

Key Points
A cyanine-based fluorescent probe specific for PRMT1 has been developed to categorize cells according to PRMT1 expression levels.
Bone marrow transplantation assays confirm that LT-HSCs express low levels of PRMT1.

Abstract
Dynamic regulation of histone modification enzymes such as PRMT1 (protein arginine methyltransferase 1) determines the ordered epigenetic transitions in hematopoiesis. Sorting cells according to the expression levels of histone modification enzymes may further define subpopulations in hematopoietic lineages with unique differentiation potentials that are presently defined by surface markers. We discovered a vital near infrared dye, E84, that fluoresces brightly following binding to PRMT1 and excitation with a red laser. The staining intensity as measured by flow cytometry is correlated with the PRMT1 expression level. Importantly, E84 staining has no apparent negative effect on the proliferation of the labeled cells. Given that long-term hematopoietic stem cells (LT-HSCs) produce low levels of PRMT1, we used E84 to sort LT-HSCs from mouse bone marrow. We found that SLAM (the signalling lymphocyte activation molecule family) marker–positive LT-HSCs were enriched in the E84low cell fraction. We then performed bone marrow transplantations with E84high or E84low Lin−Sca1+Kit+ (LSK) cells and showed that whole blood cell lineages were successfully reconstituted 16 weeks after transplanting 200 E84low LSK cells. Thus, E84 is a useful new tool to probe the role of PRMT1 in hematopoiesis and leukemogenesis. Developing E84 and other small molecules to label histone modification enzymes provides a convenient approach without modifying gene loci to study the interaction between hematopoietic stem/progenitor cell epigenetic status and differentiation state.
Introduction
Using fluorochrome-conjugated antibodies to label cell surface antigens is a widely applied approach for identifying and sorting specific cell populations via flow cytometry. However, the expression of a specific gene in an immunophenotypically defined cell population is often heterogeneous. Traditionally, a coexpression partner such as green fluorescent protein or β-galactosidase is used to monitor gene expression patterns in different tissues.1 However, these methods may not recapitulate actual biological changes, given that target genes could be functionally altered by the insertion of reporter sequences. Intracellular protein labeling with antibodies can be achieved but requires permeabilization of the cell membrane, which makes these stained cells unusable for live cell assays. Chemical-labeling technology has several advantages over traditional techniques for monitoring in vivo gene functions. Using membrane permeable fluorescent dyes to label subcellular structures is also commonly used (eg, probes for cytoskeleton, mitochondria, or lysosome).2 CFSE [5(6)-carboxyfluorescein N-hydroxysuccinimidyl ester] dye has been used successfully for labeling live cells and tracking cell proliferation by covalently conjugating with cellular proteins via lysine residues.3 Enzyme-specific chemical probes have been developed for kinases and proteases.4 Little is reported regarding intracellular dyes that are both target-protein specific and cell viability maintaining. Here we report a novel small molecule that displays enhanced fluorescence when bound to protein arginine methyltransferase 1 (PRMT1).
PRMT1 is the most abundantly expressed arginine methyltransferase enzyme (∼100-fold higher than other asymmetric arginine methyltransferases at protein levels) and accounts for most cellular arginine methylation activity.5,6 Of note, PRMT1 expression fluctuates with low levels in hematopoietic stem cells, high levels in progenitor cells, and again low levels in terminally differentiated megakaryocytes.7 Therefore, chemical probes for PRMT1 may be used to define unknown subpopulations with unique epigenetic signatures in blood lineages. Here we report the development of E84 as a novel probe for determining such subpopulations. The staining intensity of E84 is correlated with the PRMT1 expression level. E84 staining can further divide Lin−Sca1+c-Kit+ (LSK) cells into 2 distinct groups. The E84low cells can be transplanted into bone marrow and establish long-term hematopoiesis, which implies that the E84low population contains more long-term hematopoietic stem cells (LT-HSCs). We also discovered that LT-HSCs as defined by Lin−c-Kit+Sca1+CD150+CD48− are more enriched in the E84 low population. Thus, we have discovered a novel method to isolate HSCs based on specific epigenetic status.
Materials and methods
Cell culture
293T cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (FBS; Life Technologies, Grand Island, NY). DNA transfection with calcium phosphate was performed as previously reported.7 The pcDNA3 plasmid was added to balance the total amount of DNA in the transfection. A murine leukemic cell line (6133 cells) and human leukemia cell line (MV4-11) were cultured in RPMI 1640 medium containing 10% FBS, 100 U/mL penicillin/streptomycin. Additionally, 10 ng/mL of murine stem cell factor (Peprotech, Rocky Hill, NJ) was supplemented for 6133 cells.8 The 6133 stable cell lines that conditionally overexpress PRMT1 were established by spinfection with a TetOn lentiviruses expressing PRMT1. V1 and V2 are the 2 isoforms of PRMT1. Ectopic PRMT1 expression was induced by adding 50 ng/mL of doxycycline, 24 hours before analysis.
Mouse progenitor colony formation assays were performed using MethoCult GF M3434 medium according to the manufacturer’s protocol (STEMCELL Technologies, Canada). One thousand E84high or E84low LSK cells were seeded into 1.1 mL of M3434 medium in a 3.5-cm dish. The colonies were counted after 12 days of culture.
Cell viability was measured using the CellTiter-Glo Viability Assay Kit (Promega, Madison, WI). Five hundred to 1000 cells were seeded in individual wells of a 96-well plate with 100 μL of culture medium per well. Luminescent signal, proportional to cell viability, was measured using a microplate reader (Biotek, Winooski, VT).
Apoptosis was measured by annexin V levels via fluorescence-activated cell sorter (FACS) analysis. cells (5 × 104 to 1 × 105) were washed with phosphate-buffered saline (PBS) and resuspended in 100 μL of binding buffer (10 mM HEPES [N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid] pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2), then 5 μL of anti-annexin V–fluorescein isothiocyanate antibody (BD Pharmingen, Franklin Lakes, NJ) was added to the cell suspension. After a 15-minute incubation, the cells were further washed for FACS analysis.
E84 staining
Near infrared dye E84 was designed and synthesized by M.H.9 The 5 mM E84 stock solution was stored in dimethyl sulfoxide at −20°C. Exponential phase cells were harvested and washed with ice-cold PBS (Sigma-Aldrich, St. Louis, MO). E84 at a final concentration of 10 nM was incubated with 5 × 105 cells per 100 μL of PBS on ice for 30 minutes. Following 2 more washes with PBS, cells were FACS analyzed using a BD LSRFortessa cytometer (BD, Franklin Lakes, NJ), and fluorescence was measured in the allophycocyanin (APC) channel (setting: laser, 640 nM; filter set, 650LP + 670/14). FACS data were processed using FlowJo software (Version 10.4). Cell sorting was performed with a BD FACSAria II cell sorter.
Virus production and stable cell line selection
To produce lentiviruses, viral vectors were cotransfected with envelope vector pMD2.G and packaging vector pSPAX2 into 293T cells. Viruses were harvested and concentrated with 8% PEG6000 (Sigma-Aldrich).10 Concentrated viruses were used to infect target cell lines at multiplicity of infection 100. Stable cell lines were selected with 1 μg/mL of puromycin. Human PRMT1 short hairpin RNA sequence (CCGGTGAGCGTTCC TAGGCGGTTTCCTC GAGGA AACCGCCTA GGAACGCTC ATTTTTG) in the plasmid TRCN0000310243 (Sigma-Aldrich) was cloned into the pLKO-SF-RFP (red fluorescent protein) vector described previously.11 Acute myeloid leukemia line MV4-11 was transduced with lentiviral vectors coexpressing shPRMT1 or shCtrl plus RFP, and 3 days after transduction, stable cell lines were established by sorting RFP positive cells.
Intracellular staining of PRMT1
Blood cells were washed twice with PBS and resuspended in 4% paraformaldehyde in PBS at 2 million cells per mL. The mixture was incubated for 20 minutes on ice and then washed twice with PBS. Then the cell pellet was gently resuspended in permeability buffer (eBioscience cat. no. 00-8333, containing 0.5% saponin; ThermoScientific, Waltham, MA) at 2 million cells per mL. PRMT1 antibody (PRMT1 [A33] antibody #2449; Cell Signaling Technology, Danvers, MA) was added at a 50-fold dilution. After incubation for 50 minutes, the cells were washed twice with 0.1% saponin. Secondary antibody (goat anti-mouse immunoglobulin G, Alx488, 1:100) in 0.5% saponin was added for 30 minutes at room temperature. Cells were washed with FACS buffer (1× PBS/5% FBS/0.02% NaN3) and then resuspended in FACS buffer for analysis.
Western blotting
A whole cell lysate was prepared by resuspending 1 million cells in 100 μL of sodium dodecyl sulfate (SDS) sample buffer and then sonicating with a Bioruptor (Diagenode, Denville, NJ) to sheer chromatin. The lysate was loaded onto a 10% SDS–polyacrylamide gel electrophoresis gel for protein resolution, and a PVDF membrane was used to transfer proteins from the SDS–polyacrylamide gel electrophoresis gel. Protein bands were visualized using chemiluminescence and quantitated using Adobe Photoshop.
Real-time PCR assay
Total RNA was prepared using a Direct-Zol RNAprep Kit (Zymo Research, Irvine, CA). Complementary DNA was generated with a Verso complementary DNA synthesis kit (ThermoScientific) and random hexamer primers. Real-time polymerase chain reaction (PCR) assays were performed using Absolute Blue qPCR SYBR green Mix (ThermoScientific) and a ViiA 7 Real-Time PCR System (Applied Biosystems, Waltham, MA). Relative quantity of gene expression was calculated by the ΔΔCt method. The housekeeping gene glyceraldehyde-3-phosphate dehydrogenase was used as an internal control for normalization.
Bone marrow transplantation assay
Bone marrow cells from CD45.1 C57BL/6 mice were collected and stained with the combination of E84 and antibodies against mouse lineages: (Lin mix of CD11b/Mac1, Gr-1, B220, CD3, Ter119), c-Kit, and Sca-1. Lineage mix antibodies are conjugated with biotin, which was further stained with streptavidin-APC-Cy7; c-Kit-Pacific Blue and Sca-1-PE-Cy7 were antibodies directly conjugated to fluorochromes. In the gate of LSK cells, cells in the highest 25% and lowest 25% of E84 intensity were separately sorted. The sorted cells were washed and resuspended in Iscove modified Dulbecco medium. Two hundred sorted cells combined with 200 000 CD45.2+ bone marrow cells were IV injected into lethally irradiated (9 Gy) CD45.2 C57BL/6 recipient mice. Peripheral blood was collected at 72 hours, 4 weeks, 8 weeks, 12 weeks, and 16 weeks postinjection to measure the frequency of CD45.1 cells. Mice were euthanized at 16 weeks postinjection to determine reconstitution of CD45.1+ blood lineages in recipient mouse bone marrow. Antibodies used for flow cytometry: biotin-Ter119 (BD Pharmingen, 51-09082); biotin-Gr-1 (BD Pharmingen, 51-01212); biotin-B220 (BD Pharmingen, 51-01122); biotin-CD11b (BD Pharmingen, 51-01712); biotin-CD3 (BD Pharmingen. 51-01082); SA-APC-Cy7 (BD Pharmingen, 554063); PECy7- ScaI (eBioscience, 25-5981-81) and eFluor450-cKit (eBioscience, 48-1171-82); and fluorescein isothiocyanate–CD45.2 (BD Pharmingen, 553775) and APC-CD45.1 (eBioscience, 17-0454-81).
Results
E84 staining intensity is correlated with intracellular PRMT1 protein level
E84 has an emission wavelength between 650 and 750 nm with an excitation peak at ∼620 nm.9 The fluorescence intensity of E84 is enhanced up to sixfold when it binds to PRMT1.9 These in vitro data suggest that E84 may be used to label intracellular PRMT1 protein. We incubated 293T cells with E84 for 30 minutes on ice followed by extensive washing and then measured fluorescence in the APC channel (Figure 1A). The 293T cells transfected with plasmids expressing either isoform of PRMT1 (V1 or V2) have approximately sevenfold higher fluorescence signals than the 293T cells transfected with vector plasmid. The change in fluorescence intensity is correlated with western blotting results as well as the in vitro PRMT1-E84 binding result.9 To further demonstrate that E84 fluorescence intensity is correlated with PRMT1 expression levels, we knocked down PRMT1 in the leukemia cell line MV4-11. Flow cytometry analysis revealed that E84 fluorescence intensity in PRMT1 KD cells has a peak (MFI: 574) separated from the peak (MFI: 1162) in control cells (Figure 1B). In parallel, we confirmed PRMT1 knockdown in sorted cells by western blot analysis (Figure 1B, right panel). We also performed intracellular staining using an anti-PRMT1 antibody. The MFI was shifted from 704 to 346 after PRMT1 was knocked down (Figure 1B). Thus, E84 staining can be used to indicate intracellular PRMT1 levels on par with PRMT1 antibody intracellular staining.

The intensity of E84 staining is correlated with the PRMT1 protein level. (A) E84 staining of 293T cells. One million control 293T cells or 293T cells that overexpress PRMT1 (isoforms V1 or V2) were incubated in 10 nM E84 in PBS for 30 minutes and then subjected to FACS analysis. Mean fluorescence intensity (MFI) values are indicated in a plot representative of 3 independent experiments. Western blots for detecting PRMT1 in 293T cells are to the right. PRMT1 was normalized to tubulin, and ratios are presented below the PRMT1 blot. (B) MV4-11 cells that express control short hairpin RNA or shPRMT1 were stained with E84 (left panel) and anti-PRMT1 antibody (middle panel); western blots of PRMT1 (right panel) show the efficiency of PRMT1 knockdown. (C) E84 staining of 6133 cells correlates with endogenous PRMT1 expression. Left panel: 6133 cells were stained with E84 and analyzed by FACS. Middle panel: PRMT1 protein expression levels in E84-high and E84-low cells. The 6133 cells were sorted into 2 populations based on E84 staining intensity for western blotting with an anti-PRMT1 antibody. Tubulin was used as a loading control. Triangles indicate increasing amounts of lysate loaded. Right panel: mRNA levels of PRMT1, PRMT3, and PRMT6 in E84-high and E84-low cells were measured using real-time PCR assays. The data are shown as mean ± standard deviation. *P < .05. (D) E84 staining of 6133 cells expressing PRMT1 from a doxycycline-inducible promoter. Left panel: contour plots gated according to E84 staining. Right panels: E84 histogram plots of 6133 cell lines. MFI values are in the plots. Representative results from at least 3 independent experiments are presented. Western blots measure the PRMT1 protein levels in 6133 cells with and without doxycycline induction. The relative PRMT1 protein levels were quantitated and normalized to tubulin protein. ns, not significant.
The intensity of E84 staining is correlated with the PRMT1 protein level. (A) E84 staining of 293T cells. One million control 293T cells or 293T cells that overexpress PRMT1 (isoforms V1 or V2) were incubated in 10 nM E84 in PBS for 30 minutes and then subjected to FACS analysis. Mean fluorescence intensity (MFI) values are indicated in a plot representative of 3 independent experiments. Western blots for detecting PRMT1 in 293T cells are to the right. PRMT1 was normalized to tubulin, and ratios are presented below the PRMT1 blot. (B) MV4-11 cells that express control short hairpin RNA or shPRMT1 were stained with E84 (left panel) and anti-PRMT1 antibody (middle panel); western blots of PRMT1 (right panel) show the efficiency of PRMT1 knockdown. (C) E84 staining of 6133 cells correlates with endogenous PRMT1 expression. Left panel: 6133 cells were stained with E84 and analyzed by FACS. Middle panel: PRMT1 protein expression levels in E84-high and E84-low cells. The 6133 cells were sorted into 2 populations based on E84 staining intensity for western blotting with an anti-PRMT1 antibody. Tubulin was used as a loading control. Triangles indicate increasing amounts of lysate loaded. Right panel: mRNA levels of PRMT1, PRMT3, and PRMT6 in E84-high and E84-low cells were measured using real-time PCR assays. The data are shown as mean ± standard deviation. *P < .05. (D) E84 staining of 6133 cells expressing PRMT1 from a doxycycline-inducible promoter. Left panel: contour plots gated according to E84 staining. Right panels: E84 histogram plots of 6133 cell lines. MFI values are in the plots. Representative results from at least 3 independent experiments are presented. Western blots measure the PRMT1 protein levels in 6133 cells with and without doxycycline induction. The relative PRMT1 protein levels were quantitated and normalized to tubulin protein. ns, not significant.
Next, we tested the E84 staining on a murine megakaryocytic leukemia cell line (6133).8 Compared with unstained control in the FACS plot of Figure 1C, E84-stained 6133 cells have 2 unique populations (Figure 1C, left panel). We thus sorted 6133 cells into the 2 populations with distinct PRMT1 expression levels for RNA isolation and protein extraction. Western blots confirmed that the E84high cells expressed higher levels of PRMT1 protein than the E84low cells (Figure 1C, middle panel). In addition, E84high cells expressed more PRMT1 messenger (mRNA), but no difference in PRMT3 and PRMT6 mRNA levels was detected by real-time PCR analysis (Figure 1C, right panel). Given that E84 can bind to PRMT6 and PRMT3 that are homologous to PRMT1, this result confirms that the intracellular E84 staining does not reflect the intracellular protein levels of PRMT3 or PRMT6, which are extremely low as compared with PRMT1 expression level.
To further demonstrate the correlation of E84 staining with PRMT1 expression levels, we made 2 stable 6133 cell lines that express PRMT1 variant 1 or 2 upon doxycycline induction. Without doxycycline induction, both cell lines had 2 separated populations with distinct intensities of E84 staining (Figure 1D, top left panel). Upon doxycycline induction, the 2 populations merge and shift to higher fluorescence intensities (Figure 1D, bottom left panel). Histograms of 6133 cell lines showed that overexpression enhances the MFI close to 10-fold. Because of the leak of the inducible system, the E84-low populations are smaller in Figure 1D before induction as compared with the parental 6133 cells in Figure 1C. Western blotting (Figure 1D, bottom left panel) confirmed the enhanced expression of PRMT1 upon induction. The data suggest that E84 staining is more sensitive than antibody staining to detect changes in PRMT1 protein levels.
E84 is a vital fluorescent dye
Intensively washed, E84-stained 6133 cells were seeded back to growth medium and monitored for cell proliferation. E84-stained cells proliferated at a rate comparable with the parental cells treated with dimethyl sulfoxide for 4 days (Figure 2A). The intensity of intracellular E84 staining dramatically dropped in the first day and continued to drop on day 2 (Figure 2B). The E84 concentration used for staining is 10-fold lower than the concentration used for inhibiting cell proliferation, and cells are washed after 30 minutes of staining. Both steps in the staining protocol minimize E84 effect on cell proliferation. We have tested the cell proliferation status of a couple of cell lines following E84 staining (supplemental Figure 1), and to date no proliferation changes have been observed.
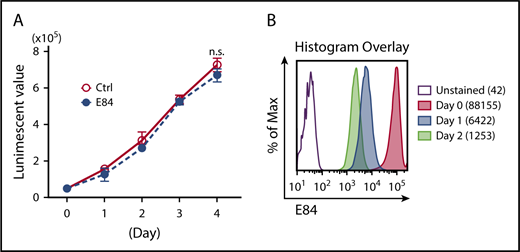
E84 staining does not block the proliferation of labeled cells. (A) Growth curves of E84-labeled 293T cells. Cells (5 × 105) were incubated in 10 nM E84 solution (or PBS control) for 30 minutes. After labeling, cells were washed with medium and cultured for 4 days. Cell viability was measured every day using the CellTiter-Glo Luminescent Cell Viability Assay Protocol (Promega). (B) MFI of E84-stained cells in culture was measured by FACS daily. After day 3, E84 staining was not detected. Representative results from 3 independent experiments are presented.
E84 staining does not block the proliferation of labeled cells. (A) Growth curves of E84-labeled 293T cells. Cells (5 × 105) were incubated in 10 nM E84 solution (or PBS control) for 30 minutes. After labeling, cells were washed with medium and cultured for 4 days. Cell viability was measured every day using the CellTiter-Glo Luminescent Cell Viability Assay Protocol (Promega). (B) MFI of E84-stained cells in culture was measured by FACS daily. After day 3, E84 staining was not detected. Representative results from 3 independent experiments are presented.
E84 staining can be used to sort LT-HSCs
We have reported that LT-HSCs, relative to both short-term HSCs (ST-HSCs) and progenitor cells, have low PRMT1 expression.7 Thus, we asked whether E84 staining can be used to enrich for LT-HSCs. Mouse LSK cells from bone marrow were further separated into SLAM marker–positive and –negative fractions (Figure 3A, left panel). We discovered that LT-HSCs have relatively lower E84 staining, whereas both ST-HSCs and multipotent progenitors (MPPs) have high E84 staining (Figure 3A, right panel). We then determined the frequencies of LT-HSCs, ST-HSCs, and MPPs by analyzing the 25% highest vs the 25% lowest E84-stained LSK cells (Figure 3B). E84high LSK cells contained more ST-HSCs and MPPs compared with E84low LSK cells that had a high percentage of LT-HSCs (Figure 3C). To determine whether E84 may inhibit cell proliferation, we sorted the E84-high and E84-low populations out of LSK cells for growth in liquid culture. The 2 populations displayed little difference in proliferation rate (Figure 3D) and no difference in terms of apoptosis as detected by annexin V staining (Figure 3E). Therefore, we further substantiate that E84 is not toxic when used in accordance with the current staining protocol. Of note, E84high LSK cells yielded fewer colonies than E84low cells in the colony formation assays (Figure 3F), which implies that difference in the epigenetic status as defined by PRMT1 expression level determines the colony formation capability of progenitor cells.
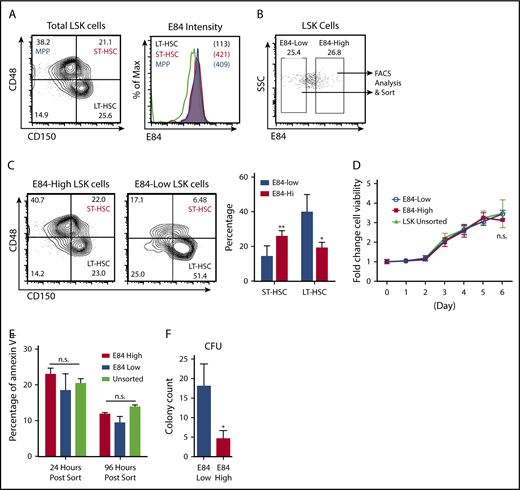
E84-low LSK cells from bone marrow are enriched for HSCs. (A) Flow cytometry analysis of hematopoietic stem/progenitor cells (HSPCs) based on E84 staining. Bone marrow cells were stained using E84, LSK, and SLAM markers to detect LT-HSCs (LSK+CD150+CD48−), ST-HSCs (LSK+CD150+CD48+), and MPPs (LSK+CD150−CD48+). Left panel: gating of LSK cells into LT-HSCs, ST-HSCs, and MPPs; right panel: histogram of E84-stained LT-HSCs, ST-HSCs, and MPPs. (B) Gating of the 25% least and 25% most intensely E84-stained LSK cells for subsequent sorting. (C) Sorted cells were analyzed for SLAM markers (CD48 and CD150) and gated accordingly. The plot is the summary of 3 independent experiments. (D) Sorted cells were cultured in Iscove modified Dulbecco medium with 10% FBS supplemented with interleukin-3/interleukin-6/stem cell factor for cell viability assays. (E) Annexin V staining of sorted cells in culture D. (F) Sorted cells were used for colony formation assays; colony-forming units (CFUs) from 3 independent assays were counted and plotted. The data are shown as mean ± standard deviation. *P < .05; **P < .01.
E84-low LSK cells from bone marrow are enriched for HSCs. (A) Flow cytometry analysis of hematopoietic stem/progenitor cells (HSPCs) based on E84 staining. Bone marrow cells were stained using E84, LSK, and SLAM markers to detect LT-HSCs (LSK+CD150+CD48−), ST-HSCs (LSK+CD150+CD48+), and MPPs (LSK+CD150−CD48+). Left panel: gating of LSK cells into LT-HSCs, ST-HSCs, and MPPs; right panel: histogram of E84-stained LT-HSCs, ST-HSCs, and MPPs. (B) Gating of the 25% least and 25% most intensely E84-stained LSK cells for subsequent sorting. (C) Sorted cells were analyzed for SLAM markers (CD48 and CD150) and gated accordingly. The plot is the summary of 3 independent experiments. (D) Sorted cells were cultured in Iscove modified Dulbecco medium with 10% FBS supplemented with interleukin-3/interleukin-6/stem cell factor for cell viability assays. (E) Annexin V staining of sorted cells in culture D. (F) Sorted cells were used for colony formation assays; colony-forming units (CFUs) from 3 independent assays were counted and plotted. The data are shown as mean ± standard deviation. *P < .05; **P < .01.
The E84low population contains LT-HSCs in bone marrow transplantation assays
LSK cells contain 10% cells that can be transplantable.12,13 We performed bone marrow transplantation of 200 E84low or E84low CD45.1 LSK cells along with 200 000 CD45.2 bone marrow helper cells into lethally irradiated mice to analyze long-term multilineage reconstitution (Figure 4A). We found that over the course of 16 weeks, E84low LSK cells were able to repopulate peripheral blood with multiple lineages, whereas the E84hi LSK cells failed to reconstitute multilineages (Figure 4B). At 16 weeks posttransplantation, cells from recipient bone marrow were isolated for blood lineage analysis. E84low cells reconstituted the whole blood system (Figure 4C). Combined with colony formation assay results in Figure 3, these data demonstrated that E84 is a viable dye to further separate mouse bone marrow HSPCs to functionally different populations, also suggesting that real long-term stem cells have relatively low expression of Prmt1.
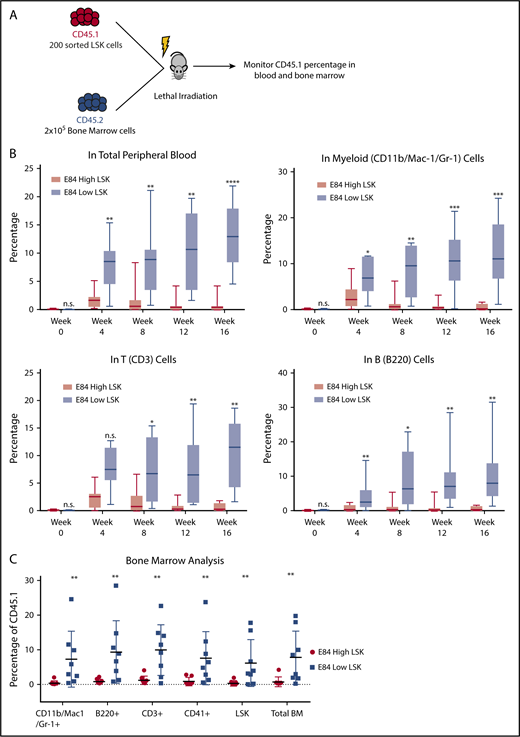
Bone marrow transplantation of E84-high and E84-low mouse LSK cells. (A) Schematic of bone marrow transplantation. Two hundred sorted E84-high or E84-low CD45.1 LSK cells were transplanted along with 2 × 105 CD45.2 whole bone marrow cells into lethally irradiated mice (E84-low, n = 7; E84-high, n = 8). (B) Percentage of CD45.1 cells of different lineages in peripheral blood over the course of 4 months. (C) Percentage of CD45.1 cells in different lineages in recipient mouse bone marrow at 16 weeks posttransplantation. Lineage-specific surface markers: Mac-1/Gr-1 for myeloid lineage; B220 for B cell lineage, and CD3 for T cell lineage. The data are shown as mean ± standard deviation. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Bone marrow transplantation of E84-high and E84-low mouse LSK cells. (A) Schematic of bone marrow transplantation. Two hundred sorted E84-high or E84-low CD45.1 LSK cells were transplanted along with 2 × 105 CD45.2 whole bone marrow cells into lethally irradiated mice (E84-low, n = 7; E84-high, n = 8). (B) Percentage of CD45.1 cells of different lineages in peripheral blood over the course of 4 months. (C) Percentage of CD45.1 cells in different lineages in recipient mouse bone marrow at 16 weeks posttransplantation. Lineage-specific surface markers: Mac-1/Gr-1 for myeloid lineage; B220 for B cell lineage, and CD3 for T cell lineage. The data are shown as mean ± standard deviation. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Discussion
E84, a cyanine-based compound, fluoresces brightly when bound to PRMT1 and excited with a red laser. We take advantage of this feature to label live cells for flow cytometry analysis. We showed that the fluorescence intensity of the stained cells is correlated with the intracellular PRMT1 level. We used FACS to sort cell populations with different intensities of E84 staining and further demonstrated that staining intensity depends on PRMT1 expression level. However, the fluorescence intensity of E84 binding to PRMT1 is not strictly linear. In addition, E84 binds to PRMTs other than PRMT1 in vitro,9 and therefore, it is important to validate the correlation of staining intensity with PRMT1 expression levels by other methods such as western blotting or FACS analysis with intracellular antibody staining when a new cell or tissue is to be studied. Although E84 was reported originally as a PRMT1 inhibitor capable of killing certain types of leukemia cells, we find that proliferation rates of E84-labeled cells do not change when stained with low concentration of E84. A short incubation period combined with a decreased E84 dose (at least 10-fold lower than the concentration for inhibiting cell proliferation) and thorough washing of extracellular E84 after staining may contribute to the observed low to no toxicity. In addition, the E84 stain quickly dissipates >10-fold within the first day, which may also contribute to the nontoxicity of E84 staining (Figure 2).
Since E84 is a novel live cell dye that specifically targets PRMT1, it has many potential applications in both basic and applied medical research. E84 staining may replace the use of anti-PRMT1 antibodies in many antibody-based protein detection techniques to visualize PRMT1 expression levels if absolute quantification is not a priority. In live cells, E84 or in the E84 derivatives in the future may be able to in situ label PRMT1 and provide information about the dynamic change of expression and subcellular localization of PRMT1. Given that high PRMT1 expression is often associated with short survival time for patients, copiously expressed PRMT1 could be a possible marker for cancer progression. One potential use of E84 is to stain patient samples as a medical diagnostic tool for identifying cells with aberrant PRMT1 expression and evaluating patient prognosis. Cell populations can be isolated by FACS based on E84 staining levels and be subjected to subsequent live cell analysis such as colony formation assays, proliferation assays, or transplantation in order to investigate whether high PRMT1 expression exists in cancer stem cells. Therefore, this dye will be a useful tool to study the role of PRMT1-related pathways.
The role of PRMT1 in tumorigenesis has been actively pursued for more than a decade now.14 PRMT1 has been shown to be an oncogene in breast cancer, colon cancer, and leukemia. Given that cancerous tissue consists of a group of heterogeneous cells and that cancer stem cells make up a small fraction of cancer cells, to further dissect cancer tissue to identify cancer stem cells will help us to understand tumorigenesis and to design therapies to target cancer stem cells. Currently cancer stem cells are isolated according to surface markers.15 Tools to directly isolate cancer stem cells according to biological functions are still rare. Metabolic labeling of aldehyde dehydrogenase can be used to separate HSPCs.16 Epigenetic reprogramming by histone modification enzymes has been shown to be crucial in the transition from normal cells to malignant cells. However, molecular probes for histone modification enzymes have not been reported. E84 is a first of its kind molecular probe for PRMT1, an enzyme critical for tumorigenesis in many types of cancers. Immunophenotypically defined cell populations may contain many different epigenetic states. The E84 staining method introduced here can divide homogeneous surface marker–defined cell populations into subgroups and provides a novel approach to study the proliferation and differentiation pathways of defined epigenetic variants. Further development of novel molecular probes for epigenetic enzymes will provide new avenues for hematological research.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Ravi Bhatia and Robert Welner for helpful discussions and The University of Alabama at Birmingham (UAB) Flow Cytometry Core for cell sorting assistance. The mouse protocol for bone marrow transplantation was approved in the protocol (IUCAC #10182).
This work was supported by grants from the Leukemia Research Foundation and the National Cancer Institute, National Institutes of Health (grant 1R21CA202390) and the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (grant 1R01DK110574) (X.Z.); by the National Institute of General Medical Sciences, National Institutes of Health (grant R01GM126154) (Y.G.Z.); and by the National Heart, Lung, and Blood Institute, National Institutes of Health (grants HL092215 and HL136165), the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (grant DK100847), and the Veterans Administration (BX000369 and BX003617) (Y.C.). National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health (grant P30 AR048311) and National Institute of Allergy and Infectious Diseases, National Institutes of Health (grant P30 AI027667) support the core facility (UAB Flow Cytometry Core).
Authorship
Contribution: H.S. designed and performed the experiments, analyzed data, and wrote the manuscript; C.-W.S., X. He, S.-M.L., H.H., and K.M.P. performed experiments; T.M.T., X. Han, C.A.K., L.L., M.H., Y.C., and Y.G.Z. provided reagents, analyzed data, and designed experiments; and X.Z. conceived the research project, designed experiments, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xinyang Zhao, Department of Biochemistry and Molecular Genetics, The University of Alabama at Birmingham, Shelby Building Room 703, 1825 University Blvd, Birmingham, AL 35294-0111; e-mail: zhaox88@uab.edu.

