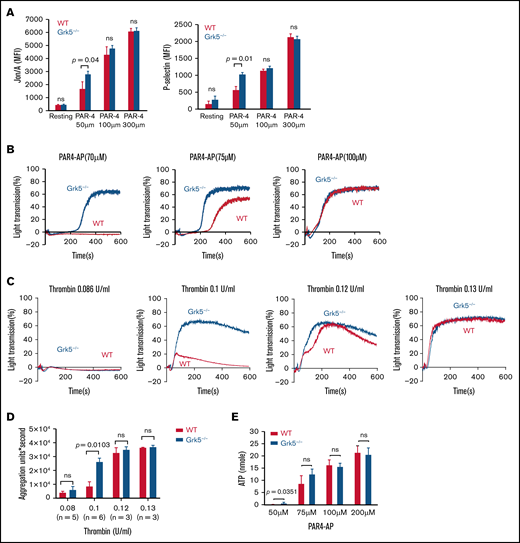
Increased integrin activation, α-granule exocytosis, aggregation, and ATP release in platelets from Grk5−/− mice. (A) Platelets from Grk5−/− and littermate control (WT) mice were stained with antibodies to activate ɑIIbβ3 (Jon/A; left panel) or P-selectin (right panel) and measured by flow cytometry. Platelets were stimulated with Par-4–AP (AYPGKF) (n = 3). Platelet reactivity in response to Par-4–AP (n = 5) (B), representative aggregation traces (C), and summarized data (D) for platelets stimulated with thrombin (mean ± SEM; n = 3-6 mice at each tested concentration). (E) ATP release for platelets from Grk5−/− and littermate control mice (WT) stimulated with Par-4–AP (AYPGKF). The results of 3 experiments (mean ± SEM) are summarized. MFI, mean fluorescence intensity; ns, not significant.
Increased integrin activation, α-granule exocytosis, aggregation, and ATP release in platelets from Grk5−/− mice. (A) Platelets from Grk5−/− and littermate control (WT) mice were stained with antibodies to activate ɑIIbβ3 (Jon/A; left panel) or P-selectin (right panel) and measured by flow cytometry. Platelets were stimulated with Par-4–AP (AYPGKF) (n = 3). Platelet reactivity in response to Par-4–AP (n = 5) (B), representative aggregation traces (C), and summarized data (D) for platelets stimulated with thrombin (mean ± SEM; n = 3-6 mice at each tested concentration). (E) ATP release for platelets from Grk5−/− and littermate control mice (WT) stimulated with Par-4–AP (AYPGKF). The results of 3 experiments (mean ± SEM) are summarized. MFI, mean fluorescence intensity; ns, not significant.
We used single end point luminescence assays15 to measure the ATP released from platelets in response to agonism by Par-4 agonist peptide and ADP. Increased ATP release was observed in platelets from Grk5−/− mice compared with WT mice upon activation using a low dose of Par-4 agonist peptide (P = .04). However, there was no evidence for a difference in the response to agonism by ADP (supplemental Figure 10). These results are compatible with the notion that platelets without Grk5 secrete a greater number of δ-granules when activated via the thrombin receptor Par-4 compared with normal WT platelets (Figure 4E).
ADP activates platelets by binding to Gq-coupled P2Y1 and Gi2-coupled P2Y12 receptors.24 ADP P2Y12 signaling triggers activation/phosphorylation of the Gi2-mediated serine/threonine kinase Akt.25 To determine whether deletion of Grk5 affects P2Y12-dependent signaling, we compared phosphorylation of Akt in WT and Grk5−/− platelets in response to ADP stimulation in the presence or absence of the P2Y1 antagonist (MRS2500) or the P2Y12 antagonist (cangrelor) (supplemental Information). Upon ADP stimulation, Grk5−/− platelets exhibited greater phosphorylation of Akt than did WT platelets, but the increase was not statistically significant (supplemental Figure 11). Greater phosphorylation of Akt from Grk5−/− platelets vs WT platelets was still observed upon blocking P2Y1 with MRS2500 (supplemental Figure 11C). However, in the presence of the P2Y12 antagonist cangrelor, the phosphorylation of Akt in response to stimulation by ADP was abolished in WT and Grk5−/− platelets (supplemental Figure 11A).
Grk5 regulates the hemostatic response to injury in vivo
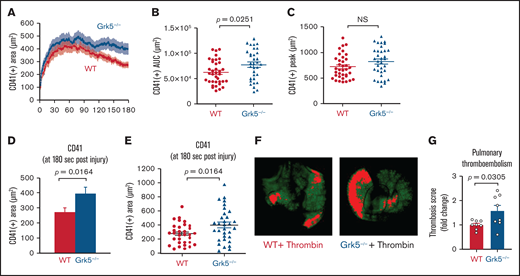
We used the laser-induced injury model in cremaster muscle arterioles to investigate whether enhanced in vitro platelet responsiveness to Par-4 receptor activation in Grk5−/− mice had an effect on thrombus formation following vascular damage.16,23 In this model, the core of the hemostatic plug consists of P-selectin–positive platelets bound by fibrin that is surrounded by a loosely packed shell of less-activated P-selectin–negative platelets. We measured platelet accumulation for 180 seconds following injury using fluorescently labeled anti-CD41 (Figure 5). Initially, platelets accumulated at similar rates in Grk5−/− and WT mice (Figure 5A). However, we observed greater accumulation at the late stage in Grk5−/− mice compared with WT mice, as demonstrated by the increase in the mean area under the curve (P = .03) and mean total platelet accumulation at 180 seconds postinjury (P = .016) (Figure 5B,D-E; supplemental Figure 12). Because there was no difference in mean P-selectin deposition between the 2 groups (supplemental Figure 13), increased thrombus formation was due to an expansion of the P-selectin–negative shell region, rather than the P-selectin–positive core region. Deleting Grk5 did not affect fibrin accumulation, suggesting that thrombin generation at the site of injury is normal in Grk5−/− mice (supplemental Figure 13). To exclude possible effects of Grk5 deletion on cells within the vascular wall, we performed additional laser injury studies in irradiated WT mice reconstituted with hematopoietic cells harvested from WT or Grk5−/− fetal livers. The results showed that the increase in platelet accumulation at the site of vascular injury was similar in reconstituted Grk5−/− mice to that seen in whole-body Grk5−/− mice (supplemental Figure 14).
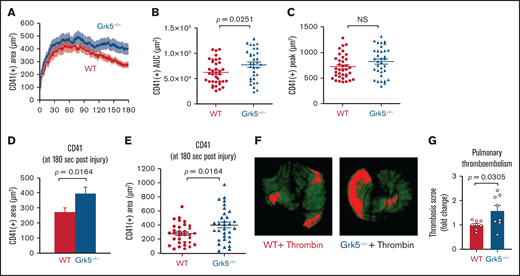
Increased platelet accumulation at the site of vascular injury in Grk5−/− mice. (A) Platelet accumulation was visualized by CD41 staining after laser injuries in cremaster muscle arterioles and recorded using confocal intravital fluorescence microscopy. (B) Area under CD41 vs time curve. (C) Peak of the CD41+ area at 180 seconds after injury. Bar graph (D) and dot plots (E) showing CD41 deposition at 180 seconds after injury. All data are mean ± SEM; 36 injuries in 3 WT mice and 36 injuries in 4 Grk5−/− mice. (F) Representative images of anti-GPIX–labeled thrombi in lungs harvested from WT and Grk5−/− mice. Images were acquired using the Odyssey LiCor imaging system. (G) Thrombosis score, representing the mean thrombus area and number of thrombi, for 8 WT mice vs 8 Grk5−/− mice. Group means were compared using the Student t test; P ≤ .05 was considered statistically significant. AUC, area under the curve; NS, not significant.
Increased platelet accumulation at the site of vascular injury in Grk5−/− mice. (A) Platelet accumulation was visualized by CD41 staining after laser injuries in cremaster muscle arterioles and recorded using confocal intravital fluorescence microscopy. (B) Area under CD41 vs time curve. (C) Peak of the CD41+ area at 180 seconds after injury. Bar graph (D) and dot plots (E) showing CD41 deposition at 180 seconds after injury. All data are mean ± SEM; 36 injuries in 3 WT mice and 36 injuries in 4 Grk5−/− mice. (F) Representative images of anti-GPIX–labeled thrombi in lungs harvested from WT and Grk5−/− mice. Images were acquired using the Odyssey LiCor imaging system. (G) Thrombosis score, representing the mean thrombus area and number of thrombi, for 8 WT mice vs 8 Grk5−/− mice. Group means were compared using the Student t test; P ≤ .05 was considered statistically significant. AUC, area under the curve; NS, not significant.
Absence of Grk5 increases thrombin-induced pulmonary thromboembolism
Finally, we evaluated the role of Grk5 in thrombin signaling using a thrombosis model. Thrombin-induced pulmonary thromboembolism is a model of systemic occlusive pulmonary microthrombi that are composed of platelet aggregates and fibrin.26,27 To determine whether deletion of Grk5 would increase the incidence of thromboembolism formation induced by platelet activation, Grk5−/− mice and their WT control littermates were injected IV with thrombin. In the absence of Grk5, there was a 1.6-fold increase in thrombus formation in the lung of Grk5−/− mice compared with their WT control littermates (Figure 5F-G).
Discussion
We performed GWAS analyses of platelet reactivity phenotypes in subsets of 1562 healthy blood donors, recapitulating genetic association signals previously reported by us and by other investigators (in ARHGEF3 and PEAR1 for ADP, in GP6 for CRP-XL, and in F2RL3 for PAR-4–specific agonist peptide). Additionally, we identified a new association between the variant rs10886430 in GRK5 and platelet reactivity to a PAR-1 agonist peptide, corroborating the recent report of an association with platelet reactivity to thrombin measured by LTA.10 Our analyses clarified the molecular mechanism distal to the allelic variation in GRK5. The direct consequence of the polymorphism almost certainly generates differential binding affinity of the transcription factors GATA1 and MEIS1 to an MK-specific enhancer element, causing allelic differences in the abundance of GRK5 transcript in platelets.
Interestingly, the association of rs10886430 with platelet reactivity colocalizes with an association with MPV reported by Astle et al.4 The G allele, which is associated with lower platelet GRK5 transcript levels and greater platelet reactivity via PAR-1, is also associated with greater MPV (P = 6.6 × 10−18). To exclude the possibility that the platelet reactivity effect is mediated entirely through variation in platelet volume, we regressed MPV from the phenotype prior to performing our GWAS.
Rodriquez et al identified the same genetic variant, rs10886430, associated with platelet reactivity to thrombin; by generating GRK5-knockdown platelets derived from MK progenitor cell lines and using a GRK5 inhibitor in human platelets, they showed that GRK5 regulates signaling primarily via PAR-4.10 In contrast, our study provides compelling evidence that GRK5 also regulates thrombin signaling via PAR-1. The platelet reactivity flow cytometry measurement in our study differs from the standard LTA because amplification of the main subsidiary activation pathways is inhibited by aspirin, hirudin, and apyrase, when activating platelets with PAR-specific peptides. Hence, we can examine the genome-wide association results in a pathway-specific manner excluding effects brought about by subsidiary activation. Furthermore, the association of PAR-1 is supported by coimmunoprecipitation of GRK5 with PAR-1 upon platelet activation by thrombin. Although not at genome-wide significance, our results also suggest that rs10886430 is associated with platelet reactivity via PAR-4. Together, these results likely indicate that, in human platelets, thrombin signaling via PAR-1 and PAR-4 is regulated by GRK5. The current challenges for studying this pathway are due to the lack of specific anti–PAR-4 antibodies. Future work will look to elucidate the contribution of GRK5 regulation in human platelet thrombin signaling once specific reagents are available.
In humans, the protease-activated GPCRs PAR-1 and PAR-4 are the receptors for thrombin. Cleavage of their exodomains by thrombin releases tethered ligands, which bind in a cognate manner to the receptor. The synthetic PAR-1 and PAR-4 agonist peptides used in this study mimic this specific mode of receptor activation. Low concentrations of thrombin initiate platelet activation via PAR-1, which is quickly terminated. PAR-4 requires higher thrombin concentrations, resulting in a later activation response, but one that is sustained.15 Phosphorylation of the thrombin PAR receptors by GPCR kinases, including GRK5, induces the GRK–β-arrestin cascade, resulting in the termination of signaling by the internalization and destruction of the active receptor in the lysosomes. Studies indicate that the signaling kinetics of PAR-1 and PAR-4 differ, at least in part, as a result of the rate or extent of signaling-dependent phosphorylation.28 Our results suggest that GRK5 levels and, thus, receptor phosphorylation, in platelets may have a lesser effect on PAR-4 than on PAR-1 receptor signaling, supporting the conclusions of these previous studies of receptor kinetics in human platelets.
In contrast to human platelets, mouse platelets express Par-3 and Par-4, but not Par-1. Thrombin signaling in mouse platelets is mediated by Par-3–facilitated cleavage of Par-4, which responds quickly to thrombin stimulation. Thus, in the context of thrombin receptor signaling, mouse Par-4 has similar kinetic properties to human PAR-1.29 Our study shows that mice that are unable to express Grk5 exhibit greater platelet activation sensitivity, principally downstream of Par-4–dependent signaling pathways. This observation is consistent with the association of the human G allele of rs10886430 with reduced levels of GRK5 transcripts and increased platelet reactivity to a PAR-1–specific agonist peptide. Recent data generated by us show that, like GRK5, GRK6 is involved in PAR-1–mediated signaling in human platelets.15 In keeping with this observation, deletion of Grk6 in mice enhances Par-4–dependent signaling. Thus, mouse Par-4 signaling is regulated by Grk6 in a manner that is similar to human PAR-1.15 The change in platelet reactivity, as measured by flow cytometry and ATP release, caused by knockout of Grk5 is less than that caused by knockout of Grk6. This is likely due to the lower abundance of Grk5 vs Grk6 in mouse platelets (supplemental Figure 15).30,31
Inhibition of platelet activation is known to reduce the risk of secondary thrombotic ischemia in patients with vascular disease. Therefore, it seems reasonable to hypothesize that variations in the sensitivity of platelets to activation by thrombin generates variations in the population’s risks of thrombotic diseases. Indeed, the G allele of rs10886430 has been associated with a greater risk for thrombotic disease in the 0.5 million participants in the UK Biobank, most notably with regard to the risk of pulmonary embolism (odds ratio, 1.28; P = 8.05 × 10−13).32 Associations have also been identified with the risk of venous thromboembolism in the INVENT Consortium (odds ratio, 1.12; P = 2.2 × 10−12) and in the Million Veteran Program (P = 6.7 × 10−11).33 In addition, Mendelian randomization analysis of individuals in the UK Biobank suggests a causal role for thrombin-induced platelet reactivity in thrombotic disease, most notably pulmonary embolism.10 The observation that Grk5−/− mice exhibit more rapid platelet accumulation during in vivo thrombus formation upon vascular damage compared with WT mice supports the hypothesis that these population associations are mediated through variations in platelet reactivity. This suggests that associations with platelet reactivity, as measured by our in vitro assays, can act as a good model for associations of platelet-mediated thrombotic pathologies of the venous circulation.
GRK5 is ubiquitously transcribed. It is expressed across the hematopoietic lineages and in endothelial cells.34 It is also expressed in most of the tissues surveyed by the GTEx studies.35 The desensitization of GPCRs is not a mechanism that is unique to the MK lineage, and interactors of GRK5, which include transmembrane, cytosolic, and nuclear GPCRs, as well as non-GPCR proteins, are involved in multiple signaling pathways, including the cell cycle, apoptosis, cell motility, and inflammation pathways. This may explain why variants in the GRK5 locus have been associated with many biomedically relevant quantitative traits and disease risks affecting a range of physiological systems. They include height, type 2 diabetes, lung function, and many neurological and behavioral traits, such as the risk of alcohol misuse and posttraumatic depression. This pleiotropy suggests that the therapeutic manipulation of GRK5 expression is likely to avoid off-target effects only if it can be achieved with cell-type specificity. For example, GRK5 activity has been shown to be increased in heart failure models and is under investigation as a potential target for pharmacological inhibition.36 However, our results and those of Rodriquez et al imply that inhibition of GRK5 in platelets would be prothrombotic and proinflammatory10 . On the other hand, further investigation of the platelet-specific binding partners of GRK5 as candidate targets for novel antithrombotic drugs may be warranted.
To conclude, by activating platelets through specific signaling pathways, we confirmed previously published associations and identified a new genetic association with platelet reactivity mediated via the thrombin GPCR PAR-1 in GRK5. The platelet eQTL, epigenetic analyses of MKs, and functional work in human platelets and mice evidence the molecular and biological mechanism mediated by the inhibitory effect of GRK5 on PAR-1. This improves our understanding of the signaling pathways associated with thrombin signaling and provides evidence for the distinct and wide-reaching consequences of dysregulation of GPCR-coupled receptor kinases.
Acknowledgments
The authors thank Xi Chen for technical assistance with aggregation assays, Stephen Garner for support and critical review of the manuscript, Tao Jiang for input on genotyping quality control methods, and Nemunas Antanavicius for input into a preliminary analysis of the PFC data. They also thank NIHR BioResource volunteers for their participation, and gratefully acknowledge NIHR BioResource centres, NHS Trusts, and staff for contributions. The authors thank the National Institute for Health Research, NHSBT, and Health Data Research UK as part of the Digital Innovation Hub Program.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant R01 HL144574 (P.M.) and British Heart Foundation grants RG/20/7/34866 and RG/15/2/31224 (J.M.G). K.D. is as an HSST trainee supported by NHS Health Education England. This research was conducted using the UK Biobank resource (application 36509)
The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care.
Authorship
Contribution: K.D., X.Z., H.M., C.K., J.B., P.L.T., M.C., J.V.M., K. Wedderburn, K. Waller, B.V., and N.K. performed experiments; K.D., N.S.G., R.K., H.V., J.L.D., W.J.A., and P.M. analyzed data; S.A. and K.S., provided research support; S.M., J.M.G., and J.Y. provided reagents; and K.D., W.H.O., J.M.G., W.J.A. and P.M., designed the study and wrote the manuscript. All authors reviewed the manuscript.
Conflict-of-interest disclosure: J.Y. is a full-time employee of Bristol Myers Squibb. The remaining authors declare no competing financial interests.
Correspondence: Kate Downes, Department of Haematology, University of Cambridge, Cambridge CB2 0PT, United Kingdom; e-mail: kate.downes@addenbrookes.nhs.uk; and Peisong Ma, Cardeza Foundation for Hematologic Research, Department of Medicine, Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA; e-mail: Peisong.Ma@jefferson.edu.