

Key Points
Using novel CRISPR-generated transgenic mice, we precisely mapped the structural requirements for HPA-1a alloantibody binding.
This approach should enable design of diagnostic and therapeutic reagents to treat platelet-specific alloimmune disorders.

Abstract
Antibodies to platelet-specific antigens are responsible for 2 clinically important bleeding disorders: posttransfusion purpura and fetal/neonatal alloimmune thrombocytopenia (FNAIT). The human platelet-specific alloantigen 1a/1b (HPA-1a/1b; also known as PlA1/A2) alloantigen system of human platelet membrane glycoprotein (GP) IIIa is controlled by a Leu33Pro polymorphism and is responsible for ∼80% of the cases of FNAIT. Local residues surrounding polymorphic residue 33 are suspected to have a profound effect on alloantibody binding and subsequent downstream effector events. To define the molecular requirements for HPA-1a alloantibody binding, we generated transgenic mice that expressed murine GPIIIa (muGPIIIa) isoforms harboring select humanized residues within the plexin-semaphorin-integrin (PSI) and epidermal growth factor 1 (EGF1) domains and examined their ability to support the binding of a series of monoclonal and polyclonal HPA-1a–specific antibodies. Humanizing the PSI domain of muGPIIIa was sufficient to recreate the HPA-1a epitope recognized by some HPA-1a–specific antibodies; however, humanizing distinct amino acids within the linearly distant but conformationally close EGF1 domain was required to enable binding of others. These results reveal the previously unsuspected complex heterogeneity of the polyclonal alloimmune response to this clinically important human platelet alloantigen system. High-resolution mapping of this alloimmune response may improve diagnosis of FNAIT and should facilitate the rational design and selection of contemplated prophylactic and therapeutic anti–HPA-1a reagents.
Introduction
Alloantibodies to platelet-specific antigens are responsible for 2 clinically important bleeding disorders: posttransfusion purpura (PTP) and fetal/neonatal alloimmune thrombocytopenia (FNAIT; variously referred to in the literature as NATP or NAIT; Newman et al1 provide a review). PTP is a rare syndrome in which a multiparous woman, after receiving a blood transfusion, enigmatically clears not only the transfused platelets but her own as well, leading to severe thrombocytopenia, bruising, and petechiae. Unlike PTP, FNAIT is a fairly common disorder, leading to severe fetal and/or neonatal thrombocytopenia in ∼1 in 1000 to 1 in 2000 live births.2,3 Although many infants recover uneventfully, FNAIT is a leading cause of severe thrombocytopenia in the fetus and neonate, with nearly half experiencing bleeding serious enough to require transfusion with antigen-negative platelets.4 The most destructive consequences of FNAIT, however, are intracranial hemorrhage and intrauterine death as early as 20 to 24 weeks of gestation.2,5,6 Despite advances in treatment, FNAIT remains a leading cause of intracranial hemorrhage in full-term infants,4,7-10 often leading to lifelong disability.
Work performed in many laboratories over the past 60 years has led to the identification of >30 distinct heritable human platelet-specific alloantigen (HPA) systems (HPAs 1-30), located on 5 different glycoproteins, currently recognized by the Platelet Nomenclature Committee of the International Society of Blood Transfusion and the International Society on Thrombosis and Haemostasis.11 Of these, the HPA-1a (also known as PlA1) epitope most commonly provokes PTP and FNAIT, responsible for ∼80% of the cases in which an alloantibody can be detected,12 and it has accordingly been the most extensively studied.
The HPA-1a/1b alloantigen system is controlled by a Leu33Pro polymorphism in platelet membrane glycoprotein (GP) IIIa13,14 (the β3 integrin subunit of the αIIbβ3 platelet fibrinogen receptor), with Pro33 (HPA-1b) homozygous individuals who also carry the HLA-DRB3*0101 allele of the major histocompatibility complex most at risk for developing an alloimmune response to the Leu33 (HPA-1a) form of GPIIIa.15-17 Polymorphic amino acid 33 is located within a heavily disulfide-bonded knot-like structure known as the plexin-semaphorin-integrin (PSI) domain, which itself lies between the hybrid and integrin epidermal growth factor 1 (EGF, I-EGF1) domains of GPIIIa18 (Figure 1). Interestingly, although some maternal anti–HPA-1a alloantibodies, classified as type I antibodies, bind normally to a mutant form of GPIIIa in which the disulfide bond linking the PSI and EGF1 domains has been disrupted, others (type II) lose reactivity,19 demonstrating that the alloimmune response to HPA-1a is heterogeneous and that sequences within the linearly distant EGF domain might be required to form a high-affinity antibody binding site on GPIIIa for at least some maternal anti–HPA-1a antibodies (shown schematically in Figure 1 and described more fully in “Discussion”).
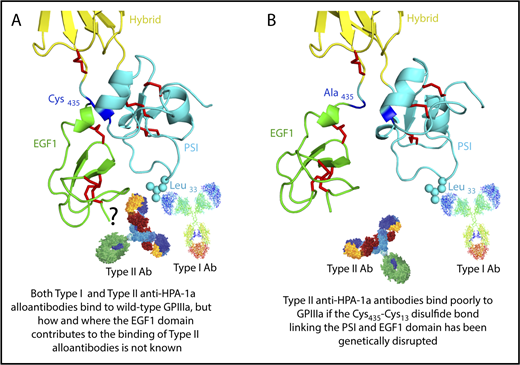
Three-dimensional structure of the human GPIIIa PSI and EGF1 domains. (A) Note that the PSI domain lies between the hybrid and EGF1 domains of GPIIIa and that polymorphic amino acid 33, which controls expression of the HPA-1a (PlA1) epitope, is directly opposite the linearly distant but conformationally close EFG1 domain. (B) Mutation to alanine of Cys435, which links the EGF1 domain to the PSI domain via a disulfide bond with Cys13, has previously been shown to result in the loss of binding of some, but not all, maternal anti–HPA-1a alloantibodies, leading to speculation that nonpolymorphic amino acids in EGF1 constitute part of the epitope for these so-called type II antibodies.
Three-dimensional structure of the human GPIIIa PSI and EGF1 domains. (A) Note that the PSI domain lies between the hybrid and EGF1 domains of GPIIIa and that polymorphic amino acid 33, which controls expression of the HPA-1a (PlA1) epitope, is directly opposite the linearly distant but conformationally close EFG1 domain. (B) Mutation to alanine of Cys435, which links the EGF1 domain to the PSI domain via a disulfide bond with Cys13, has previously been shown to result in the loss of binding of some, but not all, maternal anti–HPA-1a alloantibodies, leading to speculation that nonpolymorphic amino acids in EGF1 constitute part of the epitope for these so-called type II antibodies.
On the basis of an analysis of the 3-dimensional structure data of GPIIIa in the region of the molecule surrounding polymorphic amino acid 33, we designed and generated transgenic mice that expressed murine GPIIIa (muGPIIIa) isoforms harboring select humanized residues within the PSI and EGF1 domains and examined their ability to support the binding of a series of monoclonal and polyclonal HPA-1a–specific antibodies. Our results reveal the previously unsuspected complex heterogeneity of the polyclonal alloimmune response to this clinically important human platelet alloantigen system. High-resolution mapping of this alloimmune response may improve diagnosis of FNAIT and should facilitate the rational design and selection of contemplated prophylactic and therapeutic anti–HPA-1a reagents.
Materials and methods
Antibodies
Three antibodies with specificity for the Leu33 allelic isoform of human GPIIIa were used in this study: the murine monoclonal antibody (mAb) SZ21,20 the human mAb 26.4,21 derived from an immortalized B cell from an HPA-1a alloimmunized woman who had an infant affected by FNAIT, and B2G1,22 a humanized immunoglobulin G (IgG) derived from an scFv fragment isolated by phage display from an HPA-1a alloimmunized woman. Human maternal anti–HPA-1a antisera were provided by Richard Aster, Dan Bougie, and Brian Curtis (Blood Research Institute, BloodCenter of Wisconsin, Milwaukee, WI). Blood samples were obtained from healthy volunteers as approved by the Institutional Review Board of the Medical College of Wisconsin BloodCenter of Wisconsin after obtaining informed consent in accordance with the Declaration of Helsinki. A murine mAb, PSIB1, which binds both the human and mouse β3 integrin PSI domains, and the binding of which is unaffected by the Leu33Pro polymorphism,23 was kindly provided by Heyu Ni (University of Toronto). mAb AP2, which recognizes a complex-dependent epitope on GPIIb-IIIa but does not interfere with HPA-1a antibody binding,24 was provided by Robert Montgomery (Blood Research Institute, BloodCenter of Wisconsin).
One-step generation of mice expressing the APLD humanized form of muGPIIIa
Guide RNAs (gRNAs) were designed using the CRISPR Design Tool (http://crispr.mit.edu/) to minimize off-target effects and selected to precede a 5′-NGG protospacer-adjacent motif. To generate the vector coexpressing Cas9 and single gRNA targeting ITGB3 exon 3 (TTCTCCTTCAGGTTAC-ATCG), a pair of oligos (5′-CACCGTTCTCCTTCAGGTTACATCG-3′ and 5′-AAACCGATGTAACCTGAAGGAGAAC-3′) were annealed and cloned into the BbsI site of the Cas9 expression plasmid px459 (Addgene, Cambridge, MA). A single-stranded oligodeoxynucleotide (ssODN), 200 nucleotides in length, with the sequence 5′-GCCAGGGGGAGGTGACTTACCAGGCAGGAGGCACAGCCGCCCTAGCTCTG-ATGTTGACCTTTCCCTCGGGCTCTTCTCTTCATAGGCCTTGCCTCTGGGATCCCCACGCTGTGACCTGAAGGAGAACCTGCTGAAGGACAATTGTGCTCCAGAGTCTATTGAGTTCCCAGTCAGTGAGGCCCAGATCCTGGAGGCTAGGC-3′, was synthesized by Integrated DNA Technologies (IDT, Coralville, IA). This oligo corresponds to the antisense strand of the murine β3 gene and contains 5 nucleotide substitutions that result in the introduction of 4 human amino acid substitutions into the PSI domain of the murine β3 integrin subunit. The ssODN also contains 4 silent mutations, 2 of which introduce a diagnostic BamH1 restriction site into the plasmid, while the other 2 mutate the sequence to avoid repetitive digestion of the humanized murine β3 gene by Cas9.
Mice were maintained in the Biological Resource Center at the Medical College of Wisconsin. All animal protocols were approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee. C57BL/6N female mice were superovulated and mated with C57BL/6N male mice, and fertilized eggs were collected from the oviducts. The px459 plasmid (10 ng/μL) and ssODN (5 ng/μL) were injected into the pronuclei of fertilized oocytes. Injected zygotes were cultured in potassium simplex optimization medium with amino acids at 37°C in 5% carbon dioxide and 95% humidified air overnight. Two-cell stage embryos were then transferred into the oviducts of pseudopregnant female mice. Genomic DNA isolated from the tails of the pups was genotyped by polymerase chain reaction (PCR) and subsequent sequence analysis. The region surrounding the targeted locus was amplified using GPIIIa forward primer 5′-AACCATGGAAGGACCATGAC-3′ and GPIIIa reverse primer 5′-CACCCCAGTCCTATCCTG-TG-3′. PCR reactions were carried out using Herculase II Fusion polymerase (Agilent, Waldbronn, Germany). PCR products were purified using QiaQuick Spin Column, digested with BamHI (New England Biolabs Inc., Ipswich, MA), analyzed on 2% agarose gels, and sequenced to confirm that the DNA double-strand break had been faithfully repaired.
One-step generation of mice expressing the APLDQ humanized form of muGPIIIa
The CRISPR/Cas9 microinjection cocktail, including gRNA (CTCCTCAGAGCACTCACACA), ssODN (5′-AGCCTTCCAGCCCACGCTGCAACAATGGGAACGGGACTTTTGAGTGTGGGGTGTGCCGCTGTGACCAG-GGCTGGCTGGGGTCCCAATGCGAGTGCTCTGAGGAGGATTACCGACCCTCTCAGCAGGAAGAGTGCAGCCCCAAGGAGGGCCAGCCCATCTGCAGCCA-3′), and Cas-9 protein, was injected into the cytoplasm of fertilized APLD GPIIIa oocytes (supplemental Figure 1). Mice born from the microinjection were screened for the presence of the desired point mutation by PCR and subsequent sequencing analysis. The region surrounding the targeted locus was amplified using GPIIIa forward primer 5′-GAGAAGG-AGCAGTCTTTCA-CTATCAAGCC-3′ and GPIIIa reverse primer 5′-GCAGGAGAAGTCATCGCACTCAC-3′.
Introduction of amino acid substitutions into murine and human GPIIIa plasmids
The complementary DNA expression vector pCMV3-mouse ITGB3, encoding muGPIIIa, was purchased from Creative Biogene (Shirley, NY). Nucleotide substitutions were introduced into this plasmid using a Quick-Change site-directed mutagenesis kit (Stratagene, La Jolla, CA) to convert T30→A, S32→P, Q33→L, and N39→D, resulting in a plasmid encoding muGPIIIa containing a completely humanized PSI domain, termed APLD muGPIIIa. Using this as a template, additional mutations were introduced in the codons encoding M470 and P446 within the murine EGF1 domain to humanize them to Q470 and H446, respectively, with the resulting constructs referred to as APLDQ, APLDH, and APLDQH. Conversely, G463P464→DQ, H446→P, and Q470→M mutations were introduced into the human ITGB3 expression vector pcDNA3-human ITGB3 to generate plasmids encoding human GPIIIa with D463Q464, P446, or M470 within the human EGF1 domain. Primers used to introduce these mutations are listed in Table 1. All constructs and mutations were confirmed by nucleotide sequencing.
Oligonucleotide primers used for site-directed mutagenesis
| Mutation | Orientation | Primer sequence |
|---|---|---|
| Mouse GPIIIa | ||
| T30A S32PQ33L | Forward | 5′-gtgctcagatgagGcCttgCctcTgggctcaccccgatg-3′ |
| Reverse | 5′-catcggggtgagcccAgagGcaaGgCctcatctgagcac-3′ | |
| N39D | Forward | 5′-gggctcaccccgatgtGacctgaaggagaacctg-3′ |
| Reverse | 5′-caggttctccttcaggtCacatcggggtgagccc-3′ | |
| M470Q | Forward | 5′-gaccagggctggctggggtccCAgtgtgagtgctctgaggagg-3′ |
| Reverse | 5′-cctcctcagagcactcacacTGggaccccagccagccctggtc-3′ | |
| P446H | Forward | 5′-gaccccagccagccagggccACagcggcacac-3′ |
| Reverse | 5′-ggtgtgccgctGTggccctggctggctggggtcc-3′ | |
| Human GPIIIa | ||
| Q470M | Forward | 5′-gggcctggctggctgggatccATGtgtgagtgctcagaggaggac-3′ |
| Reverse | 5′-gtcctcctctgagcactcacaCATggatcccagccagccaggccc-3′ | |
| H446P | Forward | 5′-ctgaacctaatagccCtcgctgcaacaatgg-3′ |
| Reverse | 5′-ccattgttgcagcgaGggctattaggttcag-3′ | |
| G463D P464Q | Forward | 5′-gtggggtatgccgttgtgACcAGggctggctgggatcccag-3′ |
| Reverse | 5′-ctgggatcccagccagccCTgGTcacaacggcataccccac-3′ |
| Mutation | Orientation | Primer sequence |
|---|---|---|
| Mouse GPIIIa | ||
| T30A S32PQ33L | Forward | 5′-gtgctcagatgagGcCttgCctcTgggctcaccccgatg-3′ |
| Reverse | 5′-catcggggtgagcccAgagGcaaGgCctcatctgagcac-3′ | |
| N39D | Forward | 5′-gggctcaccccgatgtGacctgaaggagaacctg-3′ |
| Reverse | 5′-caggttctccttcaggtCacatcggggtgagccc-3′ | |
| M470Q | Forward | 5′-gaccagggctggctggggtccCAgtgtgagtgctctgaggagg-3′ |
| Reverse | 5′-cctcctcagagcactcacacTGggaccccagccagccctggtc-3′ | |
| P446H | Forward | 5′-gaccccagccagccagggccACagcggcacac-3′ |
| Reverse | 5′-ggtgtgccgctGTggccctggctggctggggtcc-3′ | |
| Human GPIIIa | ||
| Q470M | Forward | 5′-gggcctggctggctgggatccATGtgtgagtgctcagaggaggac-3′ |
| Reverse | 5′-gtcctcctctgagcactcacaCATggatcccagccagccaggccc-3′ | |
| H446P | Forward | 5′-ctgaacctaatagccCtcgctgcaacaatgg-3′ |
| Reverse | 5′-ccattgttgcagcgaGggctattaggttcag-3′ | |
| G463D P464Q | Forward | 5′-gtggggtatgccgttgtgACcAGggctggctgggatcccag-3′ |
| Reverse | 5′-ctgggatcccagccagccCTgGTcacaacggcataccccac-3′ |
Altered sequences are in upper case.
Expression of WT and mutant αllbβ3 isoforms
HEK 293FT cells were transfected with a plasmid encoding human αIIb together with a plasmid encoding wild-type (WT) or mutant forms of murine or human GPIIIa. HEK 293FT cells were grown in 6-well plates in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum without antibiotics 1 day before transfection to obtain 80% to 90% confluency at the time of transfection. Cells were transfected with 1 µg of each plasmid and 5 µL of Lipofectamine 2000 (Invitrogen) in 250 µL of Opti-MEM I Reduced Serum Medium. After transfection, cells were grown for an additional 48 hours at 37°C to allow for protein expression.
Flow cytometry
Flow cytometric analysis of antibody binding to transiently transfected HEK 293FT cells was performed 48 hours posttransfection using an Accuri C6 flow cytometer (BD Biosciences). Nontransfected cells were used as negative control. Antibody binding was detected using fluorescein isothiocyanate–labeled goat (Fab′)2 anti-human IgG and fluorescein isothiocyanate–labeled goat (Fab′)2 anti-mouse IgG, as appropriate. Data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
Inhibition of PAC-1 binding to human αIIbβIII by anti–HPA-1a alloantibodies
HEK 293FT cells were transfected with WT human αIIbβIII plus EGFP. The cells were preincubated with mAb SZ21, B2G1, or 26.4 at 2.5 μg/mL or with purified total IgG from normal control, PTP, or FNAIT sample at a 1:50 dilution at room temperature for 30 minutes and then incubated for another 30 minutes after adding 2.5 μg/mL PAC-1 with 0.2 mM of Ca+2 and 2 mM of Mn+2. The cells were stained separately with the murine mAb AP3 to detect total β3 surface expression to enable normalization of the binding and competition data. EGFP+ cells were analyzed by flow cytometry after staining with Alexa Fluor 647–conjugated goat anti-mouse IgM (for PAC-1) or Alexa Fluor 647–conjugated goat anti-mouse IgG (for AP3). The mean fluorescence intensity of PAC-1 binding was normalized to β3 expression and presented as a percentage of control in the absence of anti–HPA-1a alloantibodies.
Modified antigen capture enzyme-linked immunosorbent assay
A total of 8 × 107 washed human or murine platelets were incubated at room temperature for 1 hour with human FNAIT alloantisera that had been diluted at a 1:5 ratio, washed, and then lysed in 200 μL of ice-cold lysis buffer (20 mM of Tris [pH, 7.4], 150 mM of sodium chloride, 1% Triton X-100, 1 mM of ethylenediamine-tetraacetic acid, and 10 mM of N-ethylmaleimide, containing a protease inhibitor cocktail; Thermo Fisher Scientific, Waltham, MA). Lysates were added to microtiter wells that had been coated with anti-mouse CD41 (eBioscience, San Diego, CA) to capture immune complexes from mouse platelets or mAb AP2 to capture immune complexes from human platelets. Bound immune complexes were detected using alkaline phosphatase–conjugated anti-human IgG (Jackson ImmunoResearch Laboratories, West Grove, PA).
Molecular modeling and docking
The model of the variable region of B2G1 Fab was generated using the Rosetta Antibody Protocol.25-29 The structures of the PSI and I-EGF1 domains from the crystal structure of αIIbβIII30 (Protein Data Bank code 3FCS) were docked into the CDR loop regions of antibody B2G1 using the ClusPro protein-protein docking server.31-35 Residues A30, P32, and L33 were defined as the docking sites on integrin β3. Noncomplementarity-determining regions were automatically masked using antibody mode.36
Statistics
Data shown are means ± standard errors of the mean. Statistical comparisons were made using an unpaired 2-tailed Student t test. Differences were considered statistically significant at P < .05.
Results
Recreating the HPA-1a epitope in the PSI domain of murine platelet GPIIIa
As illustrated in Figures 1 and 2A, polymorphic amino acid Leu33 is located at the end of a long flexible loop extending from the PSI domain of GPIIIa. Previous studies incorporating a series of amino acid substitutions into a small construct composed of muGPIIIa N-terminal residues 1 to 66 demonstrated that humanizing T30A, S32P, Q33L, and N39D (shown schematically in Figure 2A) are required to reconstitute binding of the type I HPA-1a–selective mAb SZ21 and at least several human polyclonal anti–HPA-1a alloantisera.37 On the basis of these data, a CRISPR strategy was devised (Figure 2B) to introduce a repair template into exon 3 of the murine ITGB3 locus that would encode these 4 amino acid substitutions. From 60 zygotes microinjected with a plasmid construct (Figure 2C) encoding the gRNA shown in Figure 2B, the Cas9 endonuclease, and the APLD homology directed repair template, 1 female offspring gave the appropriately confirmed genotype (Figure 2D-F) and was designated the APLD mouse.
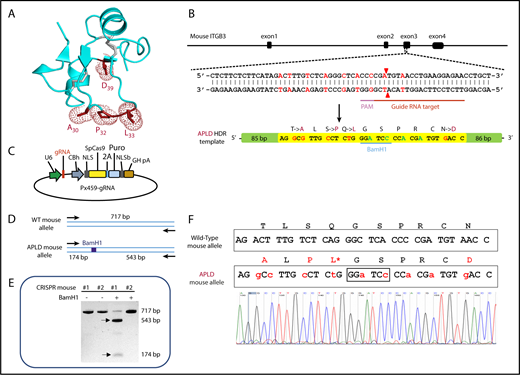
CRISPR-mediated generation of the APLD humanized transgenic mouse. (A) Three-dimensional structure of the GPIIIa PSI domain, showing the location of the residues that were mutated in the murine protein to humanize the 22 to 40 amino acid loop. (B) Schematic illustration of the ITGB3 locus, showing the location of the gRNA binding site (red bar), the protospacer adjacent motif (PAM) sequence (magenta bar), and the Cas9 cleavage site (red arrow heads). A 200-bp APLD homology directed repair (HDR) template was designed to introduce the 4 desired amino acid substitutions (mutated nucleotides labeled in red) and a diagnostic BamH1 restriction site (silent mutation nucleotides labeled in blue) flanked by 80 nucleotide homology arms. The HDR template also introduces nucleotides (green) that encode silent mutations to prevent recleavage by Cas9. (C) The 20-bp gRNA shown in panel B, designed to target the Cas9 nuclease to the ITGB3 gene, was cloned into the BbsI site of the CRISPR vector px459, which also encodes both Cas9 and a puromycin-resistance gene. Pronuclei of C57BL/6N fertilized eggs were microinjected with the px459 plasmid along with the HDR template to generate the humanized APLD mouse. (D) PCR strategy designed to report the incorporation of the HDR template within a 717-bp region surrounding the targeted site of the murine ITGB3 gene. The introduced BamH1 is marked by a blue box. (E) Genotyping of 2 representative pups. Genomic DNA from the pups’ tails was PCR amplified and digested with BamH1 to identify correctly targeted APLD alleles. The PCR product of pup 1 cut with BamH1, demonstrating successful incorporation of the HDR oligo. The arrows indicate the expected BamH1 digestion products. (F) The ITGB3 locus surrounding the genomic editing site was PCR amplified from genomic DNA of pup 1 and subjected to DNA sequence analysis, confirming precise homozygous integration of the human sequence into both alleles of murine ITGB3.
CRISPR-mediated generation of the APLD humanized transgenic mouse. (A) Three-dimensional structure of the GPIIIa PSI domain, showing the location of the residues that were mutated in the murine protein to humanize the 22 to 40 amino acid loop. (B) Schematic illustration of the ITGB3 locus, showing the location of the gRNA binding site (red bar), the protospacer adjacent motif (PAM) sequence (magenta bar), and the Cas9 cleavage site (red arrow heads). A 200-bp APLD homology directed repair (HDR) template was designed to introduce the 4 desired amino acid substitutions (mutated nucleotides labeled in red) and a diagnostic BamH1 restriction site (silent mutation nucleotides labeled in blue) flanked by 80 nucleotide homology arms. The HDR template also introduces nucleotides (green) that encode silent mutations to prevent recleavage by Cas9. (C) The 20-bp gRNA shown in panel B, designed to target the Cas9 nuclease to the ITGB3 gene, was cloned into the BbsI site of the CRISPR vector px459, which also encodes both Cas9 and a puromycin-resistance gene. Pronuclei of C57BL/6N fertilized eggs were microinjected with the px459 plasmid along with the HDR template to generate the humanized APLD mouse. (D) PCR strategy designed to report the incorporation of the HDR template within a 717-bp region surrounding the targeted site of the murine ITGB3 gene. The introduced BamH1 is marked by a blue box. (E) Genotyping of 2 representative pups. Genomic DNA from the pups’ tails was PCR amplified and digested with BamH1 to identify correctly targeted APLD alleles. The PCR product of pup 1 cut with BamH1, demonstrating successful incorporation of the HDR oligo. The arrows indicate the expected BamH1 digestion products. (F) The ITGB3 locus surrounding the genomic editing site was PCR amplified from genomic DNA of pup 1 and subjected to DNA sequence analysis, confirming precise homozygous integration of the human sequence into both alleles of murine ITGB3.
Specific amino acids within the EGF1 domain of GPIIIa are required to support the binding of type II HPA-1a antibodies
Previous studies have shown that the immune response to HPA-1a is both polyclonal and heterogeneous, with some alloantisera containing subpopulations that require, in addition to polymorphic amino acid 33, discontinuous sequences within still-to-be-characterized regions of the linearly distant EGF1 domain.19,38 As shown in Figure 3A, the prototypical type I HPA-1a–specific mAb SZ21 binds readily to APLD but not WT muGPIIIa, confirming re-creation of its epitope within the murine PSI domain. To gain further insight into the structural requirements necessary for the binding of antibody populations likely to exist in more complex polyclonal human maternal anti–HPA-1a alloantisera, we examined the ability of 16 different human FNAIT alloantisera to bind to muGPIIIa, APLD muGPIIIa, or human GPIIIa that had been immobilized in microtiter wells. As shown in Figure 3B, 3 of the 5 representative alloantisera reacted with APLD muGPIIIa, whereas 2 others did not, consistent with the notion that these alloantisera contain a preponderance of so-called type II anti–HPA-1a alloantibodies19 that require residues outside the humanized PSI domain for their binding. The reactivities and specificities of additional human anti–HPA-1a alloantisera are shown in supplemental Figure 2.
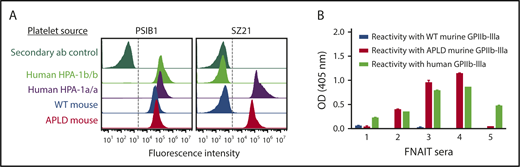
The APLD humanized murine PSI domain supports the binding of many, but not all, human anti–HPA-1a alloantisera. (A) Flow cytometric analysis of the binding of the HPA-1a–selective murine mAb SZ21 to human and mouse platelets. Note that SZ21 binds to human HPA-1a+ but not HPA-1b+ human platelets, demonstrating its alloselectivity, and to APLD but not WT murine platelets. The PSI domain–specific mAb PSIB1 was used as a positive control for expression of GPIIb-IIIa and, as shown, binds all PSI domains, regardless of species or HPA allotype. (B) Antigen capture enzyme-linked immunosorbent assay of anti–HPA-1a maternal alloantisera binding to human and murine forms of GPIIb-IIIa. Five different human FNAIT alloantisera were incubated with human or murine platelet of the indicated phenotype. Platelet/antibody complexes were then detergent lysed and added to microtiter wells that had been coated with either anti-mouse CD41 to capture immune complexes from mouse platelets or mAb AP2 to capture immune complexes from human platelets. Note that human alloantisera 2, 3, and 4 react similarly with human GPIIb-IIIa and APLD murine GPIIb-IIIa, whereas alloantisera 1 and 5 do not react with murine APLD GPIIb-IIIa, suggesting that the preponderance of the HPA-1a–specific alloantibodies present in these polyclonal sera have more complex epitope requirements. None of the FNAIT alloantisera react with WT murine GPIIb-IIIa, as expected.
The APLD humanized murine PSI domain supports the binding of many, but not all, human anti–HPA-1a alloantisera. (A) Flow cytometric analysis of the binding of the HPA-1a–selective murine mAb SZ21 to human and mouse platelets. Note that SZ21 binds to human HPA-1a+ but not HPA-1b+ human platelets, demonstrating its alloselectivity, and to APLD but not WT murine platelets. The PSI domain–specific mAb PSIB1 was used as a positive control for expression of GPIIb-IIIa and, as shown, binds all PSI domains, regardless of species or HPA allotype. (B) Antigen capture enzyme-linked immunosorbent assay of anti–HPA-1a maternal alloantisera binding to human and murine forms of GPIIb-IIIa. Five different human FNAIT alloantisera were incubated with human or murine platelet of the indicated phenotype. Platelet/antibody complexes were then detergent lysed and added to microtiter wells that had been coated with either anti-mouse CD41 to capture immune complexes from mouse platelets or mAb AP2 to capture immune complexes from human platelets. Note that human alloantisera 2, 3, and 4 react similarly with human GPIIb-IIIa and APLD murine GPIIb-IIIa, whereas alloantisera 1 and 5 do not react with murine APLD GPIIb-IIIa, suggesting that the preponderance of the HPA-1a–specific alloantibodies present in these polyclonal sera have more complex epitope requirements. None of the FNAIT alloantisera react with WT murine GPIIb-IIIa, as expected.
To determine the structural requirements for binding of type II anti–HPA-1a antibodies, we examined the binding of the prototypical type II antibody mAb 26.4 to murine APLD platelets. As shown in Figure 4A, similar to human alloantiseras 1 and 5 in Figure 3B, mAb 26.4 was unable to bind murine platelets expressing APLD GPIIIa. Close examination of the interface between the PSI and EGF1 domains (Figure 4B) revealed that a loop extending out from the EGF1 domain of human GPIIIa brings amino acid Q470 into close proximity with polymorphic residue Leu33. This residue is a methionine in muGPIIIa (Ser469 is conserved in both species). To determine whether Q470 forms a part of the epitope recognized by type II anti–HPA-1a antibodies, we further modified the sequence of muGPIIIa, starting with our APLD mouse, by introducing a homology directed repair that would change M470→Q (described in “Materials and methods”) in the murine EGF1 domain. mAb 26.4 now bound readily to platelets from this second-generation HPA-1a humanized transgenic mouse, which we designated the APLDQ mouse (Figure 4A). In contrast, the binding of mAb SZ21 was not enhanced by additional humanization of the EGF1 domain, consistent with its being classified as a type I antibody, the epitope of which is entirely contained within the PSI domain. Unexpectedly, platelets from the APLDQ mouse were completely unreactive with an HPA-1a–specific mAb, termed B2G1, that had been isolated by phage display from an HPA-1a alloimmunized woman,22 demonstrating additional unsuspected complexity in the specificities of antibody subpopulations that can exist in polyclonal maternal anti–HPA-1a alloantisera.
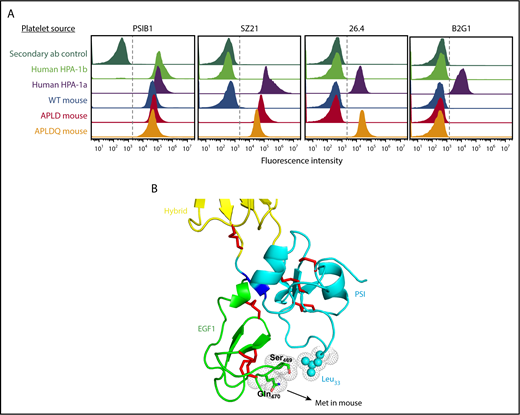
Structural requirements for binding of type II anti–HPA-1a antibodies. (A) Flow cytometric analysis of the reactivity of HPA-1a–specific mAbs with human and mouse platelets. Platelets from the indicated species and having the indicated phenotypes were reacted with mAbs SZ21, 26.4, and B2G1. Note that the type II mAb 26.4 requires that muGPIIIa be humanized from Met to Gln at residue 470 of the EGF1 domain, which is spatially close to the PSI domain, as depicted in panel B. Another type II HPA-1a–specific mAb, B2G1, remains unreactive with APLDQ platelets, highlighting the complexity of binding specificities that are likely present in the polyclonal humoral response to the Leu33Pro polymorphism that controls formation of the HPA-1a epitope.
Structural requirements for binding of type II anti–HPA-1a antibodies. (A) Flow cytometric analysis of the reactivity of HPA-1a–specific mAbs with human and mouse platelets. Platelets from the indicated species and having the indicated phenotypes were reacted with mAbs SZ21, 26.4, and B2G1. Note that the type II mAb 26.4 requires that muGPIIIa be humanized from Met to Gln at residue 470 of the EGF1 domain, which is spatially close to the PSI domain, as depicted in panel B. Another type II HPA-1a–specific mAb, B2G1, remains unreactive with APLDQ platelets, highlighting the complexity of binding specificities that are likely present in the polyclonal humoral response to the Leu33Pro polymorphism that controls formation of the HPA-1a epitope.
Figure 5A highlights the amino acid differences between the murine vs human PSI and EGF1 domains of GPIIIa. As shown, in addition to the Q470M difference that is spatially close to polymorphic residue 33, there are 6 additional amino acid differences in EGF1 between the 2 species. Molecular docking analysis of B2G1with the EGF1 and PSI domains of GPIIIa (Figure 5B) revealed that of these 6 amino acids, only H446 and Q470 are predicted to be at the antibody/antigen interface together with L33. Accordingly, expression of an APLDQ isoform of muGPIIIa with an additional Pro446→His amino acid substitution supported B2G1 binding. Conversely, substituting human H446 with a proline residue resulted in complete loss of B2G1 binding, while both B2G1 and mAb 26.4 lost reactivity with human GPIIIa if Q470 was substituted with a methionine residue. In contrast, none of the HPA-1a–specific antibodies were affected by mutation of G463D and P464Q (Figure 5C), consistent with them not being present at the antibody/antigen interface (Figure 5B). Taken together, these data demonstrate that a variable number of spatially close nonpolymorphic amino acids form multiple epitopes, each centered around polymorphic residue 33, that together comprise the target recognition sites recognized by polyclonal antibody subpopulations present in anti–HPA-1a antisera.
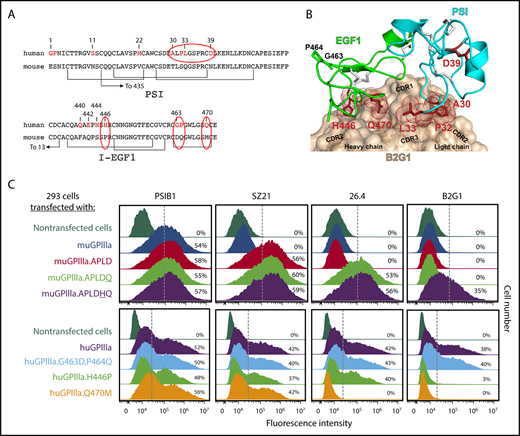
Multiple amino acids within I-EGF1 can contribute to the binding of type II anti–HPA-1a antibodies. (A) Comparison of human vs murine PSI and I-EGF1 domain sequences, with differences highlighted in red. Note especially the APLD sequences in the PSI domain and the Q470M, H446P, G463D, and P464Q differences within EGF1. (B) Structural model of the variable region of antibody B2G1 bound with the β3 PSI and I-EGF1 domains. The antibody is shown as a tan surface with the CDR loops indicated, and the side chains of integrin β3 residues at the antigen-antibody interface are shown as sticks and dots. Note that interface interacting residues include not only polymorphic amino acid 33 but also P32 in the PSI domain and H446 and Q470 of I-EGF1. Also note that G463 and P464 are nowhere near the interface. (C) Top: HEK 293 cells transiently transfected with plasmids expressing human GPIIb and a muGPIIIa isoform that had been mutated to express the indicated humanized amino acid substitution were incubated with the indicated antibodies and subjected to flow cytometric analysis. The PSI domain–specific mAb PSIB1 was used as a control for transfection efficiency. Note that mAb 26.4 requires Q470 for its binding, whereas B2G1 requires both Q470 and H446, as predicted in the docking model in panel B. Bottom: HEK 293 cells transfected with plasmids expressing human GPIIb and a human GPIIIa isoform that had been mutated to express the indicated mouse amino acids were subjected to flow cytometric analysis using the indicated antibodies. Note that the Q470→M mutation results in loss of binding of both 26.4 and B2G1, whereas the H446→P amino acid substitution affects only B2G1.
Multiple amino acids within I-EGF1 can contribute to the binding of type II anti–HPA-1a antibodies. (A) Comparison of human vs murine PSI and I-EGF1 domain sequences, with differences highlighted in red. Note especially the APLD sequences in the PSI domain and the Q470M, H446P, G463D, and P464Q differences within EGF1. (B) Structural model of the variable region of antibody B2G1 bound with the β3 PSI and I-EGF1 domains. The antibody is shown as a tan surface with the CDR loops indicated, and the side chains of integrin β3 residues at the antigen-antibody interface are shown as sticks and dots. Note that interface interacting residues include not only polymorphic amino acid 33 but also P32 in the PSI domain and H446 and Q470 of I-EGF1. Also note that G463 and P464 are nowhere near the interface. (C) Top: HEK 293 cells transiently transfected with plasmids expressing human GPIIb and a muGPIIIa isoform that had been mutated to express the indicated humanized amino acid substitution were incubated with the indicated antibodies and subjected to flow cytometric analysis. The PSI domain–specific mAb PSIB1 was used as a control for transfection efficiency. Note that mAb 26.4 requires Q470 for its binding, whereas B2G1 requires both Q470 and H446, as predicted in the docking model in panel B. Bottom: HEK 293 cells transfected with plasmids expressing human GPIIb and a human GPIIIa isoform that had been mutated to express the indicated mouse amino acids were subjected to flow cytometric analysis using the indicated antibodies. Note that the Q470→M mutation results in loss of binding of both 26.4 and B2G1, whereas the H446→P amino acid substitution affects only B2G1.
Discussion
Early studies aimed at characterizing the molecular nature of the HPA-1a epitope found that tryptic or chymotryptic proteolytic fragments of GPIIIa, ranging from 17 kDa39 to 66 kDa40 in size, could bind HPA-1a–specific alloantibodies. Later studies by Beer and Coller41 found that the 66-kDa polypeptide is composed of the 17-kDa amino terminal fragment of GPIIIa (now known to contain the PSI domain) disulfide bonded to a larger 50-kDa fragment containing residues 348 to 654 (now known to contain the EGF1 domain). After the discovery that the formation of the HPA-1a epitope is controlled by a Leu33Pro amino acid substitution at the amino terminus of GPIIIa,13,14 small synthetic peptides surrounding this polymorphic residue were synthesized but were unable to bind HPA-1a alloantibodies,42 likely as a result of the inability of linear peptides to fold and adopt the proper tertiary conformation, because there are 7 cysteine residues within the first 55 amino acids of GPIIIa that form a complex disulfide-bonded knot-like structure. Interestingly, a somewhat larger recombinant protein composed of the first 66 amino acids of GPIIIa (ie, the entire PSI domain) produced in prokaryotic λgt22 bacteriophage plaques was able to react with 4 different anti–HPA-1a sera from PTP patients,43 thereby localizing the HPA-1a epitope to the amino terminal 7 kDa of GPIIIa surrounding polymorphic amino acid 33.
Two studies published in the mid 1990s revealed that the HPA-1 epitope recognized by a subset of HPA-1a antibodies might be more complex. Valentin et al19 used site-directed mutagenesis to disrupt the disulfide bond linking the PSI domain to the EGF1 domain of GPIIIa and found that although some anti–HPA-1a alloantibodies continued to bind well, nearly one third lost some or all reactivity with the mutant protein. On the basis of these findings, the authors proposed that HPA-1a antibodies can be classified as type I or type II based upon their dependence on noncontiguous linear sequences present in the PSI and EGF1 domains. This concept was supported by the work of Stafford et al,44 who found that ∼20% of 121 maternal anti–HPA-1a alloantibodies reacted with recombinant fragments of GPIIIa only when the fragment contained both the PSI and EGF1 domains. Honda et al38 detected the presence of type II antibodies that reacted with chimeric proteins composed of Xenopus GPIIIa molecules containing various patches of human GPIIIa sequences only when the Xenopus protein contained human amino acids 26 to 38 as well as amino acids 287 to 490.
HPA-1a antibody titer alone has not consistently been found to correlate with the severity of clinical outcome,45,46 and additionally, dividing HPA-1a–specific alloantibodies into type I vs type II disappointingly provided neither a diagnostic nor prognostic advantage.44 Recently, however, Santoso et al47 reported that a specific population of anti–HPA-1a alloantibodies preferentially bind GPIIIa when it is complexed with the αv rather than αIIb integrin subunit, present on endothelial cells, and that such antibodies are strongly associated with the development of intracranial hemorrhage in FNAIT. These findings have several important implications. First, they strongly suggest that identifying and distinguishing between distinct populations of anti–HPA-1a antibodies that invariably exist within all maternal polyclonal anti–HPA-1a antisera may be the keys for predicting the risk of thrombocytopenia and bleeding in cases of FNAIT. Second, they demonstrate that the influence of the local conformation surrounding polymorphic amino acid residue 33 has a profound effect on determining the core target recognition site for alloantibody binding and its subsequent effector consequences. Our finding that the binding of 2 different type II monoclonal anti–HPA-1a antibodies can be distinguished from one another by their requirement for distinct amino acids within the EGF1 domain of GPIIIa (Figure 5) lends further support to the notion that antibody/epitope recognition involves more than simply the polymorphic amino acid and likely varies among antibody subpopulations that comprise virtually any alloimmune response. Mapping the polyclonal immune response to HPA-1a using cells expressing muGPIIIa containing specific mouse→human amino acid substitutions, together with the growing number of HPA-1a–specific monoclonal antibodies, may enable high-resolution analysis of alloantibody subpopulations to provide a predictive diagnostic benefit. Interestingly, preliminary studies (supplemental Figure 3) indicate that type I and type II alloantibody populations have distinct effects on the ability of platelets to interact with their ligand. Whereas type I antibodies have only minimal effects, type II antibodies significantly block the binding of the fibrinogen mimetic PAC-1 to the GPIIb-IIIa complex, perhaps by restraining extension of GPIIIa during the integrin activation process.48 Additional studies in this regard are the subject of an extensive planned clinical investigation.
That individual antibody populations within a given polyclonal serum have different surface topographical requirements explains why they are able to induce varying pathophysiological effects. In the world of histocompatibility testing, there is growing evidence that, in addition to genotypic matching of cell surface antigens, phenotypic determination of the antibody/epitope repertoire of the recipient, including for those epitopes contributed by residues in discontinuous positions that cluster together on the molecular surface, may be an important predictor of transplantation success.49 Structure-based matching has already been validated as a strategy to improve platelet transfusion support in refractory thrombocytopenic patients.50,51 It is possible, therefore, that precision medicine–based diagnostic regimens that consider not only polymorphic differences but also the contact areas of alloantibody subpopulations will be needed to offer a more precise dissection of the polyclonal nature of the immune response, allowing more accurate prediction of the risk of thrombocytopenia, bleeding, and intracranial hemorrhage.
The polyclonal nature of the response generated by the clinically important Leu33Pro polymorphism in GPIIIa is complex and remains a fascinating area of investigation, with implications for both prophylaxis and therapy. Given the polyclonal nature of HPA-1–specific antibodies and the likelihood that any maternal antiserum contains antibodies that come at polymorphic amino acid 33 from different angles and bind with different topographical distributions and with different affinities as a result of the involvement of additional residues, we suspect that a mixture of HPA-1–specific mAbs, rather than any single mAb, may be required to block the binding of polyclonal maternal antibodies and prevent clearance of fetal platelets from circulation. Identification of 2 residues (H446 and Q470) within EGF1 as both necessary and sufficient for binding of the type II anti–HPA-1a alloantibody does not rule out the possibility that residues within or outside of EGF1 might be required to support the binding of still-to-be-characterized type II alloantibodies. For example, D39 within the PSI domain and R93 at the hybrid/PSI interface have both been reported to affect the binding of human anti–HPA-1a antibodies,37,52 whereas other antibodies are specific for the bent conformation of the integrin, likely because of their requirement for both the PSI and EGF1 domains, as described in this study.53 Our atomic-level dissection demonstrating an increasingly wide range of antibody subpopulations present within the alloantisera of HPA-1a alloimmunized individuals highlights the challenge of developing single reagents with narrow epitope specificities to inhibit alloantibody-mediated platelet destruction. We will have to get lucky. It is possible, however, that prophylactic delivery of humanized anti–HPA-1a–specific mAbs, introduced into the maternal circulation during pregnancy or shortly after childbirth, could be used to clear neonatal platelets that have passed through the mother, thereby preventing or lessening development of the alloimmune response in the first place. An ounce of prevention may be worth a pound of cure.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Debra Newman (Blood Research Institute, BloodCenter of Wisconsin) and David Wilcox (Medical College of Wisconsin) for helpful comments, Cedric Ghevaert for mAb B2G1, Heyu Ni (University of Toronto) for supplying the PSI domain–specific mAb PSIB1, and Richard Aster and Dan Bougie (Blood Research Institute, BloodCenter of Wisconsin) for supplying human FNAIT alloantisera. The authors also thank the Transgenic Mouse Core Laboratory for its help in generating APLD mice.
This work was supported by grants HL-130054 and R35 HL139937 (P.J.N.) and HL-131836 (J.Z.) from the National Heart, Lung, and Blood Institute of the National Institutes of Health and by grants HNF1354-17 (B.S.) and SFP1281-16 (M.T.A.) from Helse Nord RHF.
Authorship
Contribution: H.Z., M.T.A., A.M.M.T., J.Z., and H.W. designed and performed experiments and analyzed data; B.R.C. and B.S. provided essential reagents and analyzed and interpreted data; J.Z. and P.J.N. designed experiments and analyzed data; and H.Z., M.T.A., A.M.M.T., J.Z., and P.J.N. wrote the paper.
Conflict-of-interest disclosure: B.R.C. consults for Prophylix Pharma AS. B.S. is a founder and stock owner of Prophylix Pharma AS. The remaining authors declare no competing financial interests.
Correspondence: Peter J. Newman, Blood Research Institute, BloodCenter of Wisconsin, 638 North 18th St, Milwaukee, WI 53201; e-mail: peter.newman@bcw.edu.

