

Key Points
DGKζ is a novel negative regulator of GPVI-mediated platelet activation.
Loss of DGKζ upregulates surface expression of GPVI in platelets and megakaryocytes.

Abstract
Diacylglycerol kinases (DGKs) are a family of enzymes that convert diacylglycerol (DAG) into phosphatidic acid (PA). The ζ isoform of DGK (DGKζ) has been reported to inhibit T-cell responsiveness by downregulating intracellular levels of DAG. However, its role in platelet function remains undefined. In this study, we show that DGKζ was expressed at significant levels in both platelets and megakaryocytes and that DGKζ-knockout (DGKζ-KO) mouse platelets were hyperreactive to glycoprotein VI (GPVI) agonists, as assessed by aggregation, spreading, granule secretion, and activation of relevant signal transduction molecules. In contrast, they were less responsive to thrombin. Platelets from DGKζ-KO mice accumulated faster on collagen-coated microfluidic surfaces under conditions of arterial shear and stopped blood flow faster after ferric chloride–induced carotid artery injury. Other measures of hemostasis, as measured by tail bleeding time and rotational thromboelastometry analysis, were normal. Interestingly, DGKζ deficiency led to increased GPVI expression on the platelet and megakaryocyte surfaces without affecting the expression of other platelet surface receptors. These results implicate DGKζ as a novel negative regulator of GPVI-mediated platelet activation that plays an important role in regulating thrombus formation in vivo.
Introduction
Platelets circulate in blood vessels, serving as cellular sentries for physical or chemical vascular injury. Should the endothelial cell lining become damaged, platelets interact via their glycoprotein Ib (GPIb)–V-IX receptor complex with collagen-bound von Willebrand factor,1 tethering them to the subendothelial lining, and cell surface GPVI can interact with collagen. GPVI/collagen interactions initiate signaling events that lead to the generation of second messengers, such as Ca2+ and diacylglycerol (DAG).2 DAG is generated from phosphatidyl inositol 4,5 bisphosphate by the β isoform of phospholipase C (PLCβ) downstream of G protein–coupled receptors and by the γ2 isoform of PLC (PLCγ2) downstream of (hem) immunoreceptor tyrosine–based activation motif receptors.3 DAG is also formed through dephosphorylation of phosphatidic acid (PA) by PA phosphatase.4 DAG binds to and activates multiple isoforms of the serine/threonine protein kinase PKC, which in turn phosphorylates a wide range of substrates, ultimately leading to integrin activation, granule secretion, and other platelet activation events that stabilize the hemostatic thrombus, sometimes referred to as the platelet plug.3,5 Therefore, regulation of intracellular DAG level is an important early event in the platelet activation process.
Diacylglycerol kinases (DGKs) are a family of enzymes thought to suppress cellular activation by reducing the level of DAG via conversion of DAG to PA.6-8 The DGK family consists of 10 isozymes (α, β, γ, δ, η, κ, ε, ζ, ι, and θ) that are divided into 5 classes based on structure. DGK proteins have been isolated from human platelets,9 and reverse transcription polymerase chain reaction (RT-PCR) analysis showed that human platelets express most of the isozymes, with the exception of DGKβ and -θ.10 Type I DGKs (DGKα, -β, and -γ) have been the most studied, largely because of the availability of inhibitors, and have been reported to have functional roles in human platelets. For example, treating human platelets with the type I DGK inhibitor R59949 resulted in enhanced platelet aggregation in response to vasopressin, collagen, and U46619.11,12 Although R59949 had no effect on thrombin-mediated platelet aggregation, it reduced calcium mobilization after thrombin stimulation, suggesting that type I DGKs differentially regulate GPVI- vs thrombin-mediated platelet activation.10 R59022, another type 1 DGK inhibitor, has variously been shown to suppress13 or potentiate14 thrombin-induced platelet aggregation. However, discrepant findings from these studies have resulted in confusion regarding the actual role of the various DGK isozymes in platelet activation.
DGKζ is a type IV DGK that is widely expressed in different cell types and tissues.15-19 T cells are known to express high levels of DGKζ,7,20 and DGKζ-knockout (DGKζ-KO) mice exhibit a mild increase in the number of CD8+ T cells, together with elevated intracellular levels of DAG and enhanced Ras-ERK signaling after T-cell receptor engagement.19,21,22 DGKζ-KO T cells also show increased upregulation of activation markers, proliferation, and enhanced responses to pathogens. Interestingly, RNA-sequencing analysis revealed that DGKζ is the most abundant DGK isozyme in mouse platelets and the third most abundant in human platelets.23 However, whether DGKζ is expressed in platelets at the protein level and whether it plays any role in thrombosis and hemostasis are unknown. Therefore, the purpose of the present study was to examine these issues and determine the functional consequences of DGKζ deficiency.
Methods
Mice
DGKζ-KO mice (C57BL/6J background) have previously been described.19 Wild-type (WT) C57BL/6J mice were obtained from littermate controls or purchased from The Jackson Laboratory (Bar Harbor, ME). Mice were maintained in the Biological Resource Center at the Medical College of Wisconsin. All animal protocols were approved by the Medical College of Wisconsin Institutional Animal Care and Use Committee. All studies were performed using mice of either sex between the ages of 10 and 20 weeks.
Blood parameter analysis
Blood was drawn from the inferior vena cava, and blood cell counts were measured using the scil Vet ABC Hematology Analyzer (scil Corporation, Gurnee, IL).
Platelet flow cytometry
Platelet surface receptor expression was measured in whole blood after staining with the indicated fluorescein isothiocyanate–conjugated antibodies using the BD Accuri C6 Plus (BD Biosciences). Data were analyzed using FlowJo software (Tree Star, Ashland, OR). The extent of agonist-induced platelet activation was assessed by staining agonist-stimulated washed platelets with activation-specific markers (fluorescein isothiocyanate–conjugated anti–P-selectin or phycoerythrin-conjugated Jon/A).
Mouse platelet isolation
Washed platelets were prepared as previously described24 with modification in Tyrode’s buffer (137 mM of sodium chloride, 2.5 mM of potassium chloride, 0.36 mM of sodium dihydrogen phosphate dihydrate, 13.8 mM of sodium bicarbonate, 20 mM of N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, and 0.1% glucose). Washed platelets were resuspended in Tyrode’s buffer to an appropriate concentration for the experiments.
Western blotting
Washed platelets were stimulated with collagen-related peptide (CRP; 1 μg/mL) at 37°C with stirring for the indicated times, and reactions were terminated by adding an equal volume of ice-cold 2× lysis buffer (300 mM of sodium chloride, 20 mM of tris[hydroxymethyl]aminomethane, 2 mM of EGTA, 2 mM of EDTA, and 2% NP40 [pH, 7.5]). The samples were diluted with an equal volume of 2× sample buffer (4% sodium dodecyl sulfate, 10% 2-mercaptoethanol, 20% glycerol, and 50 mM of tris[hydroxymethyl]aminomethane [pH, 6.8]), separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred to a poly(vinylidene difluoride) membrane. Western blotting was performed with the indicated antibodies. Bands were quantitated by densitometry using IMAGE J software (National Institutes of Health, Bethesda, MD).
Platelet aggregation and secretion
Aggregation was monitored by light transmission with a Born Lumi-Aggregometer (Chrono-log Corporation). Platelet adenosine triphosphate release was measured using luciferin/luciferase reagent (Chrono-lume; Chrono-log Corporation).
Platelet spreading
Eight-chamber glass tissue culture slides (Corning, Corning, NY) were coated with 1% bovine serum albumin (BSA), 50 μg/mL of collagen, 50 μg/mL of CRP, or 30 μg/mL of fibrinogen at 4°C overnight. Wells were blocked with 1% BSA. Washed platelets (200 μL; 2 × 107/mL) in Tyrode’s buffer supplemented with 1 mM of calcium chloride were allowed to spread for the indicated times at 37°C. Adherent platelets were fixed with 5% neutral buffered formalin and permeabilized with 0.1% Triton-X100. The fixed platelets were stained with phalloidin-TRITC (10 μg/mL). Images were taken with an inverted microscope (Nikon, Melville, NY). Platelet spreading area was analyzed using IMAGE J software, and results are reported as the mean area of spread platelets (μm2 per platelet).
Tail bleeding assay
Blood loss was measured in 10- to 15-week-old WT and DGKζ-KO mice after excision of a tail at a distance of 1 mm from the tip. The time to cessation of bleeding was recorded.
ROTEM analysis
Whole blood was collected from the vena cava and diluted at a ratio of 1:9 (volume/volume) with 3.8% sodium citrate. Thrombocytopenic blood was prepared as previously described.25 Hemostasis in mouse whole blood at the described platelet count was measured by rotational thromboelastometry (ROTEM) using the ROTEM delta (ROTEM, Durham, NC).
FeCl3-induced thrombus formation
Mice were anesthetized, and the right carotid arteries were detached from surrounding tissues. Vascular injury was induced by applying a filter paper saturated with 10% ferric chloride (FeCl3) to the top of the vessel for 3 minutes. A microvascular flow probe attached to a transit-time perivascular flowmeter (Transonic, Ithaca, NY) was positioned on the carotid artery to monitor blood flow. Blood flow was recorded from immediately after removing the filter paper until 3 minutes after full occlusion. The time to first occlusion was defined by blood flow of <0.05 mL per minute.
In vitro platelet adhesion and thrombus formation in microfluidic chambers
Whole blood was collected and anticoagulated with heparin/PPACK and labeled with mepacrine (Calbiochem). Thrombus formation on collagen was performed as previously described24 on collagen (50 μg/mL) at an arterial sheer rate of 1500 s−1. Images of platelet adhesion and thrombus formation were acquired by epifluorescence microscopy at a rate of 1 frame per second. Platelet accumulation was analyzed using IMAGE J software, and results are reported as the mean area covered by platelet aggregates (%). For 3-dimensional image analysis, microchannels were washed with phosphate-buffered saline at the end of flow analysis and fixed, permeabilized, and stained with phalloidin-TRITC. Z stack images were taken using step changes of 1.16 μm with an FV1000 Olympus laser scanning confocal microscope (Olympus, Tokyo, Japan) and analyzed using Slidebook 6 software (Intelligent Imaging Innovations, Denver, CO). Results were reported as thrombus volume and height.
Mouse megakaryocyte isolation and culture
Bone marrow cells were collected and resuspended in ammonium-chloride-potassium buffer (0.15 M of ammonium chloride, 1 mM of potassium bicarbonate, and 0.1 mM of disodium EDTA [pH, 7.3]) to remove red cells. Cells expressing Gr-1, CD11b, B220, and CD16/32 were depleted using magnetic Dynabeads. The remaining cells were cultured in complete Stempro medium (Gibco, Grand Island, NY) supplemented with 20 ng/mL of murine stem cell factor (SCF) for 2 days and then further cultured in the presence of 20 ng/mL of murine SCF and 50 ng/mL of murine thrombopoietin (TPO). After 5 days of culture in SCF/TPO-supplemented medium, mature megakaryocytes were enriched by BSA gradient.
Quantitative RT-PCR
RNA was isolated from mature megakaryocytes with the RNeasy Mini Plus Kit (Qiagen, Germantown, MD). RNA was then converted to complementary DNA using the SuperScript III Kit (Invitrogen). RT-PCR was performed in triplicate using the PrimeTime Std qPCR Assay (IDT, San Jose, CA) with Brilliant II SYBR Green QPCR Master Mix (Agilent Technologies, Santa Clara, CA) in an ABI 7500 (Applied Biosystems, Foster City, CA). Transcripts for hypoxanthine-guanine phosphoribosyltransferase, integrin αIIb, GPIbα, GPVI, integrin α2, FcRγ chain, and DGKζ were analyzed. Relative gene expression levels were calculated using the comparative CT method, using hypoxanthine-guanine phosphoribosyltransferase for normalization.
Statistics
All experiments were performed >3 times, with data shown as the mean ± standard error of the mean (SEM). Statistical analysis was performed using unpaired Student t test using Prism 6.0 software (GraphPad, San Diego, CA).
Results
DGKζ-KO platelets are hyperreactive to GPVI agonists
Splice variants of DGKζ having different N-termini have been identified in T cells and macrophages.19,26 As shown in Figure 1A, only the short isoform was present in WT mouse platelets, and this was absent in DGKζ-KO mouse platelets. However, WT mouse megakaryocytes expressed both the long and short isoforms, both of which were absent in DGKζ-KO megakaryocytes (Figure 1A). Expression of other DGK isozyme expressions (α, γ, and ε) were comparable between WT and DGKζ-KO platelets (supplemental Figure 1A). DGKζ-KO mice had normal platelet counts and mean platelet volumes, indicating that they have normal thrombopoiesis (Figure 1B). No differences in other blood parameters, such as white or red blood cell count, were observed (Table 1).
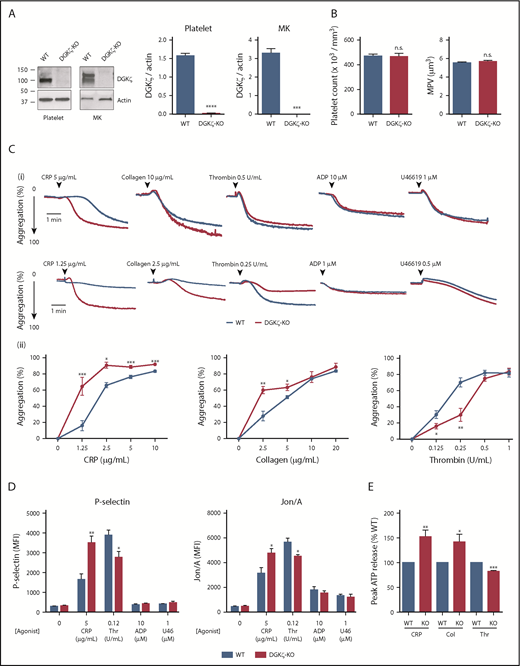
DGKζ negatively regulates GPVI-mediated platelet activation but enhances thrombin (Thr)-mediated platelet activation. (A) Whole lysates of washed platelets and megakaryocytes (MKs) from WT and DGKζ-KO mice were analyzed by western blotting for DGKζ and actin. The blot shown on the left is representative of 3 to 4 independent experiments. Quantification of band densities (right) is reported as the mean ± SEM (n = 3-4). (B) Platelet counts (left) and mean platelet volumes (MPVs; right) observed in WT and DGKζ-KO mice. Values represent means ± SEM (n = 20). (Ci) Aggregation of washed platelets from WT and DGKζ-KO mice was measured by lumiaggregometry in response to high (top) and low (bottom) doses of CRP, collagen (Col), Thr, adenosine 5′-diphosphate (ADP), and U46619. Each aggregation tracing is representative of 3 to 5 independent experiments. (ii) Dose-response curve of maximal platelet aggregation after CRP, Col, and Thr stimulation. Values represent the mean ± SEM of maximal platelet aggregation observed in 3 to 4 independent experiments. (D) P-selectin exposure (left) and Jon/A binding as a reporter of activation of integrin αIIbβ3 (right) were measured by flow cytometry in washed platelets after 20 minutes of stimulation with CRP, Thr, ADP, and U46619 (U46) at the indicated concentrations. Values represent the mean ± SEM of mean fluorescence intensity (MFI) observed in 5 independent experiments. (E) Quantification of peak adenosine triphosphate (ATP) released from platelets stimulated with CRP (2.5 μg/mL), Col (5 μg/mL), and Thr (0.5 U/mL) relative to WT. ATP release was measured by lumiaggregometry. Values represent the mean ± SEM observed in 3 to 5 independent experiments. Statistical analysis was performed by the unpaired Student t test. *P < .05, **P <.01, ***P < .001, ****P < .0001 of DGKζ-KO as compared with WT. n.s., not significant.
DGKζ negatively regulates GPVI-mediated platelet activation but enhances thrombin (Thr)-mediated platelet activation. (A) Whole lysates of washed platelets and megakaryocytes (MKs) from WT and DGKζ-KO mice were analyzed by western blotting for DGKζ and actin. The blot shown on the left is representative of 3 to 4 independent experiments. Quantification of band densities (right) is reported as the mean ± SEM (n = 3-4). (B) Platelet counts (left) and mean platelet volumes (MPVs; right) observed in WT and DGKζ-KO mice. Values represent means ± SEM (n = 20). (Ci) Aggregation of washed platelets from WT and DGKζ-KO mice was measured by lumiaggregometry in response to high (top) and low (bottom) doses of CRP, collagen (Col), Thr, adenosine 5′-diphosphate (ADP), and U46619. Each aggregation tracing is representative of 3 to 5 independent experiments. (ii) Dose-response curve of maximal platelet aggregation after CRP, Col, and Thr stimulation. Values represent the mean ± SEM of maximal platelet aggregation observed in 3 to 4 independent experiments. (D) P-selectin exposure (left) and Jon/A binding as a reporter of activation of integrin αIIbβ3 (right) were measured by flow cytometry in washed platelets after 20 minutes of stimulation with CRP, Thr, ADP, and U46619 (U46) at the indicated concentrations. Values represent the mean ± SEM of mean fluorescence intensity (MFI) observed in 5 independent experiments. (E) Quantification of peak adenosine triphosphate (ATP) released from platelets stimulated with CRP (2.5 μg/mL), Col (5 μg/mL), and Thr (0.5 U/mL) relative to WT. ATP release was measured by lumiaggregometry. Values represent the mean ± SEM observed in 3 to 5 independent experiments. Statistical analysis was performed by the unpaired Student t test. *P < .05, **P <.01, ***P < .001, ****P < .0001 of DGKζ-KO as compared with WT. n.s., not significant.
Blood parameters of WT and DGKζ-KO mice (n = 20)
| Blood parameter | Mean ± SEM | ||||
|---|---|---|---|---|---|
| Platelet count, 106/mL | Platelet volume, μm3 | WBC count, 106/mL | RBC count, 109/mL | Hematocrit, % | |
| WT | 469.7 ± 16.2 | 5.5 ± 0.1 | 2.9 ± 0.3 | 7.0 ± 0.2 | 33.4 ± 0.8 |
| DGKζ-KO | 466.8 ± 24.2 | 5.7 ± 0.1 | 2.7 ± 0.2 | 6.9 ± 0.2 | 33.9 ± 0.9 |
| Blood parameter | Mean ± SEM | ||||
|---|---|---|---|---|---|
| Platelet count, 106/mL | Platelet volume, μm3 | WBC count, 106/mL | RBC count, 109/mL | Hematocrit, % | |
| WT | 469.7 ± 16.2 | 5.5 ± 0.1 | 2.9 ± 0.3 | 7.0 ± 0.2 | 33.4 ± 0.8 |
| DGKζ-KO | 466.8 ± 24.2 | 5.7 ± 0.1 | 2.7 ± 0.2 | 6.9 ± 0.2 | 33.9 ± 0.9 |
RBC, red blood cell; WBC, white blood cell.
To determine the consequences of DGKζ deficiency for platelet function, the responsiveness of DGKζ-KO vs WT platelets after stimulation with platelet agonists was assessed. As shown in Figure 1C, platelet aggregation in response to collagen or the GPVI-specific agonist CRP was potentiated in DGKζ-KO relative to WT platelets. In contrast, the response to a submaximal concentration of thrombin or the PAR4-activating peptide (AYPGKG-NH2; supplemental Figure 1B) was reduced in DGKζ-KO relative to WT platelets. Responses to ADP and the thromboxane A2 mimetic U46619 were unaffected by DGKζ deficiency. Consistent with the aggregation results, P-selectin exposure, activation of integrin αIIbβ3 (Jon/A binding), and dense granule secretion assessed by adenosine triphosphate were elevated after CRP stimulation but reduced after thrombin stimulation and were the same after ADP or U46619 stimulation in DGKζ-KO relative to WT platelets (Figure 1D-E; supplemental Figure 1C). Calcium mobilization after CRP stimulation was also enhanced in DGKζ-KO platelets (supplemental Figure 1D). As reported in type I DGK inhibitor–treated human platelets, thrombin-mediated calcium mobilization was reduced in DGKζ-KO platelets.
To determine whether other DGKs have an additive effect on platelet activation, we treated WT and DGKζ-KO platelets with a type I DGK inhibitor. Treatment with the type I DGK inhibitor R59022 did not affect CRP- or thrombin-mediated platelet activation, suggesting that DGKζ is a major isozyme regulating mouse platelet activation (supplemental Figure 1E). To determine whether the differential effect of DGKζ disruption on GPVI- and PAR-mediated platelet activation is due to the differential role of DAG in these pathways, platelet aggregation in DAG-treated platelets was analyzed. DAG treatment potentiated both CRP- and thrombin-mediated platelet activation, suggesting that DAG is a positive regulator for both pathways (supplemental Figure 1F).
Consistent with the aggregation and secretion results, DGKζ-KO platelets spread faster and to a greater extent on both collagen and CRP and, somewhat unexpectedly, on immobilized fibrinogen as well (Figure 2A-B). Spreading on either of these substrates is known to be enhanced by secreted ADP.3,27 In the case of GPVI, ADP is released as a result of interactions between GPVI/FcRγ chain and collagen. In αIIbβ3/fibrinogen interactions, it is released as a consequence of αIIbβ3-mediated outside-in signal amplification, which, like signaling through GPVI, utilizes immunoreceptor tyrosine–based activation motif–mediated activation of the Syk→PLCγ2 pathway that generates IP3 and DAG.24,28 Addition of ADP or thrombin to either WT or DGKζ-KO platelets (supplemental Figure 2) normalized the spreading response, demonstrating that the enhanced spreading of DGKζ-KO platelets is due to enhanced signal amplification.

DGKζ-KO platelets exhibit enhanced platelet spreading on substrates for both GPVI and integrin αIIbβ3. Washed platelets from WT and DGKζ-KO mice were allowed to adhere on immobilized CRP (50 μg/mL), collagen (50 μg/mL), or fibrinogen (30 μg/mL) for the indicated times at 37°C. After washing with phosphate-buffered saline to remove unbound platelets, adherent platelets were fixed and stained with phalloidin-TRITC. (A) Images of spread platelets representative of 3 independent experiments are shown. Bars represent 10 μm. (B) The average area covered by individual platelets was quantified from at least 4 images per substrate and 250 to 500 platelets per time point. Spreading area is reported as the mean ± SEM. Statistical analysis was performed using the unpaired Student t test. ****P < .0001 of DGKζ-KO as compared with WT.
DGKζ-KO platelets exhibit enhanced platelet spreading on substrates for both GPVI and integrin αIIbβ3. Washed platelets from WT and DGKζ-KO mice were allowed to adhere on immobilized CRP (50 μg/mL), collagen (50 μg/mL), or fibrinogen (30 μg/mL) for the indicated times at 37°C. After washing with phosphate-buffered saline to remove unbound platelets, adherent platelets were fixed and stained with phalloidin-TRITC. (A) Images of spread platelets representative of 3 independent experiments are shown. Bars represent 10 μm. (B) The average area covered by individual platelets was quantified from at least 4 images per substrate and 250 to 500 platelets per time point. Spreading area is reported as the mean ± SEM. Statistical analysis was performed using the unpaired Student t test. ****P < .0001 of DGKζ-KO as compared with WT.
DGKζ-KO mice exhibit faster time vessel occlusion after arterial injury
Given the opposing effects of DGKζ deficiency on in vitro measures of GPVI- and thrombin-mediated platelet activation, we investigated the consequences of DGKζ deficiency for hemostasis in vivo. Tail resection bleeding time is highly dependent on thrombin-mediated platelet activation,29,30 and ROTEM analyses used to support decisions made before surgery or to monitor anticoagulant therapy are initiated by addition of tissue factor to drive thrombin-mediated platelet activation as well. As shown in Figure 3A, DGKζ-KO and WT mice had similar tail bleeding times, and blood from these mice showed no significant difference in ex vivo hemostatic plug formation in ROTEM chambers (Figure 3B), either under conditions of a normal platelet count (200 × 106/mL) or thrombocytopenia (50 × 106/mL). Taken together, these findings demonstrate that DGKζ deficiency does not affect hemostasis initiated by thrombin. Notably, however, DGKζ deficiency markedly improved time to vessel occlusion after FeCl3-induced carotid artery injury (Figure 3C-D), a GPVI-dependent event initiated by exposure of collagen at the site of injury.31,32 Supplemental Figure 3 shows that the endothelial lining was disrupted and that collagen was exposed at the site of oxidative injury under the conditions employed in our FeCl3 injury model. In vitro, DGKζ-KO platelets adhered to a collagen-coated surface more rapidly and to a greater extent under arterial shear (Figure 4A-B), whereas the extent of thrombus growth after initial adhesion (a measure of thrombotic tendency) was not different from that of WT platelets (Figure 4C). These data are consistent with the platelet aggregation results (Figure 1) showing enhanced platelet reactivity to collagen.
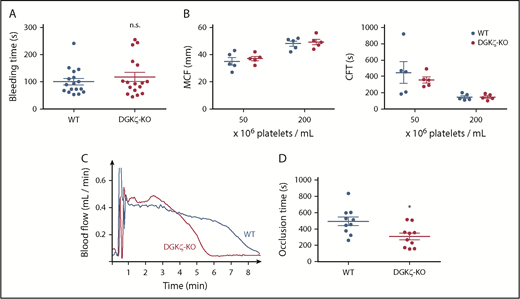
DGKζ-KO mice exhibit normal hemostasis but faster time to platelet plug formation after arterial injury. (A) Bleeding assay was performed by tail tip amputation followed by immersion in phosphate-buffered saline at 37°C. Symbols represent times to cessation of bleeding for individual animals, and error bars represent the mean ± SEM (n = 10-12). (B) ROTEM was performed using whole blood collected from WT and DGKζ-KO mice and adjusted to the indicated platelet count. Results are shown as maximal clot firmness (MCF; n = 5; left) and clot formation time (CFT; n = 5; right). Symbols represent MCF and CFT for individual animals, and bars represent the mean ± SEM. (C-D) The right carotid artery was injured by applying 10% FeCl3 for 3 minutes. Blood flow was monitored with a Doppler flow probe until 3 minutes after full occlusion. (C) Representative flow traces for WT and DGKζ-KO mice. (D) Symbols represent times to occlusion in individual animals, and bars represent the mean ± SEM (n = 10). Statistical analysis was performed by the unpaired Student t test. *P < .05 of DGKζ-KO as compared with WT.
DGKζ-KO mice exhibit normal hemostasis but faster time to platelet plug formation after arterial injury. (A) Bleeding assay was performed by tail tip amputation followed by immersion in phosphate-buffered saline at 37°C. Symbols represent times to cessation of bleeding for individual animals, and error bars represent the mean ± SEM (n = 10-12). (B) ROTEM was performed using whole blood collected from WT and DGKζ-KO mice and adjusted to the indicated platelet count. Results are shown as maximal clot firmness (MCF; n = 5; left) and clot formation time (CFT; n = 5; right). Symbols represent MCF and CFT for individual animals, and bars represent the mean ± SEM. (C-D) The right carotid artery was injured by applying 10% FeCl3 for 3 minutes. Blood flow was monitored with a Doppler flow probe until 3 minutes after full occlusion. (C) Representative flow traces for WT and DGKζ-KO mice. (D) Symbols represent times to occlusion in individual animals, and bars represent the mean ± SEM (n = 10). Statistical analysis was performed by the unpaired Student t test. *P < .05 of DGKζ-KO as compared with WT.

DGKζ deficiency improves platelet adhesion to collagen under conditions of flow. Whole blood collected from WT and DGKζ-KO mice was labeled with mepacrine and flowed over collagen-coated microchannels. (A) Representative images of platelet coverage under conditions of arterial shear (1500 s−1). Images were taken under ×20 objective. (B) Surface area coverage at the indicated time points was quantified and reported as the mean ± SEM (n = 6). (C) Microchannels were washed after whole blood perfusion for 300 seconds and fixed, permeabilized, and stained with phalloidin-TRITC; Z stack images were then taken. (i) Representative Z stack images. Bars represent 100 μm. The volume (ii) and height (iii) of each platelet thrombus at 300 seconds were quantified and reported as the mean ± SEM (n = 5; 76-116 thrombi per sample). Statistical analysis was performed by the unpaired Student t test. *P < .05, **P < .01, ***P < .001 of DGKζ-KO as compared with WT.
DGKζ deficiency improves platelet adhesion to collagen under conditions of flow. Whole blood collected from WT and DGKζ-KO mice was labeled with mepacrine and flowed over collagen-coated microchannels. (A) Representative images of platelet coverage under conditions of arterial shear (1500 s−1). Images were taken under ×20 objective. (B) Surface area coverage at the indicated time points was quantified and reported as the mean ± SEM (n = 6). (C) Microchannels were washed after whole blood perfusion for 300 seconds and fixed, permeabilized, and stained with phalloidin-TRITC; Z stack images were then taken. (i) Representative Z stack images. Bars represent 100 μm. The volume (ii) and height (iii) of each platelet thrombus at 300 seconds were quantified and reported as the mean ± SEM (n = 5; 76-116 thrombi per sample). Statistical analysis was performed by the unpaired Student t test. *P < .05, **P < .01, ***P < .001 of DGKζ-KO as compared with WT.
Increased GPVI expression in DGKζ-KO platelets and megakaryocytes
To investigate the mechanism underlying enhanced GPVI-mediated platelet reactivity, we evaluated the effect of DGKζ deficiency on platelet surface receptor expression. Interestingly, flow cytometric analysis revealed that the GPVI expression level on the platelet surface was nearly 50% higher in DGKζ-KO (mean fluorescence intensity, 3156) relative to WT platelets (mean fluorescence intensity, 2075), whereas surface expression of integrin α2, integrin αIIb, PECAM-1, GPIbα, and CLEC-2 was not affected (Figure 5A; supplemental Figure 4). Western blot analysis confirmed the increase in GPVI levels in DGKζ-KO platelets, which interestingly did not have increased levels of the GPVI-associated FcRγ chain subunit (Figure 5B). These results indicate that DGKζ deficiency specifically augments GPVI protein expression without affecting the expression of other receptors.
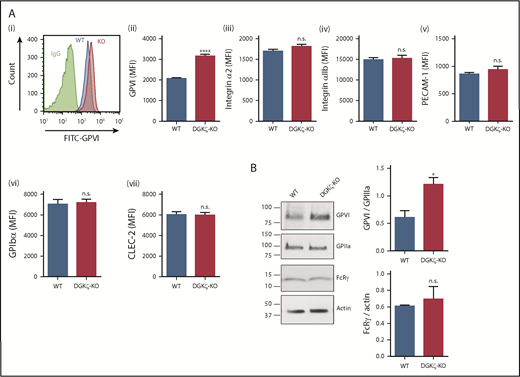
Increased expression of GPVI in DGKζ-KO platelets. (A) Platelet surface receptor expression of GPVI (i-ii), integrin α2 (iii), integrin αIIb (iv), PECAM-1 (v), GPIbα (vi), and CLEC-2 (vii) was measured in diluted whole blood samples from WT and DGKζ-KO mice by flow cytometry. Mean fluorescence intensity (MFI) is reported as the mean ± SEM (n = 10). (B) Whole lysates of washed platelets from WT and DGKζ-KO mice were analyzed by western blotting for GPVI, GPIIIa, FcRγ chain, and actin. The blots are representative of 4 independent experiments. Quantification of band densities (right) is reported as the mean ± SEM (n = 4). Statistical analysis was performed using the unpaired Student t test. *P < .05, ****P < .0001 of DGKζ-KO as compared with WT platelets. FITC, fluorescein isothiocyanate; IgG, immunoglobulin G.
Increased expression of GPVI in DGKζ-KO platelets. (A) Platelet surface receptor expression of GPVI (i-ii), integrin α2 (iii), integrin αIIb (iv), PECAM-1 (v), GPIbα (vi), and CLEC-2 (vii) was measured in diluted whole blood samples from WT and DGKζ-KO mice by flow cytometry. Mean fluorescence intensity (MFI) is reported as the mean ± SEM (n = 10). (B) Whole lysates of washed platelets from WT and DGKζ-KO mice were analyzed by western blotting for GPVI, GPIIIa, FcRγ chain, and actin. The blots are representative of 4 independent experiments. Quantification of band densities (right) is reported as the mean ± SEM (n = 4). Statistical analysis was performed using the unpaired Student t test. *P < .05, ****P < .0001 of DGKζ-KO as compared with WT platelets. FITC, fluorescein isothiocyanate; IgG, immunoglobulin G.
The level of protein expression in platelets and megakaryocytes is often, but not always, similar, including expression of GPVI.33-35 To determine whether the increased level of platelet GPVI expression began during the megakaryocyte differentiation, we examined platelet surface receptor expression in mouse hematopoietic stem cell–derived megakaryocytes during differentiation. As shown in Figure 6A, surface GPVI was absent in both WT and DGKζ-KO immature megakaryocytes and gradually increased during TPO-supported maturation. However, by day 5 of differentiation, GPVI expression was significantly higher in DGKζ-KO megakaryocytes, suggesting that the enhanced GPVI expression that we saw in DGKζ-KO platelets is attributable to upregulation of GPVI expression during megakaryocytopoiesis. In contrast, we did not see any difference in the level of expression of integrin α2, integrin αIIb, or GPIbα in DGKζ-KO relative to WT megakaryocytes, consistent with that observed in DGKζ-KO platelets (Figure 5). We further investigated whether increased DAG in DGKζ-KO platelets might be causing GPVI upregulation by adding DAG to the megakaryocyte culture. As shown in supplemental Figure 5, we did not see upregulation of GPVI by DAG treatment, suggesting that DGKζ activity, rather than intracellular DAG, leads to GPVI upregulation. To examine whether GPVI expression is regulated at the transcriptional level in megakaryocytes, we determined the mRNA level of these receptors in mature megakaryocytes using quantitative PCR. In contrast to protein expression, GPVI mRNA levels did not differ significantly between WT and DGKζ-KO megakaryocytes (Figure 6B), demonstrating that transcription of GPVI is comparable. There was also no difference in mRNA levels encoding the FcRγ chain, integrin α2, integrin αIIb, or GPIbα. These results suggest that protein expression of GPVI is regulated at the level of translation or that there is decreased GPVI degradation in DGKζ-KO relative to WT megakaryocytes.

DGKζ deficiency enhances surface expression of GPVI without increased messenger RNA (mRNA) in murine megakaryocytes. (A) Bone marrow–derived hematopoietic stem cells from WT and DGKζ-KO mice were cultured in TPO plus SCF for 0, 1, 5, or 8 days to obtain megakaryocytes, and surface expression of GPVI, integrin α2, integrin αIIb, and GPIbα was measured by flow cytometry. Mean fluorescence intensity (MFI) is reported as the mean ± SEM (n = 6). (B) Mature megakaryocytes were enriched by using a BSA density gradient, and RNA was extracted. Quantitative RT-PCR was performed, and relative expression levels of GPVI, FcRγ chain, integrin α2, integrin αIIb, GPIbα, and DGKζ in DGKζ-KO megakaryocytes are shown relative to WT expression levels (n = 5). Hypoxanthine-guanine phosphoribosyltransferase was used for normalization. *P < .05, **P < .01 of DGKζ-KO as compared with WT megakaryocytes. N.D., not detectable.
DGKζ deficiency enhances surface expression of GPVI without increased messenger RNA (mRNA) in murine megakaryocytes. (A) Bone marrow–derived hematopoietic stem cells from WT and DGKζ-KO mice were cultured in TPO plus SCF for 0, 1, 5, or 8 days to obtain megakaryocytes, and surface expression of GPVI, integrin α2, integrin αIIb, and GPIbα was measured by flow cytometry. Mean fluorescence intensity (MFI) is reported as the mean ± SEM (n = 6). (B) Mature megakaryocytes were enriched by using a BSA density gradient, and RNA was extracted. Quantitative RT-PCR was performed, and relative expression levels of GPVI, FcRγ chain, integrin α2, integrin αIIb, GPIbα, and DGKζ in DGKζ-KO megakaryocytes are shown relative to WT expression levels (n = 5). Hypoxanthine-guanine phosphoribosyltransferase was used for normalization. *P < .05, **P < .01 of DGKζ-KO as compared with WT megakaryocytes. N.D., not detectable.
Increased GPVI-mediated platelet signaling in DGKζ-KO platelets
To examine the consequences of increased GPVI expression in DGKζ-KO platelets, we investigated the activation state of signaling molecules downstream of GPVI stimulation. Previous studies in T cells have shown that DAG accumulates as a consequence of DGKζ deficiency, resulting in increased ERK phosphorylation, a commonly used reporter of cytosolic DAG levels.21,22 Consistent with the findings in T cells, both the rate and extent of ERK phosphorylation were significantly greater in DGKζ-KO relative to WT platelets after addition of CRP (Figure 7A-B). Interestingly, activation of signaling molecules that are upstream of DAG generation in the GPVI signal transduction pathway, such as PLCγ2 and Akt, was also increased in DGKζ-KO platelets, suggesting that DGKζ also influences early signaling events, perhaps as a result of the increased static level of GPVI on the platelet surface. Phosphorylation of the negative regulatory tyrosine (Tyr 507) on the Src family kinase Lyn was also found to be significantly reduced in DGKζ-KO platelets, even under resting conditions, suggesting that Lyn is in more primed state. Taken together, these data demonstrate that DGKζ deficiency results in increased surface expression of GPVI and that increased expression of this adhesion and signaling receptor produces platelets that are in a potentiated, more easily activatable state.
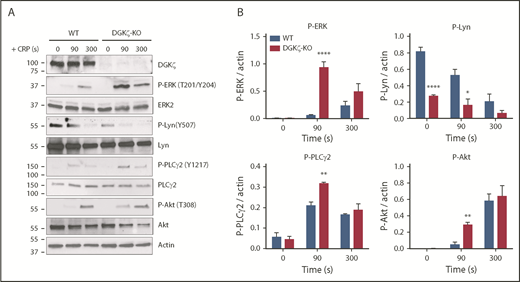
DGKζ deficiency enhances signaling in response to GPVI activation. WT and DGKζ-KO washed platelets were stimulated with 1 μg/mL of CRP for 90 and 300 seconds under stirring conditions in an aggregometer. Lysates were analyzed for DGKζ, phosphorylated ERK (P-ERK; Thr202/Tyr204), ERK2, P-Lyn (Tyr 507), Lyn, P-PLCγ2 (Tyr 1217), PLCγ2, P-Akt (Thr 308), Akt, and actin. (A) Blot shown is representative of 3 independent experiments. (B) Quantification of band densities normalized to actin over all experiments is reported as the mean ± SEM (n = 3). Statistical analysis was performed using the unpaired Student t test. *P < .05, **P < .01, ****P < .0001 of DGKζ-KO as compared with WT platelets.
DGKζ deficiency enhances signaling in response to GPVI activation. WT and DGKζ-KO washed platelets were stimulated with 1 μg/mL of CRP for 90 and 300 seconds under stirring conditions in an aggregometer. Lysates were analyzed for DGKζ, phosphorylated ERK (P-ERK; Thr202/Tyr204), ERK2, P-Lyn (Tyr 507), Lyn, P-PLCγ2 (Tyr 1217), PLCγ2, P-Akt (Thr 308), Akt, and actin. (A) Blot shown is representative of 3 independent experiments. (B) Quantification of band densities normalized to actin over all experiments is reported as the mean ± SEM (n = 3). Statistical analysis was performed using the unpaired Student t test. *P < .05, **P < .01, ****P < .0001 of DGKζ-KO as compared with WT platelets.
Discussion
DAG is an important secondary messenger that regulates platelet activation. Cytosolic levels of DAG are regulated by DGKs, which phosphorylate DAG, thereby lowering its concentration. Although the roles of DGKs in T-cell responsiveness have been well studied, DGK roles in platelet activation have not been fully examined. In the present study, we show that a specific DGK isoform, DGKζ, is expressed in platelets and megakaryocytes and that it negatively regulates GPVI-mediated platelet reactivity in mice. Although it is unclear whether DGKζ in other cell types affects hemostasis, we found that platelets from DGKζ-KO mice have enhanced responsiveness to GPVI-specific agonists and that DGKζ deficiency leads to upregulation of GPVI surface expression. This is the first study demonstrating a role for DGKζ in platelet activation.
To date, only pharmacological inhibitors have been used to examine the function of DGKs in human platelets. Although these inhibitors preferentially target type I DGKs (α, β, and γ) and have little or no effect on DGKζ activity,36 they have nevertheless shown that type I DGKs have a negative regulatory role in GPVI-mediated platelet activation and contradictory roles in regulating thrombin-mediated platelet activation, with some studies showing enhancement and others showing suppression of platelet aggregation and Ca2+ influx.10-12 These opposing results could be due to off-target effects of the inhibitors or the concentration of the agonists and inhibitors used.
Our results using DGKζ-KO mice, platelets, and megakaryocytes demonstrate that DGKζ functions to negatively regulate GPVI-mediated platelet activation, while having a mild but positive regulating influence on platelet activation induced by low-dose thrombin. The enhanced responsiveness to GPVI-specific agonists in DGKζ-KO platelets is mediated by 2 pathways, shown in supplemental Figure 6. The first is through upregulating the cell surface expression levels of GPVI, leading to enhanced activation of signaling molecules immediately downstream of this receptor, including Lyn and PLCγ2, and the other is through enhanced Ras-ERK signaling, an often-used sensitive reporter of intracellular DAG levels and enhanced PKC activation. Currently, we cannot separate the relative importance of the 2 pathways in the enhanced responsiveness of DGKζ-KO platelets, because DGKζ-specific inhibitors have not yet been developed. Their availability would enable us to determine whether short-term DGKζ inhibition, as opposed to genetic deletion from birth, can increase cell surface expression of GPVI and/or the activation of ERK signaling in mouse and human platelets.
Similar to previous studies using pharmacological inhibition by type I DGK inhibitors, we observed (Figure 1) that genetic deletion of DGKζ results in enhanced platelet aggregation to collagen and CRP, while at the same time conferring a somewhat reduced response to low-dose, but not high-dose, thrombin. These findings are consistent with the observation that tail vein bleeding times and ROTEM analysis, both initiated by relatively high doses of thrombin, are largely the same in WT and DGKζ-KO mice (Figure 3), whereas vessel occlusion time after FeCl3 vessel injury, which is thought to expose subendothelial collagen fibers, is more rapid in DGKζ-KO mice. Interestingly, mice in which the DGKε isozyme has been knocked down exhibit a prothrombotic phenotype, including enhanced platelet adhesion to endothelial cells in vitro,37 consistent with our observations of more rapid occlusion in DGKζ-KO mice.
Nevertheless, it remains unclear how DGKζ differentially regulates GPVI- vs thrombin-mediated platelet responsiveness. One speculation is that GPVI and thrombin use different subtypes of PLC, with GPVI using PLCγ2 and thrombin using PLCβ, because the subcellular location and access to substrates could affect the DGKζ activity. A similar phenotype has been reported for PKCδ, which limits secretion and aggregation in GPVI-mediated platelet activation but enhances them under thrombin stimulation,38-40 suggesting PKC activation could be differentially regulated under these pathways. Another possibility is that depletion of DGKζ reduces the available pool of PA, leading to reduced platelet responsiveness. The role of PA in enhancing thrombin-mediated platelet activation has been suggested by Marumo et al,10 who reported that treatment of human platelets with propranolol, an inhibitor of PA phosphatase, enhanced Ca2+ entry after thrombin stimulation.
One of the fascinating observations in the present study was the finding that DGKζ-KO platelets have increased expression of GPVI without affecting the total cellular expression of its noncovalently-linked partner, the FcRγ chain. This is consistent with reports showing that even though FcRγ chain–deficient platelets do not express GPVI,41 GPVI-deficient platelets have normal FcRγ chain expression,42 which is perhaps coupled with other adhesion receptors like GPIb as well.43 These results suggest that the FcRγ chain is expressed in excess of that required to traffic GPVI to the cell surface.
The biochemical explanation for increased GPVI expression remains to be explored and could theoretically involve transcriptional, posttranslational, and/or cellular trafficking and degradation levels of control. Studies in other cell types have shown that DGKζ mainly localizes in the nucleus in neurons and lung cells and actually associates with chromatin,44 suggesting that DGKζ can directly or indirectly regulate transcription. However, because DGKζ-KO megakaryocytes have increased GPVI levels without any difference in their mRNA levels (Figure 6), it is unlikely that DGKζ regulates GPVI transcription. It is also possible that surface expression levels of GPVI could be regulated by metalloproteinase-mediated shedding; however, because shedding is known to occur upon cellular activation,45 and GPVI levels are already constitutively upregulated in both resting DGKζ-KO platelets and megakaryocytes, shedding is unlikely the mechanism underlying the increased GPVI expression levels in DGKζ-KO cells. Preliminary studies in our laboratory using proteasome inhibitors suggest that DGKζ deficiency might be causing reduced GPVI degradation. Examining the cell and molecular regulation of GPVI expression levels in megakaryocytes and platelets will thus require further investigation.
Finally, there seems to be growing interest in the pathophysiology of DGKs in human health and disease. Loss-of-function mutations in DGKε have recently been implicated in atypical hemolytic-uremic syndrome,46 perhaps because of a hyperactive renal endothelium that predisposed the patient to localized thrombosis, leading to renal failure. In the field of cancer, Riese et al47 recently proposed that development of novel inhibitors targeting the α and ζ isoforms of DGK could be useful in improving the reactivity of tumor-specific T cells that are increasingly being employed in immunotherapy. The finding in the present work that such reagents also have the potential to augment platelet reactivity suggests that caution must be exerted before widespread adoption of this strategy. Additional work is thus warranted to identify the cell-type specificity and action of DGKs in thrombosis, hemostasis, vascular biology, and immunology.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the members of the Yan-Qing Ma laboratory at the Blood Research Institute for help in setting up the ferric chloride arterial injury model.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01 HL-130054 and R35 HL-139937 (P.J.N.) and by a T32 training grant from the National Heart, Lung, and Blood Institute of the National Institutes of Health (A.J.M.).
Authorship
Contribution: A.J.M. designed and performed experiments, analyzed data, created the figures, and wrote the manuscript; N.M.Z. designed and performed experiments and analyzed data; M.J.R. provided important material; D.K.N. designed experiments and wrote the manuscript; and P.J.N. designed experiments, wrote the manuscript, and supervised the work.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Peter J. Newman, Blood Research Institute, Versiti, PO Box 2178, Milwaukee, WI 53201; e-mail: peter.newman@bcw.edu.

