

Key Points
Human blood CD16+ and CD16− MDDCs exhibit unique ITGAE/CD103+RALDH2+TCF4+ and CDH1/E-cadherin+ phenotypes, respectively.
CD16+ monocytes preserve a unique transcriptional signature during differentiation into DCs, including the LPS-induced TNF production.

Abstract
Classical CD16− vs intermediate/nonclassical CD16+ monocytes differ in their homing potential and biological functions, but whether they differentiate into dendritic cells (DCs) with distinct contributions to immunity against bacterial/viral pathogens remains poorly investigated. Here, we employed a systems biology approach to identify clinically relevant differences between CD16+ and CD16− monocyte-derived DCs (MDDCs). Although both CD16+ and CD16− MDDCs acquire classical immature/mature DC markers in vitro, genome-wide transcriptional profiling revealed unique molecular signatures for CD16+ MDDCs, including adhesion molecules (ITGAE/CD103), transcription factors (TCF7L2/TCF4), and enzymes (ALDH1A2/RALDH2), whereas CD16− MDDCs exhibit a CDH1/E-cadherin+ phenotype. Of note, lipopolysaccharides (LPS) upregulated distinct transcripts in CD16+ (eg, CCL8, SIGLEC1, MIR4439, SCIN, interleukin [IL]-7R, PLTP, tumor necrosis factor [TNF]) and CD16− MDDCs (eg, MMP10, MMP1, TGM2, IL-1A, TNFRSF11A, lysosomal-associated membrane protein 1, MMP8). Also, unique sets of HIV-modulated genes were identified in the 2 subsets. Further gene set enrichment analysis identified canonical pathways that pointed to “inflammation” as the major feature of CD16+ MDDCs at immature stage and on LPS/HIV exposure. Finally, functional validations and meta-analysis comparing the transcriptome of monocyte and MDDC subsets revealed that CD16+ vs CD16− monocytes preserved their superior ability to produce TNF-α and CCL22, as well as other sets of transcripts (eg, TCF4), during differentiation into DC. These results provide evidence that monocyte subsets are transcriptionally imprinted/programmed with specific differentiation fates, with intermediate/nonclassical CD16+ monocytes being precursors for pro-inflammatory CD103+RALDH2+TCF4+ DCs that may play key roles in mucosal immunity homeostasis/pathogenesis. Thus, alterations in the CD16+/CD16− monocyte ratios during pathological conditions may dramatically influence the quality of MDDC-mediated immunity.
Introduction
Peripheral blood monocytes are myeloid cells originating from the bone marrow that infiltrate secondary lymphoid organs and peripheral tissues after a few hours/days of circulation through the blood.1,2 Monocytes contribute to tissue homeostasis and innate/adaptive immunity against pathogens, especially upon differentiation into dendritic cells (DCs) or macrophages (MΦ).3-13 In the intestine, the majority of DCs and MΦ are derived from blood monocytes.4,14 In other tissues, monocyte-derived myeloid cells coexist with self-renewing tissue-resident MΦ of embryonic/fetal origin.15,16 The proportion between these 2 pools varies from homeostasis to tissue injury and during aging.17 Insights into the existence of self-renewing tissue-resident myeloid cells were mainly provided by studies in mice models,18-20 with evidence in humans being still scarce.21
Human monocytes are classified into 3 major subsets according to their differential expression of CD14 and CD16: classical CD14++CD16−, intermediate CD14++CD16+, and nonclassical CD14+CD16++ monocytes.22 Further, other surface markers such as Slan/M-DC8 proved useful to distinguish classical vs intermediate/nonclassical monocytes.23-25 Multiple transcriptional studies support the existence of a developmental relationship between these blood monocyte subsets.26-29 Most recently, deuterium labeling in humans and complementary studies in humanized mice models demonstrated the sequential differentiation of bone marrow-derived classical monocytes into intermediate and nonclassical monocytes.2 The identification of CCR2 and CX3CR1 as chemokine receptors involved in the tissue-specific homing of classical and intermediate/nonclassical monocytes, respectively, in humans and mice5,30,31 open the path for a detailed understanding of the distinct roles played by these monocytes in tissue homeostasis and disease pathogenesis.4,13,32-35 Briefly, intermediate/nonclassical monocytes distinguish from classical monocytes by their increased frequency during various pathological conditions, as well as their ability to produce pro-inflammatory cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-1,22,33,36-41 to maintain blood vessel homeostasis (“patrolling” CX3CR1+ monocytes),35 and to sense viruses via TLR7/TLR8.3,35,42 Given that monocytes are DC precursors,3-13 these findings raise new questions relative to potential functional differences between classical and intermediate/nonclassical monocyte-derived DCs (MDDCs). Although both CD16− and CD16+ monocytes differentiate into DCs in the presence of granulocyte-MΦ colony-stimulating factor (GM-CSF) and IL-4 in vitro,43,44 CD16+ monocytes were reported to preferentially acquire DC features in a model of trans endothelial migration.45 Also, CD16+ is distinguished from CD16− MDDCs by their cytokine production, expression of costimulatory molecules, trafficking potential, and antigen presentation capacity.43-48 At steady state, it was reported that monocytes can traffic into tissues where they capture antigens while retaining their “monocyte” phenotype.34,49 Whether such tissue-infiltrating monocytes return in the blood circulation remains a yet unproved possibility.50 The latter scenario may explain the transcriptional and functional heterogeneity of CD16+/CD16− monocyte subsets in the blood, indicative of different stages of monocyte differentiation.26-29
In this study, we used a systems biology approach to explore features of CD16+ and CD16− MDDCs. Our results support a model in which CD16+ and CD16− monocytes are imprinted/programmed to give rise to transcriptionally distinct MDDC subsets that may play different roles in tolerance vs immunity. Of particular importance, our study reveals that CD16+ vs CD16− MDDCs express unique adhesion molecules (eg, CD103), enzymes (eg, RALDH2), and transcription factors (eg, transcription factor 4 [TCF4]), typically expressed by mucosal DCs, supporting a scenario in which CD16+ monocytes are precursors for mucosal DCs. These insights orient the rational use of CD16− vs CD16+ monocytes in DC-based immunotherapies.
Material and methods
Study subjects
Healthy HIV-uninfected donors were recruited at the Montreal Chest Institute, McGill University Health Centre and Centre Hospitalier de l’Université de Montréal (CHUM, Montreal, QC, Canada). Large quantities of peripheral blood mononuclear cells (PBMCs) (109-1010 cells) were collected by leukapheresis, as previously described.51 Cytomegalovirus infection was determined on detection of cytomegalovirus-specific antibodies (Abs), using chemiluminescent microparticle immunoassay.52
Ethics statement
This study, using PBMC samples from healthy HIV-uninfected subjects, was conducted in compliance with the principles included in the Declaration of Helsinki. This study received approval from the Institutional Review Board of the McGill University Health Centre and the CHUM-Research Centre, Montreal, Quebec, Canada. Written informed consents were obtained from all study participants.
Flow cytometry phenotypic analysis
Surface staining was performed as previously described,53 with the following fluorochrome-conjugated Abs: CD3 FITC (BW264/56), CD8 FITC (BW135/80), CD19 FITC (LT19), CD1c phycoerythrin (PE) (AD5-8E7; Miltenyi Biotec, Auburn, CA); CD56 FITC (HCD56; Biolegend, San Diego, CA); CCR7 PE (150503; R&D Systems, Minneapolis, MN); CD1a FITC (HI149), DC-SIGN PE (DCN46), HLA-DR APC (G46-6; BD Pharmingen, San Diego, CA); and CD16 PE-Cy5 (3G8), CD14 FITC (RMO52), and CD83 PE-Cy5 (HB15a; Beckman Coulter, Brea, CA). Cells were analyzed using a LSRII cytometer, Diva (BD Biosciences, San Jose, CA). Positivity gates were placed using fluorescence minus 1.53,54
Magnetic and fluorescence-activated cell sorting
Total monocytes were isolated from PBMCs of HIV-uninfected subjects by negative selection, using magnetic beads (MACS; Miltenyi).26 Monocytes were further stained with a cocktail of CD16, CD1c, CD3, CD8, CD19, and CD56 Abs for fluorescence-activated cell sorting (FACS). Of note, CD14 Abs were not included in the sorting cocktail in an effort to avoid any potential stimulation via CD14.55 The CD16+ (including mainly nonclassical CD14+CD16++ monocytes) and CD16− monocytes, lacking expression of the lineage markers CD3, CD8, CD19, CD56, and CD1c, were sorted by FACS (BDAria II; BD Biosciences; low pressure [20 PSI], 100 μM nozzle). Postsort quality control analysis indicated purities greater than 96% (supplemental Figure 1).
Generation of immature and mature MDDCs
Monocyte subsets were differentiated into DCs by culture in RPMI 1640 media (2% fetal bovine serum; 1% penicillin/streptomycin) containing GM-CSF and IL-4 (20 ng/mL; R&D Systems) for 6 days; cytokine-containing media were refreshed every 2 days (Figure 1A). For phenotypic analysis, MDDC maturation was induced by stimulation with Escherichia coli lipopolysaccharides (LPS; SIGMA; 100 ng/mL) for 48 hours. Immature and mature MDDCs were stained with Abs against CD14 (monocyte marker), CD1a, CD1c, and DC-SIGN (immature MDDC markers), and CD83 and CCR7 (mature MDDC markers).
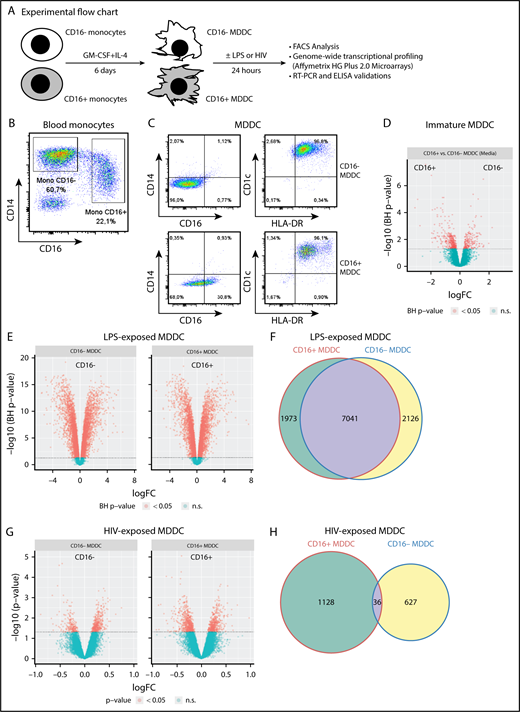
CD16+and CD16−monocytes differentiate into DCs with distinct transcriptional profiles. (A) Shown is the experimental flowchart. Briefly, total monocytes were purified by negative selection using magnetic beads. Highly pure CD16+ and CD16− monocytes were subsequently sorted by FACS on staining with CD16 Abs and a cocktail of FITC-conjugated nonmonocyte lineage-specific Abs (CD1c, CD3, CD8, CD19, and CD56; supplemental Figure 1). Immature MDDCs were generated by culturing monocyte subsets in the presence of GM-CSF and IL-4 for 6 days. Total RNA was extracted from matched CD16+ and CD16− MDDCs (n = 5) that were exposed to media, LPS, or HIV for 24 hours. Genome-wide transcriptional profiling was performed using the Affymetrix HG Plus 2.0 microarrays. (B-C) Shown are the expressions of CD14 and CD16 on total monocytes before sort (B) and the expression of CD14, CD16, CD1c, and HLA-DR on immature CD16+ and CD16− MDDCs on differentiation in vitro (C). Results in B-C are from 1 donor representative of results generated with cells from more than 10 donors. (D-H) Shown are Venn diagrams of differentially expressed genes in CD16+ vs CD16− MDDCs in response to media (D), LPS (E), and HIV (G), as well as the representation of the number of commonly and differentially expressed genes on exposure to LPS (F) and HIV (H). n.s., not significant.
CD16+and CD16−monocytes differentiate into DCs with distinct transcriptional profiles. (A) Shown is the experimental flowchart. Briefly, total monocytes were purified by negative selection using magnetic beads. Highly pure CD16+ and CD16− monocytes were subsequently sorted by FACS on staining with CD16 Abs and a cocktail of FITC-conjugated nonmonocyte lineage-specific Abs (CD1c, CD3, CD8, CD19, and CD56; supplemental Figure 1). Immature MDDCs were generated by culturing monocyte subsets in the presence of GM-CSF and IL-4 for 6 days. Total RNA was extracted from matched CD16+ and CD16− MDDCs (n = 5) that were exposed to media, LPS, or HIV for 24 hours. Genome-wide transcriptional profiling was performed using the Affymetrix HG Plus 2.0 microarrays. (B-C) Shown are the expressions of CD14 and CD16 on total monocytes before sort (B) and the expression of CD14, CD16, CD1c, and HLA-DR on immature CD16+ and CD16− MDDCs on differentiation in vitro (C). Results in B-C are from 1 donor representative of results generated with cells from more than 10 donors. (D-H) Shown are Venn diagrams of differentially expressed genes in CD16+ vs CD16− MDDCs in response to media (D), LPS (E), and HIV (G), as well as the representation of the number of commonly and differentially expressed genes on exposure to LPS (F) and HIV (H). n.s., not significant.
Confocal microscopy
The visualization of MDDC phenotype and morphology was performed by confocal microscopy, as previously described.56,57 Briefly, MDDCs were stained with mouse anti-human PE-conjugated CD1a Abs (BD Pharmingen) and Pholloidin-AF488 (Invitrogen). Epi-fluorescent and Spinning Disc confocal microscopy images were acquired out on an automated Cell Observer Z1 microscope (Carl Zeiss), using the AxioVision 4.8.2 software (Carl Zeiss).
Genome-wide transcriptional profiling
Matched CD16+ and CD16− MDDCs were generated from 5 different healthy HIV-uninfected donors and exposed to media, E coli LPS (100 ng/mL), or infectious NL4.3BaL HIV-1 virions (50 ng HIV-p24/106 MDDC) for 24 hours. The HIV NL4.3Bal molecular clone plasmid was a gift from Dana Gabuzda (Dana-Farber Cancer Institute, Boston, MA). HIV viral stocks were prepared by transfection of 293T cells with the appropriate plasmid; the concentration of HIV stocks was determined by ELISA quantification of HIV-p24 protein; and HIV infectivity was titrated on primary CD4+ T cells, as previously described.57-60 Total RNA was isolated using RNeasy columns kit (Qiagen). RNA quantity was determined by Pearl NanoPhotometer (Implen, Germany; 1-5 µg RNA/106 cells). Genome-wide analysis of gene expression was performed by Génome Québec Innovation Centre (Montreal, QC, Canada), using the Affymetrix ST 2.0 chips, covering more than 53 000 transcripts and known splice variants across the human transcriptome. Gene expression data were normalized using the robust multiarray average method, as implemented in the oligo Bioconductor package.60,61 Differentially expressed genes were adjusted using Benjamini-Hochberg method for multiple hypotheses testing, as we previously reported.57-59 This allowed the use of false discovery rate thresholds to assess whether a gene is differentially expressed or not. Differentially expressed genes were further identified (cutoff 1.3-fold; P < .05; adjusted [adj.] P < .05) and classified through gene ontology (GO), using the NetAffx web-based application (Affymetrix), whereas differentially expressed pathways were identified using ingenuity pathway analysis, gene set variation analysis,62 and gene set enrichment analysis,63 as previously described by our group.56,57 Corresponding heat maps for biological function categories were generated using programming language R.56 The clustering was performed using the complete linkage method on the Euclidean distances from the values represented in the heat maps, as we previously reported.57-59 Although these methods are known to be “unsupervised,” clustering samples with a subset of differentially expressed genes are considered as “supervised.”
Real-time reverse transcription polymerase chain reaction quantification
One-step SYBR Green real-time reverse transcription polymerase chain reaction (RT-PCR) was carried out in a LightCycler 480 II (Roche), using Qiagen reagents according to manufacturer’s recommendations, as previously described.58 Briefly, human ITGAE/CD103, CDH1/E-cadherin; ALDH1A2/RALDH2, TCF7L2/TCF4, IGSF2/CD101, and SIGLEC1/CD169 primers were purchased from Qiagen (QuantiTect Primer Assay). The expression of each gene was normalized relative to the internal control 28S rRNA levels (forward 5′-CGAGATTCCTGTCCCCACTA-3′; reverse 5′-GGGGCCACCTCCTTATTCTA-326 ).58 Each RT-PCR reaction was performed in 2 to 3 replicates.
Cytokine and chemokine quantification
Levels of TNF-α, CCL22, and CCL18 in cell culture supernatants were quantified by ELISA (R&D Systems), according to the manufacturer’s protocol.
Statistics
Statistical analyses were performed using the GraphPad Prism 6. Details are included in figure legends. Sample size calculations for microarray studies and other investigations were based on previous results.26,53,56,64,65
Accession numbers
The entire microarray dataset and technical information requested by Minimum Information About a Microarray Experiment are available at the Gene Expression Omnibus database under accession number GSE111474.
Results
Peripheral blood CD16+ and CD16− monocytes acquire typical DC features in vitro
Phenotypic features of CD16+ and CD16− MDDCs were first investigated before/after LPS-induced maturation. Highly pure CD16+ and CD16− monocytes were cultured in the presence of GM-CSF and IL-4 for 6 days (Figure 1A; supplemental Figure 1). Both CD16+ and CD16− monocytes differentiate into DCs, as reflected by high levels of CD1c and HLA-DR and the loss of CD14 expression, with CD16 expression conserved on a fraction of CD16+ MDDCs (Figure 1B-C). Further, the expression of immature (CD1a, CD1c, DC-SIGN) vs mature (CD83, CCR7) DC markers was assessed on these MDDC subsets before and after LPS-induced maturation. Both CD16+ and CD16− MDDCs expressed similarly high levels of CD1c, CD1a and DC-SIGN at immature stage, and the mature DC markers CD83 and CCR7 upon LPS exposure (supplemental Figure 2A-B). Staining with CD1a Abs and phalloidin (actin filaments) followed by confocal microscopy visualization demonstrated that both MDDC subsets acquired a typical veiled morphology, with a tendency for more protrusions/filopodia on the surface of CD16+ vs CD16− MDDCs (supplemental Figure 2C). Thus, both CD16+ and CD16− monocytes differentiate into DCs expressing classical immature/mature DC markers and typical veiled morphology.
CD16+ and CD16− MDDCs respond differently to bacterial vs viral antigens
To determine whether the distinct ability of CD16+/CD16− monocytes to sense bacterial/viral pathogens3,42 is inherited by their DC progeny, genome-wide transcriptional profiles of CD16+ and CD16− MDDCs were analyzed before/after exposure to LPS or infectious HIV-1 (Figure 1A,D-H; supplemental Figure 3). Of note, the HIV concentration used (ie, 50 ng/mL HIV-p24) was selected in preliminary experiments as being optimal in terms of efficient HIV trans infection from MDDCs to CD4+ T cells (data not shown). Differentially expressed probe sets were identified based on P values (P < .05), adjusted P values (adj. P < .05), and fold change (FC) expression ratios (Figure 1D-H; supplemental Tables 1-8). A total of 3374 probe sets were differentially expressed between CD16+ and CD16− MDDCs at immature stage (Figure 1D). Upon LPS-induced maturation, 9014 and 9167 probe sets were regulated in CD16+ and CD16− MDDCs, respectively (P < .05; Figure 1E). Among LPS-modulated probe sets, 7041 were expressed in both MDDC subsets, whereas 1973 and 2126 transcripts were differentially regulated by LPS in CD16+ and CD16− MDDCs, respectively (Figure 1F). In contrast to the high number of LPS-modulated genes, only 1164 and 663 probe sets were modulated by HIV in CD16+ and CD16− MDDCs, respectively (P < .05; Figure 1G), with 36 commonly HIV-modulated transcripts (Figure 1H). These results demonstrate that CD16+ and CD16− monocytes are precursors for transcriptionally distinct MDDC subsets able to sense bacterial/viral pathogens in unique manners.
Distinct transcriptional profiles in CD16+ and CD16− MDDCs at immature stage
Based on P values (<.05) and FC expression ratios (cutoff, 1.3), 447 and 692 probe sets were preferentially expressed in immature CD16+ and CD16− MDDCs, respectively (data not shown). When adj. P < .05 were applied, 159 and 288 probe sets were found preferentially expressed in immature CD16+ and CD16− MDDCs, respectively; new molecular markers for CD16+ MDDCs and CD16− MDDCs were identified (supplemental Tables 1 and 2).
Gene set variation analysis of differentially expressed genes identified canonical pathways (C2) and biological processes (C5) preferentially associated with CD16+ or CD16− MDDCs (supplemental Figure 4A). Among C2 canonical pathways, pathways enriched in CD16+ MDDCs were linked to nuclear receptors and T-helper pathways, as well as regulation of interferon α (IFNA) pathway; whereas pathways enriched in CD16− MDDCs were linked to DNA replication and repair and telomere maintenance (supplemental Figure 4A-B). Among C5 biological processes, pathways enriched in CD16+ MDDCs were linked to actin filament, response to extracellular stimuli, IFN-γ production, positive regulation of MAPKKK cascade, lymphocyte differentiation, regulation of lymphocyte activation, positive regulation of T-cell activation, B-cell activation, and lymphocyte activation (supplemental Figure 4B). In contrast, biological processes enriched in CD16− MDDCs included nuclease and endonuclease activity, cell cycle, DNA replication, chromatin, and microtubule motor activity (supplemental Figure 4B).
Further GO classification of differentially expressed genes in CD16+ and CD16− MDDCs revealed transcripts related to different biological functions, including: transcription factors, cytokines, adhesion molecules, chemotaxis, and cell projections (Figure 2). For the GO term transcription factors, transcripts enriched in CD16+ MDDCs included the myocyte enhancer factor 2, polycomb group ring finger 2, peroxisome proliferator-activated receptor γ, transcription factor 7-like 2 (TCF7L2 or TCF4), basic helix-loop-helix family member e41, forkhead box O1 (FOXO1), homeodomain interacting protein kinase 2, and activating transcription factor 3, whereas transcription factors enriched in CD16− MDDCs included E2F transcription factor 6 (E2F6) and the hypoxia inducible factor 1, α subunit (HIF1A; Figure 2A). The heat map in Figure 2A also reveals upregulation of transcripts for the stem cell surface markers CD34 and c-kit in CD16− vs CD16+ MDDCs. For the GO term cytokines, JAK1, CCL22, IL-21R, PTGER4, IL-15, TNFSF14, CD86, SERPINA1, and aldehyde dehydrogenase 1 family member A2 (ALDH1A2 or retinaldehyde dehydrogenase 2 [RALDH2]) transcripts were enriched in CD16+ MDDCs, whereas IL1RL2, dual oxidase 1 (DUOX1), interferon α-inducible protein 6 (IFI16), IL17RB, and CCL18 were enriched in CD16− MDDCs (Figure 2B). For the GO term adhesion molecules, CD16+ MDDCs were enriched in CD97, signaling lymphocytic activation molecule family 7, TNFRSF12A, ITGAL, paladin, RAPH1, ALCAM, LY9, and ITGAE (or CD103) transcripts, whereas C-type lectin domain family 4, member M; integrin, α 9; SIGLEC1; CDH1 (or E-cadherin); ADAM23; CDH2; EPCAM; SELL; c-kit; CD34; and IFT74 transcripts were upregulated in CD16− MDDCs (Figure 2C). The GO term chemotaxis further revealed preferential expression of CCR6 in CD16− MDDCs (Figure 2D). Finally, several transcripts associated with the GO term cell projections were differentially expressed in CD16+ vs CD16− MDDCs (Figure 2E).
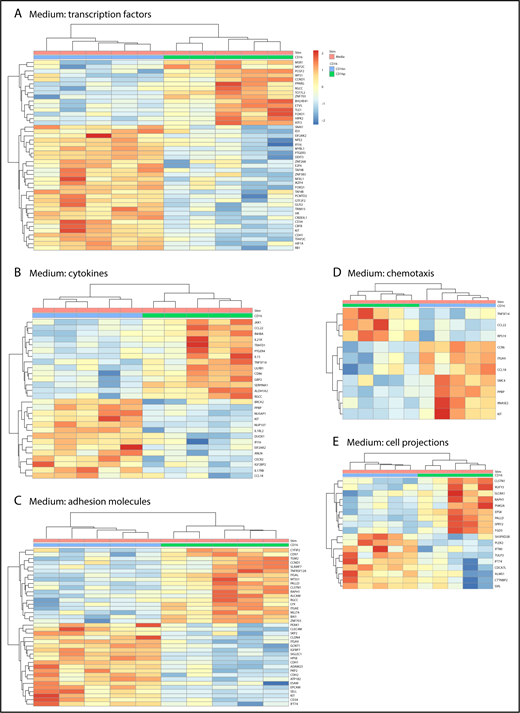
Gene ontology classification of differentially expressed genes in immature CD16+and CD16−MDDCs. Transcriptional profiling was performed as described in Figure 1A,D. Differentially expressed genes in CD16+ (green) vs CD16− (blue) MDDCs exposed to media (immature; P < .05; FC cutoff, 1.3) were classified using gene ontology in (A) transcription factors, (B) cytokines, (C) adhesion molecules, (D) chemotaxis, and (E) cell projections. Each heat map column represents data from a distinct donor for matched immature CD16+ vs CD16− MDDCs (n = 5).
Gene ontology classification of differentially expressed genes in immature CD16+and CD16−MDDCs. Transcriptional profiling was performed as described in Figure 1A,D. Differentially expressed genes in CD16+ (green) vs CD16− (blue) MDDCs exposed to media (immature; P < .05; FC cutoff, 1.3) were classified using gene ontology in (A) transcription factors, (B) cytokines, (C) adhesion molecules, (D) chemotaxis, and (E) cell projections. Each heat map column represents data from a distinct donor for matched immature CD16+ vs CD16− MDDCs (n = 5).
Transcripts differentially modulated by LPS in CD16+ vs CD16− MDDC
Large sets of transcripts were modulated by LPS in both CD16+ and CD16− MDDCs (Figure 1E-F). Nevertheless, unique transcriptional signatures induced by LPS were identified in each MDDC subset (Figure 3A; supplemental Figures 3 and 5). Using adj. P < .05 and FC cutoff 1.3 selection criteria, 261 and 221 transcripts were found upregulated and downregulated, respectively, after LPS exposure uniquely in CD16+ MDDCs (supplemental Tables 3 and 4), whereas 265 and 467 transcripts were upregulated and downregulated, respectively, by LPS preferentially in CD16− MDDCs (supplemental Tables 5 and 6). Among transcripts specifically modulated by LPS in CD16+ MDDCs, CCL8, SIGLEC1, microRNA (MIR)4439, SCIN, IL7R, PLTP, TNF, CFP (properdin, the positive regulator of the alternative pathway of complement activation66,67 ), clusterin, C2, MIR331, triggering receptor expressed on myeloid cells 139, MAP3K8, CXCL1, IL-18, BCL11A, perforin 1, MIR4436, MIR299, and retinol dehydrogenase 11 (RDH11) were upregulated, whereas lipoprotein lipase, kelch-like family member 6, CD109, TLR3, ITGAE, ABCA1, ITGAV, KLF10, and FCGR3A were downregulated (supplemental Tables 3 and 4). Among transcripts specifically modulated by LPS in CD16− MDDCs, upregulated were matrix metallopeptidase 10 (stromelysin 2, MMP10), MMP1, TNFRSF11A, IL-1A, lysosomal-associated membrane protein 1, MMP8, fibrillin 1, aldehyde dehydrogenase 1 family member L2 (ALDH1L2), IL-1 receptor antagonist, triggering receptor expressed on myeloid cells 1, and mucosa-associated lymphoid tissue lymphoma translocation gene 1 (MALT1), whereas CD24; STEAP family member 4; CD163 molecule-like 1(CD163L1); MIR146B; p21 protein (Cdc42/Rac)-activated kinase 7 (PAK7); CCL18; G protein-coupled receptor 34; cell adhesion molecule 3 (CAM3); IL-17RB; CD207; CD180; legumain; MIR3174; C-type lectin domain family 4, member M; CD69; CD163; IL-10; integrin, α 9; and myeloperoxidase transcripts were downregulated (supplemental Tables 5 and 6).
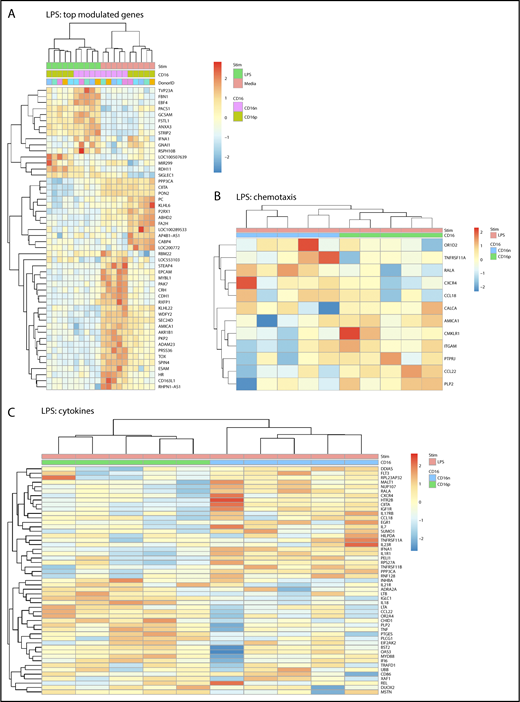
Differential gene expression in CD16+and CD16−MDDCs in response to LPS. Transcriptional profiling was performed as described in Figure 1A,E-F with matched MDDC subsets exposed to media (immature) or LPS (mature) for 24 hours. Shown are top-regulated genes in immature and mature CD16+ compared with CD16− MDDCs (A) and differentially expressed genes in CD16+ vs CD16− MDDC subsets exposed to LPS (P < .05; FC cutoff, 1.3) linked to the gene ontology terms chemotaxis (B) and cytokines (C). Heat map cells are scaled by the expression level z-scores for each probe individually. Results were generated with cells from 5 different donors identified with different color codes.
Differential gene expression in CD16+and CD16−MDDCs in response to LPS. Transcriptional profiling was performed as described in Figure 1A,E-F with matched MDDC subsets exposed to media (immature) or LPS (mature) for 24 hours. Shown are top-regulated genes in immature and mature CD16+ compared with CD16− MDDCs (A) and differentially expressed genes in CD16+ vs CD16− MDDC subsets exposed to LPS (P < .05; FC cutoff, 1.3) linked to the gene ontology terms chemotaxis (B) and cytokines (C). Heat map cells are scaled by the expression level z-scores for each probe individually. Results were generated with cells from 5 different donors identified with different color codes.
GO classification of differentially expressed genes in CD16+ and CD16− MDDCs exposed to LPS revealed transcripts linked to cell migration and cytokines. For the GO term chemotaxis, top-regulated transcripts included OR1D2, TNFRSF11A, RALA, CXCR4, and CCL18 for CD16− MDDC, and AMICA1; chemerin chemokine-like receptor 1; ITGAM; protein tyrosine phosphatase; receptor type, J; CCL22; and proteolipid protein 2 for CD16+ MDDCs (Figure 3B). For the GO term cytokines, top regulated transcript enriched in CD16− MDDCs included fms-related tyrosine kinase 3, MALT1, NUP107, IGF1R, CXCR4, IL17RB, CCL18, EGR1, IL-7, IL-23R, IFNA1, and IL-1R1, whereas in CD16+, MDDCs included IL-21R, LTB, IL-18, LTA, CCL22, TNF, MYD88, interferon α-inducible protein 6, CD86, and DUOX2 (Figure 3C). Thus, both MDDC subsets sense LPS, but in transcriptionally distinct manners, with CD16+ compared with CD16− MDDCs exhibiting a superior pro-inflammatory profile, as reflected by their upregulated expression of LTB, IL-18, LTA, TNF, CCL22, MYD88, and DUOX2 transcripts.
Transcripts differentially modulated by HIV in CD16+ vs CD16− MDDCs
Transcriptional changes observed in CD16+ or CD16− MDDCs on exposure to HIV were minor compared with those induced by LPS (Figure 1D-H; supplemental Figures 3 and 6). A number of 44 and 84 transcripts were specifically up- and downregulated, respectively, by HIV in CD16+ MDDCs (supplemental Table 7). Also, a number of 25 and 30 transcripts were specifically up- and downregulated, respectively, by HIV in CD16− MDDCs (P < .05; FC cutoff 1.3; supplemental Table 8). Heat maps in supplemental Figure 6A-B illustrate top transcripts modulated by HIV in CD16+ and CD16− MDDCs (P < .05; FC cutoff, 1.3). None of these changes in gene expression reached an adj. P < .05 (supplemental Figure 3). Of note, HIV differentially regulated the expression of multiple MIRs in both CD16+ and CD16− MDDCs. Transcripts upregulated by HIV in CD16+ MDDCs included the IFI6, MIR568, MIR4718, CD101 (a molecule potentially involved in the negative regulation of T-cell activation68 ), MIR4717, SIGLEC1, MIR3692, MIR3188, MIR4441, Kruppel-like factor 10 (KLF10), MIR4672, MIR548S, CASP6, MIR583, and MIR4491, whereas downregulated transcripts included the ρ-associated, coiled-coil containing protein kinase 1 pseudogene 1 (ROCK1P1), ubiquitin-specific peptidase 17-like family member 5, cytokine receptor-like factor 2, MIR548I3, MIR509-3, IFNA1, fms-related tyrosine kinase 3, IL-2RA, mediator complex subunit 27, activating transcription factor 3, IFNA16, and CCR4 (Figure 4A; supplemental Table 7; supplemental Figure 6A). Transcripts upregulated by HIV in CD16− MDDCs included MIR1271, MIR516A2, MIR3911, MIR196A1, S100B, MIR320C1, MIR3665, MIR3201, and MIR523; whereas downregulated transcripts included MIR4499, MIR3975, MIR4263, MIR545, MIR130B, and MIR4308 (Figure 4A; supplemental Table 8; supplemental Figure 6B). Of note, immature CD16+ MDDCs shared transcripts with HIV-exposed CD16− MDDCs (Figure 4A). When GO analysis was performed, transcripts associated to exosomes were predominant and preferentially expressed by HIV-exposed CD16+ vs CD16− MDDCs (Figure 4B). These results reveal minor but tailored effects HIV exerts on the functionality of CD16+ and CD16− MDDCs.
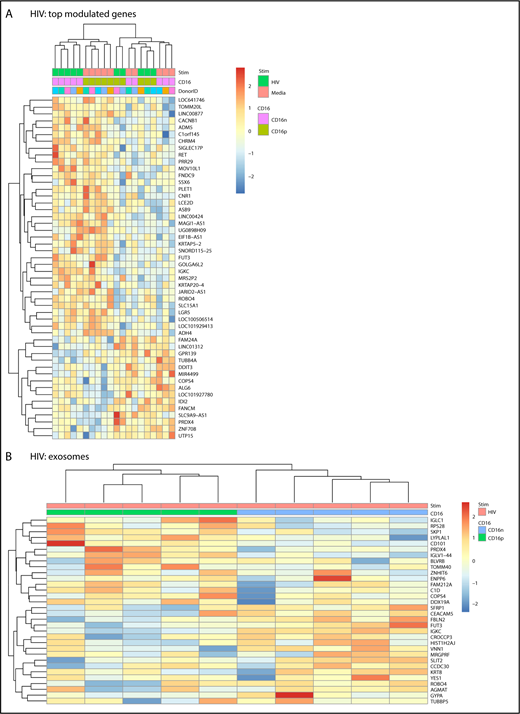
Differential gene expression in CD16+and CD16−MDDCs in response to HIV. Transcriptional profiling was performed as described in Figure 1A,G-H, with matched MDDCs subsets exposed to media (immature) or HIV for 24 hours. Shown are top differentially regulated genes in immature and HIV-exposed CD16+ (green) and CD16− MDDCs (pink) (A) and differentially expressed genes in CD16+ (green) vs CD16− (blue) MDDC subsets exposed to HIV (P < .05; FC cutoff, 1.3) linked to the gene ontology terms exosome (B). Heat map cells are scaled by the expression-level z-scores for each probe individually. Results were generated with cells from 5 different donors identified with different color codes.
Differential gene expression in CD16+and CD16−MDDCs in response to HIV. Transcriptional profiling was performed as described in Figure 1A,G-H, with matched MDDCs subsets exposed to media (immature) or HIV for 24 hours. Shown are top differentially regulated genes in immature and HIV-exposed CD16+ (green) and CD16− MDDCs (pink) (A) and differentially expressed genes in CD16+ (green) vs CD16− (blue) MDDC subsets exposed to HIV (P < .05; FC cutoff, 1.3) linked to the gene ontology terms exosome (B). Heat map cells are scaled by the expression-level z-scores for each probe individually. Results were generated with cells from 5 different donors identified with different color codes.
CD16+ MDDCs exhibit a unique pro-inflammatory molecular signature
Gene set enrichment analysis identified canonical pathways modulated in CD16+ vs CD16− MDDCs by LPS and/or HIV exposure (Figure 5). Canonical pathways linked to 20S proteasome, myeloid lineage, myeloid MΦs DC inflammatory interleukin, and inflammation II were enriched in CD16+ vs CD16− MDDCs in all experimental conditions (Figure 5A). Changes in the expression of various MIRs were also observed, including the preferential expression in CD16+ vs CD16− MDDCs of MIR1271 under constitutive conditions, MIR146b on LPS stimulation, and MIR4499 after HIV exposure (Figure 5B). Thus, distinct canonical pathways and MIR-mediated regulatory mechanisms control MDDC functions, with inflammation being a key feature of CD16+ MDDCs.
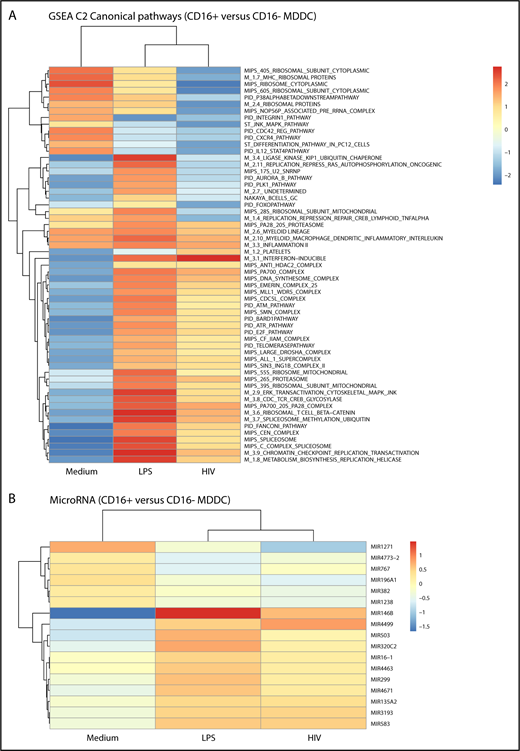
CD16+and CD16−MDDCs exposed to media, LPS, or HIV exhibit unique molecular signatures. Genome-wide transcriptional profiles were generated as described in Figure 1A,D-H for MDDC subsets exposed to media, LPS, or HIV for 24 hours. (A) Gene set enrichment analysis allowed the identification of top-regulated canonical pathways (C2), commonly and differentially expressed between CD16+ vs CD16− MDDCs exposed to media, LPS, or HIV-1. (B) Shown are top-regulated micro-RNAs (MIR) differentially expressed between CD16+ and CD16− MDDCs exposed to media, LPS, or infectious HIV virions. Results were generated with matched MDDC subsets from 5 different donors.
CD16+and CD16−MDDCs exposed to media, LPS, or HIV exhibit unique molecular signatures. Genome-wide transcriptional profiles were generated as described in Figure 1A,D-H for MDDC subsets exposed to media, LPS, or HIV for 24 hours. (A) Gene set enrichment analysis allowed the identification of top-regulated canonical pathways (C2), commonly and differentially expressed between CD16+ vs CD16− MDDCs exposed to media, LPS, or HIV-1. (B) Shown are top-regulated micro-RNAs (MIR) differentially expressed between CD16+ and CD16− MDDCs exposed to media, LPS, or infectious HIV virions. Results were generated with matched MDDC subsets from 5 different donors.
CD16+ monocytes differentiate into CD103+RALDH2+TCF4+ DCs that produce high TNF levels
The microarray results listed in supplemental Tables 1-8 and illustrated in supplemental Figure 7 were further subject to RT-PCR and ELISA validations. Results in Figure 6A reveal that CD103, RALDH2, and TCF4 are indeed transcriptional markers for immature CD16+ MDDCs, whereas CD16− MDDCs exhibited preferentially CDH1/E-cadherin transcripts. RT-PCR validations also revealed that HIV-exposed CD16+ and CD16− MDDCs are distinguished by preferential expression of CD101 and CD169, respectively (Figure 6B). Finally, results in Figure 6C illustrate that CD16+ vs CD16− MDDCs produce higher levels of TNF-α on LPS exposure. Also, CD16+ vs CD16− MDDCs produced superior levels of CCL22 at immature stage or on HIV exposure, whereas CCL18 was mainly produced by immature and mature CD16− MDDCs (Figure 6C). In contrast to HIV that did not increase the ability of MDDCs subsets to produce these cytokines/chemokines, LPS was a strong inducer of TNF-α and CCL22, but not CCL18 (Figure 6C). Thus, CD16+ monocytes are precursors for CD103+RALDH2+TCF4+ DCs with a superior pro-inflammatory and chemoattractant potential compared with CD16− MDDCs that express a CDH1+ transcriptional signature.
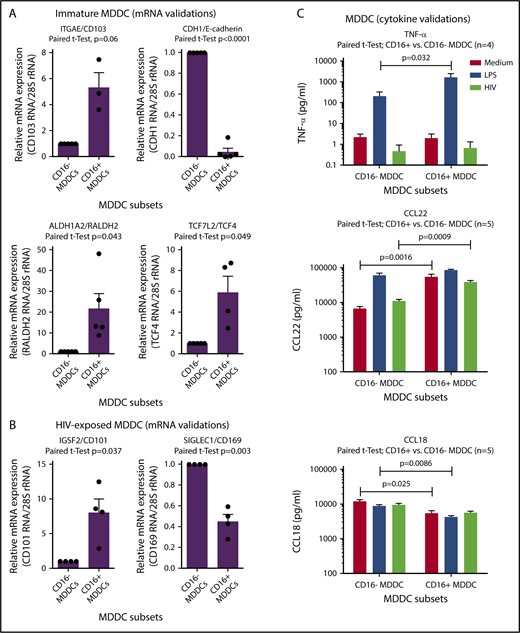
Novel functional markers for CD16+and CD16−MDDCs. MDDC subsets were generated and exposed to media (immature), LPS (mature), or HIV for 24 hours, as described in Figure 1A. Shown are real-time RT-PCR validation of relative ITGAE/CD103, CDH1/E-cadherin, ALDH1A2/RALDH2, and TCF7L2/TCF4 mRNA expression in immature MDDC subsets (A), as well as IGSF2/CD101 and SIGLEC1/CD169 in HIV-exposed MDDC subsets (B). (A-B) Results were generated with matched MDDC subsets from 3 to 5 different donors. Each symbol represents the median value of 2 to 3 RT-PCR replicates, with values for CD16− MDDCs being considered 1. Paired Student t test P values are indicated on the graphs. (C) Shown are levels of TNF-α, CCL22, and CCL18 quantified by ELISA in cell culture supernatants from matched CD16+ and CD16− MDDCs on exposure to media, LPS, or HIV (n = 4-5, mean ± SEM). Paired Student t test P values for log10 cytokine levels are indicated on the graphs.
Novel functional markers for CD16+and CD16−MDDCs. MDDC subsets were generated and exposed to media (immature), LPS (mature), or HIV for 24 hours, as described in Figure 1A. Shown are real-time RT-PCR validation of relative ITGAE/CD103, CDH1/E-cadherin, ALDH1A2/RALDH2, and TCF7L2/TCF4 mRNA expression in immature MDDC subsets (A), as well as IGSF2/CD101 and SIGLEC1/CD169 in HIV-exposed MDDC subsets (B). (A-B) Results were generated with matched MDDC subsets from 3 to 5 different donors. Each symbol represents the median value of 2 to 3 RT-PCR replicates, with values for CD16− MDDCs being considered 1. Paired Student t test P values are indicated on the graphs. (C) Shown are levels of TNF-α, CCL22, and CCL18 quantified by ELISA in cell culture supernatants from matched CD16+ and CD16− MDDCs on exposure to media, LPS, or HIV (n = 4-5, mean ± SEM). Paired Student t test P values for log10 cytokine levels are indicated on the graphs.
A conserved transcriptional signature in CD16+ monocytes and MDDCs
A meta-analysis was performed using transcripts differentially expressed in CD16+ vs CD16− monocytes (GSE1683626 ) and CD16+ vs CD16− MDDC data sets (GSE111474; Figure 1A,D-H). Results in Figure 7 reveal multiple transcripts conserved between CD16+ MDDCs and their monocyte progenitors. This indicates that CD16+ monocytes are stably imprinted with a transcriptional profile that is conserved during differentiation into DCs.
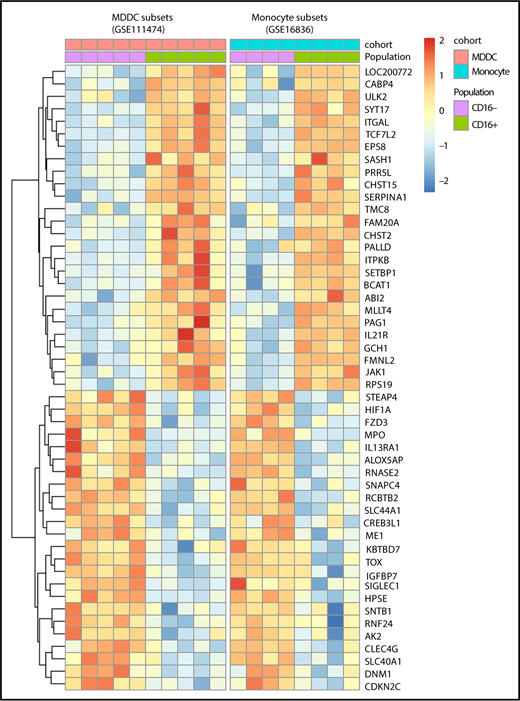
Meta-analysis identifies a transcriptional signature conserved by CD16+and CD16−monocytes during differentiation into DCs. Genes differentially expressed in CD16+ vs CD16− monocytes (GSE1683626 ) and CD16+ vs CD16− MDDCs (GSE111474, current manuscript) were subject to a meta-analysis that led to the identification of a transcriptional signature preserved during monocyte differentiation into DCs.
Meta-analysis identifies a transcriptional signature conserved by CD16+and CD16−monocytes during differentiation into DCs. Genes differentially expressed in CD16+ vs CD16− monocytes (GSE1683626 ) and CD16+ vs CD16− MDDCs (GSE111474, current manuscript) were subject to a meta-analysis that led to the identification of a transcriptional signature preserved during monocyte differentiation into DCs.
Discussion
Monocytes are precursors for DCs,11,34,69 professional antigen-presenting cells that shape self-tolerance as well as immunity to pathogens.13,70 Here, we provide evidence that intermediate/nonclassical CD16+ and classical CD16− monocytes are precursors for transcriptionally/functionally distinct DC subsets (Figure 8) that may exhibit a peculiar capacity to shape immunity against bacterial/viral pathogens. Thus, a functional specialization exists not only between CD16+ and CD16− monocytes3,5,22,42 but also between their DC progeny, supporting the idea that changes in monocyte heterogeneity during various pathological conditions have consequences on the quality of DC-mediated immune responses.13
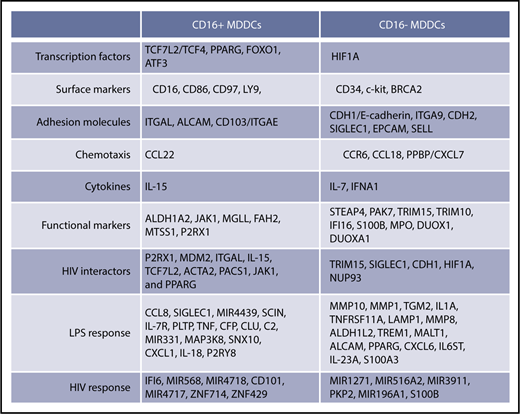
Unique transcriptional signatures discriminate CD16+from CD16−MDDCs. Shown are top-differentially expressed genes in CD16+ vs CD16− MDDCs at immature stage or on exposure to LPS and HIV. Selected top-modulated genes encode for transcriptional factors, surface markers, adhesion molecules, chemotaxis, cytokines, functional markers, HIV interactors, LPS response, and HIV response. Transcripts in bold were validated at RNA and/or protein level in Figure 6.
Unique transcriptional signatures discriminate CD16+from CD16−MDDCs. Shown are top-differentially expressed genes in CD16+ vs CD16− MDDCs at immature stage or on exposure to LPS and HIV. Selected top-modulated genes encode for transcriptional factors, surface markers, adhesion molecules, chemotaxis, cytokines, functional markers, HIV interactors, LPS response, and HIV response. Transcripts in bold were validated at RNA and/or protein level in Figure 6.
Biological processes linked to lymphocyte activation were enriched in CD16+ vs CD16− MDDCs. Indeed, CD16+ MDDCs expressed transcripts involved in immunological synapse formation, including activation molecules (CD86, CD97, LY9), adhesion molecules (ALCAM, LFA-1), and transcription factors (FOXO1, activating transcription factor 3, TCF4). CD97 was previously identified as a marker for CD16+ monocytes26 and an HIV permissiveness factor71 ; FOXO1 upregulates CCR7 and ICAM1 for complete DC maturation72 ; AFT3 has a role in Th17 polarization73 ; and TCF4, a component of the Wnt/β-catenin pathway, regulates DC functions74,75 and restricts HIV in myeloid cells.76 As opposed to CD16+ MDDCs, transcriptional profiles in CD16− MDDCs point to a less active cellular metabolism, with enriched expression of early hematopoietic precursor and stem cell transcripts (CD34, c-kit, and ITAG9, HIF1A), cell migration markers (CCR6, SELL and EPCAM), cytoskeleton (PAK7), adhesion molecules (CDH1 and CDH2) and phagocytosis (SIGLEC1). The biological processes enriched in CD16− vs CD16+ MDDCs included biological terms such as cell cycle, DNA replication, chromatin, and microtubule motor activity. CD16− compared with CD16+ MDDCs, were also enriched in transcripts involved in the protection against oxidative stress including myeloperoxidase, DUOX1, and DUOAX1.77 Interestingly, CD16− MDDCs also expressed the sensor for microbial DNA IFI16,78 previously reported to sense HIV in CD4+ T cells.79
Pioneering studies by Sánchez-Torres et al demonstrated that CD16+ vs CD16− MDDCs differ in their immunogenic potential,43 with differences in CD86 and CD11A expression that are confirmed by our transcriptional studies. One major finding of our study is that CD16+ and CD16− MDDCs exhibit unique CD103+RALDH2+TCF4+ and E-cadherin+ phenotypes, respectively. Our findings mirror evidence in mice demonstrating distinct roles of mucosal CD103+ and E-cadherin+ DCs in intestinal tolerance (Treg differentiation) and immunity (Th17 differentiation), respectively.80-82 RALDH2 is involved in the biosynthesis of the retinoic acid (RA) from vitamin A.83-85 The coexpression of TCF4 and RALDH2 in CD16+ MDDCs is consistent with the fact that TCF4 regulates RALDH2 expression in DCs.74,86 In humans, CD1c+ DCs were reported to produce RA.87 Our transcriptional profiling indicates that RA-producing DCs differentiate from CD16+ monocytes. It is known that RA-producing DCs imprint CD4+ T cells with gut-homing potential during antigenic presentation.88,89 RA was reported to promote naive T-cell differentiation into Tregs or Th17 cells, depending on the cytokinic environment.90 We demonstrated that RA acts on memory CD4+ T-cells to increases HIV permissiveness in Th17 cells via mechanistic target of rapamycin–dependent mechanisms.59,64 Thus, by producing RA, CD16+ MDDCs may either act as regulatory DCs or activate Th17 cells to promote HIV dissemination at mucosal level.13,91 Indeed, CD16+ monocytes express high levels of CX3CR1,30,31 a chemokine receptor mediating migration into the gut,92 and they may differentiate into mucosal CD103+RALDH2+ DCs. The pathogenic potential of CD16+ vs CD16− MDDCs in the context of HIV-1 infection may also be mediated by the production of the homeostatic cytokine IL-15,93,94 pro-inflammatory cytokine TNF-α41, and the chemokine CCL22, which may attract CCR4+ Th17 cells and deliver costimulatory signals essential for HIV replication.95 The pathogenicity of CD16+ MDDCs during HIV infection requires further in-depth investigations.
Despite the fact that both CD16+ and CD16− MDDCs acquire a mature CD83+CCR7+ phenotype after LPS exposure, distinct transcriptional signatures were observed in these subsets. Mature CD16+ MDDCs expressed transcripts for inflammatory cytokines/chemokines (TNF-α, IL-18, CXCL1), complement components (C2, CFP), phagocytosis and HIV capture (SIGLEC1),96 and microRNA (Mir-331, Mir-4439). In contrast, mature CD16− MDDCs expressed transcripts associated with type-1 interferon response (IL1A, IFNA1), inflammasome activation pathway (MALT197,98 ), cell migration (CXCL6 and metalloproteinases MMP1,99 MMP8100 and MMP10101 ), Th17 polarization (IL-23, IL-6ST), DC-T-cell interaction (ALCAM, TNFRSF11), and mitochondrial functions (ALDH1L2). Thus, CD16+ and CD16− MDDCs respond differently to LPS, indicative that each DC subset exhibits a distinct immunological role in immunity.
In contrast to LPS, HIV exposure induced minimal changes in the transcriptional profiles of both CD16+ and CD16− MDDCs, with no signs of DC maturation. Our results are in line with studies published by other groups demonstrating the limited capacity of MDDCs to sense HIV.102-105 Indeed, HIV was reported to inhibit the function of TANK-binding kinase 1, a kinase involved in IFN signaling pathway.106 Nevertheless, specific transcripts (ie, CD101 and SIGLEC1) and microRNAs were identified as differentially modulated by HIV in these MDDC subsets; their role in controlling the immunogenic vs pathogenic features of CD16+ MDDCs during bacterial or viral infection remains to be investigated in future studies.
Previous transcriptional characterization of monocyte subsets and their progeny provided valuable insights into their functional specialization.24,26-29 In our study, canonical pathways linked to inflammation distinguished CD16+ from CD16− MDDCs at immature stage, as well as after exposure to LPS or HIV. Noteworthy, similar to CD16+ monocytes,22,33,36-41 we demonstrate that CD16+ MDDCs are major TNF-α producers in response to LPS. Consistently, our meta-analysis revealed that transcripts differentially expressed in CD16+ vs CD16− monocytes26 were also found in the present study as markers of immature CD16+ MDDCs (eg, TCF7L2) and CD16− MDDCs (eg, HIF1α; Figure 7). Thus, CD16+ and CD16− monocytes are imprinted with a unique transcriptional program, likely via epigenetic modifications,107 that is in part preserved during differentiation into DCs. This unique transcriptional imprinting/programming may be the result of distinct cell-to-cell and/or antigens/pathogens interactions during monocyte trafficking in vivo.35,49
In this study, we did not distinguish between intermediate and nonclassical CD16+ monocytes. Future studies should use alternative markers such as M-DC8/slan23-25,41 to study the immunological features of DCs derived from these functionally distinct CD16+ monocyte subsets.24,25,41,108
In conclusion, our results provide evidence supporting the existence of a “division of labor” between CD16+ and CD16− MDDCs and offer a solid platform for future validations toward the identification of molecular determinants of DC immunogenicity/pathogenicity, with relevance for pathological conditions such as sepsis and HIV-1 infection (Figure 8). Alterations of the CD16+/CD16− monocyte ratio may significantly influence the quality of DC-mediated immune responses in the context of specific pathogens; these aspects deserve future detailed investigations. Finally, our results provide guidance for the rational use of CD16+ vs CD16− MDDCs in DC-based therapeutic strategies.
The data reported in this article have been deposited in the Gene Expression Omnibus database (accession number GSE111474).
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the Centre de recherche du Centre Hospitalier de l'Université de Montréal FACS Core Facility for expert technical support in FACS analysis and cell sorting, Mario Legault (HIV/AIDS and Infectious Diseases Network of the Fonds de Recherche Québec-Santé) for help with ethical approvals and informed consents, and Dana Gabuzda (Dana-Farber Cancer Institute, Boston, MA) for the HIV molecular clones. The authors thank Josée Girouard and Angie Massicotte (McGill University Health Centre) for their involvement in human subject recruitment for leukapheresis collection, and Natalia Fonseca do Rosario and Laurence Raymond Marchand for assay optimization, contribution to cell sorting and culture, and critical reading of the manuscript. Lastly, the authors acknowledge the major contribution to this work of all human donors via their gift of leukapheresis.
This work was supported by grants from the Canadian Institutes of Health Research (CIHR; HOP-120239) (P.A.) and by Canadian HIV Cure Enterprise Team Grant HIG-133050 from the CIHR in partnership with the Canadian Association for AIDS Research and International AIDS Society. V.S.W. received a CIHR Fellowship. J.-P.R. holds a Louis-Lowenstein Chair in Hematology and Oncology, McGill University.
Authorship
Contribution: V.S.W. generated the majority of results, analyzed data, prepared figures, and wrote the manuscript; A.C. generated results in Figure 6 and significantly contributed to manuscript revision; J.-P.G. performed microarray data analysis and generated figures; D.G. and A.G. performed flow cytometry sorting; A.C.-B. generated results in supplemental Figure 2C; Y.Z. performed experiments; C.L.T. and J.-P.R. ensured access to clinical samples/information and/or provided experimental protocols; P.A. designed research, analyzed data, and wrote the manuscript; and all authors reviewed and accepted the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Petronela Ancuta, CHUM-Research Centre, 900 rue Saint-Denis, Tour Viger R, Room R09.416, Montreal, QC H2X 0A9, Canada; e-mail: petronela.ancuta@umontreal.ca.

