

Key Points
HCK expression requires PAX5 and TLR/MYD88-directed STAT3, NF-kB, and AP1 signaling in MYD88-mutated B-cell lymphomas.
TLR/MYD88 signaling activates JunB, and JunB regulates HCK expression in MYD88-mutated B-cell lymphomas.

Abstract
Hematopoietic cell kinase (HCK) is an SRC family member that is aberrantly upregulated in B-cell neoplasms dependent on MYD88-activating mutations and supports their growth and survival. We showed herein that activation of Toll-like receptor (TLR) signaling in MYD88 wild-type B cells also triggered HCK expression, denoting on path regulatory function for HCK by MYD88. To clarify the signaling cascades responsible for aberrant HCK expression in MYD88-mutated B-cell lymphomas, we performed promoter-binding transcription factor (TF) profiling, PROMO weighted TF consensus binding motif analysis, and chromatin immunoprecipitation studies. We identified PAX5, and the mutated MYD88 downstream signaling mediators STAT3, NF-κB, and AP-1, as important drivers of HCK transcription. Knockdown of PAX5, a crucial regulatory factor required for B-cell commitment and identity, abrogated HCK transcription in MYD88-mutated lymphoma cells. Among AP-1 complex components, JunB showed greatest relevance to TLR/MYD88 signaling and HCK transcription regulation. In MYD88-mutated Waldenström macroglobulinemia and activated B-cell-diffuse large B-cell lymphoma cells, knockdown of MYD88 reduced phosphorylation of JunB but not c-Jun, and knockdown of JunB reduced HCK protein levels. Deletion of STAT3, NF-κB, and AP-1 binding sites reduced corresponding TFs binding and HCK promoter activity. Moreover, inhibitors to STAT3, NF-κB, and AP-1 reduced HCK promoter activity and messenger RNA levels, particularly in combination, in MYD88-mutated lymphoma cells. The findings provide new insights into the transcriptional regulation of HCK prosurvival signaling by mutated MYD88, and the importance of JunB as a downstream mediator of the MYD88-directed signaling apparatus.
Introduction
Hematopoietic cell kinase (HCK) is a member of the SRC family tyrosine kinases and is normally expressed in cells of myeloid and B-lymphocyte lineages. In B-lymphocyte lineages, HCK is commonly expressed in earlier B-cell progenitors and is downregulated in mature B cells.1 In contrast, HCK is aberrantly overexpressed and is activated in B-cell lymphomas (Waldenström macroglobulinemia [WM], and activated B-cell [ABC] subtype diffuse large B-cell lymphoma [DLBCL]) that represent later stages of B-cell differentiation and are characterized by activating mutations in MYD88.2 HCK triggers multiple growth and survival pathways, including BTK, PI3Kδ/AKT, and ERK1/2, which are essential to WM and ABC-DLBCL survival.2 Recent clinical trials have shown that ibrutinib, a pleiotropic inhibitor that potently inhibits HCK, produces remarkable responses in MYD88-mutated WM,3 ABC-DLBCL,4 and primary central nervous system (CNS) lymphoma.5 Mutations that abolish ibrutinib-HCK binding greatly diminish antitumor activity in MYD88-mutated lymphoma cells, highlighting the importance of HCK as an essential target of ibrutinib in MYD88-driven diseases.2 Moreover, the potent HCK inhibitor A419259 shows robust activity in MYD88-mutated WM and ABC-DLBCL cells, supporting the importance of HCK as a therapeutic target in MYD88-mutated B-cell malignancies.2 However, little is known about the transcriptional regulation of HCK in MYD88-mutated malignancies. Such information could provide important new insights into MYD88-related oncogenesis and development of targeted therapeutics. We therefore sought to clarify the transcriptional regulation of HCK in MYD88-mutated B-cell lymphomas.
Materials and methods
Cell lines and treatments
MYD88L265P-mutated BCWM.1 and MWCL-1 WM cells, TMD-8, HBL-1, and OCI-Ly3 ABC-DLBCL cells, and MYD88S222R-mutated SU-DHL2 ABC-DLBCL cells, along with MYD88 wild-type (MYD88WT) OCI-Ly7, OCI-Ly19, Ramos, RPMI-8226, and MM.1S malignant B cells, were used in these experiments. The identities of the cell lines used in this study were confirmed via STR profiling with the GenePrint 10 System (Promega, Madison, WI). LPS-EB (5 µg/mL) or 5 µM ODN-2006 (InvivoGen, San Diego, CA) was used to stimulate Toll-like receptor 4 (TLR4) or TLR9 signaling. Native or HCK promoter-driven luciferase reporter transduced BCWM.1 or TMD8 cells were treated with inhibitors to transcription factors (TFs) STAT3 (STA-21; Selleck Chemicals, Houston, TX; Galiellalactone, Tocris Bioscience, Minneapolis, MN); AP1 (SP100030; SR 11302; Tocris Bioscience), and NF-κB (ACHP; Tocris Bioscience; QNZ; Triptolide [PG490]; Selleck Chemicals) for HCK transcription or promoter activity studies.
Promoter binding TF profiling assay
To characterize TFs that bind to HCK promoter and regulate HCK transcription, a Promoter-Binding TF Profiling Array I (Signosis, Santa Clara, CA) was used following the manufacturer’s instructions. Briefly, the HCK promoter sequence was used as a competitor to a set of 48 biotin-labeled TF-binding DNA motifs. Nuclear extracts from unstimulated and LPS-stimulated BCWM.1 (24 hours) were prepared using a Nuclear Extract Kit (Active Motif, Carlsbad, CA) and mixed with biotin-labeled TF-binding DNA motifs. The composition and quantity of the TF-bound DNA motifs were determined by streptavidin-horseradish peroxidase after hybridization of eluted DNA motifs, and the resulting chemiluminescence was measured using a 2104 EnVision Multilabel Reader (Perkin Elmer, Hopkinton, MA).
Chromatin immunoprecipitation (ChIP) assay
ChIP was performed using a Magna ChIP A/G kit (EMD Millipore, Danvers, MA) per manufacturer’s instructions. MYD88-mutated and wild-type control cells were fixed with 1% formaldehyde and lysed with cell lysis buffer. Following sonication, DNA-bound protein was immunoprecipitated using ChIP-grade antibodies for c-Jun, JunB, STAT3 (Cell Signaling Technology, Danvers, MA), NF-κB-p65 (Abcam, Cambridge, MA), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH; OriGene Technologies, Rockville, MD) as control. After elution of protein/DNA complexes, coprecipitated DNA was purified for polymerase chain reaction (PCR) quantification.
Quantitative reverse transcription polymerase chain reaction (RT-PCR) and PCR
Total RNA were isolated using AllPrep DNA/RNA Mini Kit (Qiagen, Germantown, MD), and complementary DNA synthesized by SuperScript III First-Strand Synthesis SuperMix (Thermo Fisher Scientific, Waltham, MA). Quantitative detection of messenger RNA (mRNA) levels for HCK was performed using TaqMan Gene Expression Assays (ID: Hs00176654_m1; Thermo Fisher Scientific) with TaqMan Gene Expression Master Mix per manufacturer’s instructions using the ABI Prism 7500 Sequence Detection System (Thermo Fisher Scientific). For quantification of coprecipitated DNA in ChIP, a quantitative PCR was performed to determine the amount of coprecipitated DNA from different cell lines using SYBR Green Master Mix (Thermo Fisher Scientific) with primer set (HCK-pro-4F and HCK-pro-4R) (Table 1).
Primer sets used for PCR and targeted sequences for knockdown studies
| Targeted sequences | Forward primer | Reverse primer |
|---|---|---|
| STAT3-mut | GCCCCCGCCTCTAGTTCTAGAAAGTTGGCA-CCCCGGAACCTCAGGGGCTG | CAGCCCCTGAGGTTCCGGGGTGCCAACTT-TCTAGAACTAGAGGCGGGGGC |
| AP1_mut-1 | ATCCCATCCACTTGGCCCTCCAGCCCGACT | AGTCGGGCTGGAGGGCCAAGTGGATGGGAT |
| AP1_mut-2 | CGGAAGAGAAACCGCAGACCTGCCTGGAAG | AGTCGGGCTGGAGGGCCAAGTGGATGGGAT |
| AP1_mut-3 | AGGAGACGGAGGTTACGGGGAGCCGGTTAG | CTAACCGGCTCCCCGTAACCTCCGTCTCCT |
| AP1_mut-4 | CCCGCCTCTAGTTCTGTTTCCCGGCACTGG | CCAGTGCCGGGAAACAGAACTAGAGGCGGG |
| AP1_mut-5 | CTCGGGCCCGTGCAGCCACCACTTCTCTGG | CCAGAGAAGTGGTGGCTGCACGGGCCCGAG |
| AP1_mut-6 | GTTCCCGGTGGGGGCCGCCGGGTTGGCAGC | GCTGCCAACCCGGCGGCCCCCACCGGGAAC |
| NF-kB_mut-1 | TCGGGCCGCGGAGTTGCCAGAAGGGAGGGG | CCCCTCCCTTCTGGCAACTCCGCGGCCCGA |
| NF-kB_mut-2 | CGCGCACAGGGGACCGCCCGCGAGGGGTCC | GGACCCCTCGCGGGCGGTCCCCTGTGCGCG |
| NF-kB_mut-3 | GGAGACGGAACGTCGGGTAGCCCGGGGGAT | ATCCCCCGGGCTACCCGACGTTCCGTCTCC |
| NF-kB_mut-4 | TGGATTCCCGGGGAAGGCCCGTGCAGTGAC | GTCACTGCACGGGCCTTCCCCGGGAATCCA |
| NF-kB_mut-5 | GGATGGGGCTTCACAGAACCCGGGATTAC-GGTTCCCGGTGGGGGCCGGGTC | GACCCGGCCCCCACCGGGAACCGTAATCC-CGGGTTCTGTGAAGCCCCATCC |
| HCK promoter cloning | HCK-pro-F, GAGCTGGGCACAATCCCATC | HCK-In1-R, CAACTACCCCCTTCCCCAAGCTG |
| HCK promoter quantification | HCK-pro-4F, ATCCCTTGATAGGTGGGGTTAGGAT | HCK-pro-4R, TAAACTCAACTTGCCCCCAAAGATT |
| PAX5 shRNA1 | Target, GGTAATTGGAGGATCCAAA | |
| PAX5 shRNA2 | Target, GCAAGAGAGACGAAGGTAT | |
| JunB shRNA1 | Target, CGCATCAAAGTGGAGCGCAAGC | |
| JunB shRNA2 | Target, GGCCTCTCTCTACACGACTACA |
| Targeted sequences | Forward primer | Reverse primer |
|---|---|---|
| STAT3-mut | GCCCCCGCCTCTAGTTCTAGAAAGTTGGCA-CCCCGGAACCTCAGGGGCTG | CAGCCCCTGAGGTTCCGGGGTGCCAACTT-TCTAGAACTAGAGGCGGGGGC |
| AP1_mut-1 | ATCCCATCCACTTGGCCCTCCAGCCCGACT | AGTCGGGCTGGAGGGCCAAGTGGATGGGAT |
| AP1_mut-2 | CGGAAGAGAAACCGCAGACCTGCCTGGAAG | AGTCGGGCTGGAGGGCCAAGTGGATGGGAT |
| AP1_mut-3 | AGGAGACGGAGGTTACGGGGAGCCGGTTAG | CTAACCGGCTCCCCGTAACCTCCGTCTCCT |
| AP1_mut-4 | CCCGCCTCTAGTTCTGTTTCCCGGCACTGG | CCAGTGCCGGGAAACAGAACTAGAGGCGGG |
| AP1_mut-5 | CTCGGGCCCGTGCAGCCACCACTTCTCTGG | CCAGAGAAGTGGTGGCTGCACGGGCCCGAG |
| AP1_mut-6 | GTTCCCGGTGGGGGCCGCCGGGTTGGCAGC | GCTGCCAACCCGGCGGCCCCCACCGGGAAC |
| NF-kB_mut-1 | TCGGGCCGCGGAGTTGCCAGAAGGGAGGGG | CCCCTCCCTTCTGGCAACTCCGCGGCCCGA |
| NF-kB_mut-2 | CGCGCACAGGGGACCGCCCGCGAGGGGTCC | GGACCCCTCGCGGGCGGTCCCCTGTGCGCG |
| NF-kB_mut-3 | GGAGACGGAACGTCGGGTAGCCCGGGGGAT | ATCCCCCGGGCTACCCGACGTTCCGTCTCC |
| NF-kB_mut-4 | TGGATTCCCGGGGAAGGCCCGTGCAGTGAC | GTCACTGCACGGGCCTTCCCCGGGAATCCA |
| NF-kB_mut-5 | GGATGGGGCTTCACAGAACCCGGGATTAC-GGTTCCCGGTGGGGGCCGGGTC | GACCCGGCCCCCACCGGGAACCGTAATCC-CGGGTTCTGTGAAGCCCCATCC |
| HCK promoter cloning | HCK-pro-F, GAGCTGGGCACAATCCCATC | HCK-In1-R, CAACTACCCCCTTCCCCAAGCTG |
| HCK promoter quantification | HCK-pro-4F, ATCCCTTGATAGGTGGGGTTAGGAT | HCK-pro-4R, TAAACTCAACTTGCCCCCAAAGATT |
| PAX5 shRNA1 | Target, GGTAATTGGAGGATCCAAA | |
| PAX5 shRNA2 | Target, GCAAGAGAGACGAAGGTAT | |
| JunB shRNA1 | Target, CGCATCAAAGTGGAGCGCAAGC | |
| JunB shRNA2 | Target, GGCCTCTCTCTACACGACTACA |
HCK promoter clone and luciferase reporter assay
HCK promoter that covers 1 kb upstream from the transcription initiation site to the 0.5-kb intron sequence downstream of the first exon was cloned by PCR from BCWM.1 cells using primer set (HCK-pro-F and HCK-In1-R) and was cloned into TA cloning vector (pCR-TOPO, Thermo Fisher Scientific). A series of mutant clones with deleted TF-binding motif was generated using GeneArt Site-Directed Mutagenesis PLUS System (Thermo Fisher Scientific) with primer sets for STAT3-mut, AP1-mut-1 to 6, and NF-κB-mut-1 to 5 (Table 1). HCK promoter and its mutants were then cloned into a promoterless lentiviral luciferase reporter vector (pLVX-MetLuc; Takara Bio USA, Inc, Mountain View, CA) and transduced into HEK-293, BCWM.1, and TMD-8 cells. The luciferase activities were determined by luminescent measurement on equal cell numbers with Ready-To-Glow Secreted Luciferase Reporter Systems (Takara Bio USA, Inc). The luciferase reporter vector–transduced HEK-293 cells were treated with 1 ng/mL tumor necrosis factor-α (TNF-α) overnight before luciferase activity detection.
TF pull-down assays
A TF pull-down assay was performed using biotin-labeled wild-type HCK promoter or corresponding TF binding site mutant that was amplified with biotin-labeled primer set HCK-pro-F and HCK-In1-R and incubated with BCWM.1 and TMD-8 nuclear extracts. The pull-down assay was performed using EpiQuik General Protein-DNA Binding Assay Kit (Epigentek, Farmingdale, NY). Briefly, 0.2 µg biotin-labeled probe was added to 500 µg nuclear extract and rotated in 4°C for 60 minutes and then incubated with prewashed streptavidin beads for another 30 minutes at 4°C. After washing steps, bound TFs were eluted with sodium dodecyl sulfate–loading buffer and analyzed by western blot.
Lentiviral overexpression or knockdown experiments
Overexpression of MYD88L265P or MYD88WT in BCWM.1 or Ramos cell lines, or knockdown of MYD88 in BCWM.1 and TMD8 cells, was performed using the same sets of lentiviral vectors as previously described.6 Knockdown of PAX5 was performed using a lentiviral short hairpin RNA (shRNA) expression vector pLV-H1-mPGK-Green (BioSettia, San Diego, CA) designed to target PAX5 (Table 1). Knockdown of JunB was done using lentiviral shRNA expression vector pLV-EFS>EGFP (VectorBuilder Inc, Chicago, IL) designed to target JunB (Table 1). Following lentiviral transduction, GFP+ cells were selected by flow sorting, and mRNA or protein levels for HCK, PAX5, or JunB were detected. A scrambled shRNA vector was used for control purposes.
Transcriptome studies
Transcriptome profiling was conducted by the Center for Cancer Computational Biology at the Dana-Farber Cancer Institute (Boston, MA) using the NEBNext Ultra RNA Library Prep Kit (New England BioLabs, Ipswich, MA) as previously described.7 These studies were approved by the Dana-Farber/Harvard Cancer Center institutional review board, and written consent from patients for use of their samples was obtained.
Western blotting
Western blots were performed for the detection of protein phosphorylation or expression levels in cell lines or following the cell stimulation by LPS (for TLR4) or ODN-2006 (for TLR9); MYD88 overexpression or knockdown; PAX5 knockdown; JunB knockdown or pull-down with biotinylated probes using antibodies for PAX5 (Abcam); phospho-NF-kB-p65(Ser529; Rockland Immunochemicals, Limerick, PA); phospho-JunB (Ser79; OriGene Technologies); phospho-c-Jun (Ser63), phospho-STAT3 (Tyr705), STAT3, NF-kB-p65, JunB, c-Jun, JunD, HCK, MYD88 (Cell Signaling Technology). GAPDH antibody (Santa Cruz, CA) was used for determination of protein loading.
Statistical analysis
The statistical significance of differences was analyzed using 1-way analysis of variance with Tukey's multiple comparisons test by Prism software. Pairwise comparisons using Wilcoxon rank sum test adjusted for multiple hypothesis testing with the Holm-Bonferroni method were used for transcriptome data analysis. Differences were considered significant when P < .05.
Results
TLR/MYD88 signaling promotes HCK expression in B-cell lymphoma cells
In previous studies, we observed that HCK was aberrant upregulated and hyperactivated and supported the growth and survival of MYD88-mutated WM and ABC-DLBCL cells.2 To further validate the expression of HCK in WM patients, we performed transcriptome analysis of MYD88-mutated WM lymphoplasmacytic cells (LPCs) and compared HCK expression to healthy donor peripheral blood B cells, memory B cells, and bone marrow plasma cells. Our findings by transcriptome analysis validated our previous quantitative RT-PCR findings that showed HCK to be overexpressed in MYD88-mutated LPCs (Figure 1A). No differences in HCK expression were observed based on the concurrent presence or absence of CXCR4 mutations in MYD88-mutated primary LPCs. Given these findings, we sought to clarify whether HCK expression could be upregulated by native TLR/MYD88 signaling. Agonists to TLR4 (LPS-EP) and TLR9 (ODN-2006) were therefore used to stimulate MYD88 wild-type and mutated B-cell lines. Stimulation of MYD88WT cells with either a TLR4 or a TLR9 agonist strongly triggered HCK expression. Among MYD88-mutated cells, TLR4 or TLR9 stimulation increased HCK transcription in BCWM.1 WM cells that have moderate HCK expression at baseline, but had little impact on HCK transcription levels in MWCL-1 WM, or TMD-8 and HBL-1 ABC-DLBCL cells that show high levels of HCK expression at baseline (Figure 1B). The increased HCK expression induced by TLR4 or TLR9 agonist stimulation was confirmed at the protein level by western blotting in MYD88-mutated BCWM.1 cells and MYD88WT Ramos cells (Figure 1C). Importantly, western blot analysis showed that overexpression of mutated MYD88 but not wild-type MYD88 robustly increased HCK protein levels in MYD88-mutated BCWM.1 as well as MYD88WT Ramos cells (Figure 1D). The above data therefore demonstrate that HCK expression is promoted by TLR/MYD88 signaling at both the transcription and the protein levels.

TLR/MYD88 signaling promotes HCK expression in B-cell lymphoma cells. (A) HCK transcript levels by transcriptome analysis in MYD88-mutated bone marrow LPCs from WM patients (WM), peripheral CD19+ B cells (PB), and CD19+CD27+ memory B cells (MB) from healthy donors, and CD138+ bone marrow plasma cells (PC) from healthy donors. (B) Quantitative RT-PCR to detect HCK transcript levels was performed following stimulation with a TLR4 (LPS-EB), TLR9 (ODN-2006) for 24 hours in MYD88-mutated BCWM.1, MWCL-1, TMD-8, HBL-1 cells, and MYD88WT OCI-Ly7, OCI-Ly19, and Ramos cells. (C) HCK protein expression was assessed by western blot analysis following stimulation with TLR4 (LPS-EB) and TLR9 (ODN-2006) agonists for 48 hours in MYD88-mutated BCWM.1 and MYD88WT Ramos cells. (D) HCK protein expression by western blot analysis following transduction of vector only, or vectors expressing MYD88WT or MYD88L265P in BCWM.1 and Ramos cells. *P < .05; **P < .01; ***P < .001. endo-MYD88, endogenous MYD88; MYD88-flag, N-terminal flag-tagged MYD88; NS, not significant.
TLR/MYD88 signaling promotes HCK expression in B-cell lymphoma cells. (A) HCK transcript levels by transcriptome analysis in MYD88-mutated bone marrow LPCs from WM patients (WM), peripheral CD19+ B cells (PB), and CD19+CD27+ memory B cells (MB) from healthy donors, and CD138+ bone marrow plasma cells (PC) from healthy donors. (B) Quantitative RT-PCR to detect HCK transcript levels was performed following stimulation with a TLR4 (LPS-EB), TLR9 (ODN-2006) for 24 hours in MYD88-mutated BCWM.1, MWCL-1, TMD-8, HBL-1 cells, and MYD88WT OCI-Ly7, OCI-Ly19, and Ramos cells. (C) HCK protein expression was assessed by western blot analysis following stimulation with TLR4 (LPS-EB) and TLR9 (ODN-2006) agonists for 48 hours in MYD88-mutated BCWM.1 and MYD88WT Ramos cells. (D) HCK protein expression by western blot analysis following transduction of vector only, or vectors expressing MYD88WT or MYD88L265P in BCWM.1 and Ramos cells. *P < .05; **P < .01; ***P < .001. endo-MYD88, endogenous MYD88; MYD88-flag, N-terminal flag-tagged MYD88; NS, not significant.
HCK promoter-binding TF profiling and consensus TF binding motif analysis
Because the activation of the TLR/MYD88 pathway triggers HCK expression, we next performed promoter-binding TF profiling to delineate the TFs that bind to the HCK promoter in MYD88-mutated cells. We used BCWM.1 WM cells for these experiments because HCK was induced by TLR stimulation. Nuclear extracts from untreated and LPS-treated BCWM.1 cells were used for HCK promoter binding analysis, and heat maps depicting relative changes in TF binding are shown in Figure 2A. A cross-sample comparison for each single TF is also shown in Figure 2B. TFs were deemed to show significant HCK promoter binding if their signals reduced >50% following addition of HCK promoter in LPS-stimulated BCWM.1 cell nuclear extracts based on the similar justification used by others.8-10 LPS stimulation activated many TFs under TLR/MYD88 signaling and showed increased signals for specific DNA motifs binding, whereas some of these specific DNA motifs binding signals were greatly reduced in the presence of HCK promoter sequence, indicating the presence of these specific TF binding motifs on the HCK promoter (Figure 2A-B). AP-1, C/EBP, CAR, CDP, CREB, GAS/ISRE, GR/PR, NF-1, NF-κB, PAX5, and STAT3 met the criteria and showed the highest magnitude of increase following LPS stimulation of BCWM.1 cells. Among these TFs, PAX5 showed the greatest signaling attenuation following addition of HCK promoter sequence in both unstimulated and LPS-stimulated BCWM.1 WM cells. To further explore the TFs that regulate HCK transcription, we next analyzed the HCK promoter sequence with PROMO software to delineate TF consensus binding motifs by weighted analysis.11,12 The HCK promoter was predicted by PROMO to bind to PAX5, with 35 consensus PAX5 binding sites identified on the HCK promoter (Figure 2C). In addition, PROMO analysis predicted binding of STAT3 (1 binding site), NF-κB (5 sites), and AP-1 (6 binding sites) to the HCK promoter (Figure 2C).
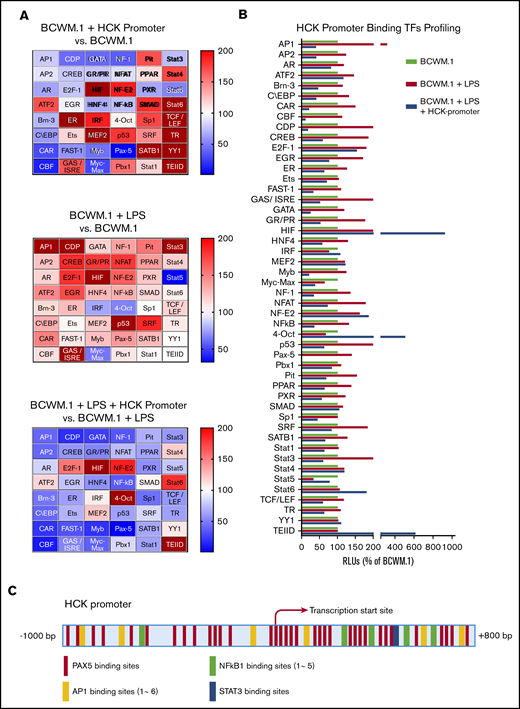
TF binding profile and consensus binding motifs for HCK promoter. (A) Findings from Promoter-binding studies to delineate TFs bound to the HCK promoter in MYD88-mutated cells. Heat maps indicate change in relative luminescence units (RLU%), which depict free unbound activated TFs in nuclear extracts from MYD88-mutated BCWM.1 cells. Top panel depicts RLU changes following the addition of HCK promoter sequence to nuclear extract of unstimulated BCWM.1 cells (BCWM.1 + HCK promoter vs BCWM.1). Middle panel depicts RLU changes in BCWM.1 cell nuclear extracts following LPS stimulation (BCWM.1 + LPS). Bottom panel depicts RLU changes in LPS-stimulated BCWM.1 cell nuclear extract following the addition of HCK promoter sequence (BCWM.1 + LPS + HCK promoter vs BCWM.1 + LPS). Blue to brown spectrum in heat maps indicates relative level increase in free activated TFs detected by Promoter-binding assay. (B) Cross-sample comparison of RLU% depicting free unbound activated TFs after addition of HCK promoter in unstimulated BCWM.1 cell nuclear extracts; LPS-stimulated BCWM.1 cell nuclear extracts; and after addition of HCK promoter sequence in LPS-stimulated BCWM.1 cell nuclear extract. (C) PROMO-weighted consensus TF-binding sites for TFs predicted to bind to the HCK promoter.
TF binding profile and consensus binding motifs for HCK promoter. (A) Findings from Promoter-binding studies to delineate TFs bound to the HCK promoter in MYD88-mutated cells. Heat maps indicate change in relative luminescence units (RLU%), which depict free unbound activated TFs in nuclear extracts from MYD88-mutated BCWM.1 cells. Top panel depicts RLU changes following the addition of HCK promoter sequence to nuclear extract of unstimulated BCWM.1 cells (BCWM.1 + HCK promoter vs BCWM.1). Middle panel depicts RLU changes in BCWM.1 cell nuclear extracts following LPS stimulation (BCWM.1 + LPS). Bottom panel depicts RLU changes in LPS-stimulated BCWM.1 cell nuclear extract following the addition of HCK promoter sequence (BCWM.1 + LPS + HCK promoter vs BCWM.1 + LPS). Blue to brown spectrum in heat maps indicates relative level increase in free activated TFs detected by Promoter-binding assay. (B) Cross-sample comparison of RLU% depicting free unbound activated TFs after addition of HCK promoter in unstimulated BCWM.1 cell nuclear extracts; LPS-stimulated BCWM.1 cell nuclear extracts; and after addition of HCK promoter sequence in LPS-stimulated BCWM.1 cell nuclear extract. (C) PROMO-weighted consensus TF-binding sites for TFs predicted to bind to the HCK promoter.
The expression and phosphorylation of TFs that bind to HCK promoter in MYD88-mutated lymphoma cells
Combining both HCK promoter-binding TFs profiling and consensus TF binding motifs analysis, we next examined protein expression and phosphorylation of PAX5, STAT3, NF-κB-p65, and AP-1 complex components in MYD88-mutated WM and ABC-DLBCL cells in which we previously showed high levels of HCK transcript and protein expression relative to MYD88WT lymphoma and myeloma cells.2 Western blot analysis showed that PAX5 was abundantly expressed in WM and ABC-DLBCL cells, but was absent in MM1S myeloma cells as expected. Importantly, either marked protein expression or higher phosphorylation or both for STAT3, NF-kB-p65, and the AP-1 components JunB and c-Jun were detected in MYD88L265P WM and ABC-DLBCL cells, as well as the expression of AP-1 component JunD in MYD88S222R-mutated SU-DHL-2 ABC-DLBCL cells that highly express HCK (Figure 3A). By comparison, MYD88WT lymphoma and myeloma cells, which show lower HCK expression, showed either lower STAT3, JunB, c-Jun, and JunD expression or lower phosphorylation for STAT3, NF-kB-p65, and JunB (Figure 3A).
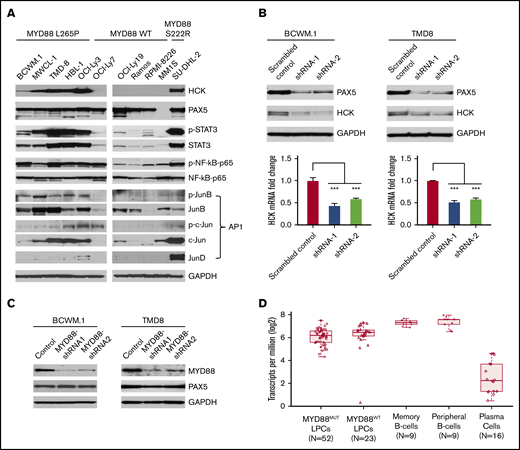
TF expression, activation, and the impact of PAX5 on HCK transcription in MYD88-mutated lymphoma cells. (A) Western blot studies depicting protein expression levels of PAX5, STAT3, NF-kB (NF-κB-p65), and AP-1 complex members (JunB, c-Jun, JunD) predicted by TF promoter-binding assay and PROMO analysis as HCK promoter binding TFs in MYD88-mutated WM and ABC-DLBCL cell lines (BCWM.1, MWCL-1, TMD-8, HBL-1, OCI-Ly3, and SU-DHL-2) and MYD88 wild-type B-cell lymphoma (OCI-Ly7, OCI-Ly19, Ramos) and myeloma cells (RPMI-8226, MM.1S). The HCK protein expression levels and the phosphorylation levels of mutated MYD88–directed TFs STAT3, NF-κB-p65, and AP-1 complex members (JunB, c-Jun) were also detected. GAPDH protein expression was used to demonstrate uniform protein loading. (B) The regulation of HCK transcription by PAX5 was assessed by lentiviral knockdown of PAX5 with 2 distinct shRNAs in MYD88-mutated BCWM.1 and TMD-8 cells and compared with scrambled control vector. Quantitative RT-PCR was performed after day 5 of lentiviral transduction. HCK protein levels and knockdown efficiencies for PAX5 were analyzed by western blot at the same time as the sample collection for HCK mRNA quantification. GAPDH was used for loading control. (C) The regulation of PAX5 by mutated MYD88 was assessed by lentiviral-mediated knockdown of MYD88 in MYD88-mutated BCWM.1 and TMD-8 cells using 2 distinct shRNAs and compared with scrambled control vector. Protein levels of PAX5 are shown, and GADPH served as a protein loading control. (D) Transcriptome analysis depicting PAX5 transcript levels in CD19-selected bone marrow LPCs from MYD88-mutated WM patients, and MYD88 wild-type WM patients; peripheral CD19-selected B cells and CD19- and CD27-selected memory B cells from healthy donors; and CD138-selected bone marrow plasma cells from healthy donors. ***P < .001.
TF expression, activation, and the impact of PAX5 on HCK transcription in MYD88-mutated lymphoma cells. (A) Western blot studies depicting protein expression levels of PAX5, STAT3, NF-kB (NF-κB-p65), and AP-1 complex members (JunB, c-Jun, JunD) predicted by TF promoter-binding assay and PROMO analysis as HCK promoter binding TFs in MYD88-mutated WM and ABC-DLBCL cell lines (BCWM.1, MWCL-1, TMD-8, HBL-1, OCI-Ly3, and SU-DHL-2) and MYD88 wild-type B-cell lymphoma (OCI-Ly7, OCI-Ly19, Ramos) and myeloma cells (RPMI-8226, MM.1S). The HCK protein expression levels and the phosphorylation levels of mutated MYD88–directed TFs STAT3, NF-κB-p65, and AP-1 complex members (JunB, c-Jun) were also detected. GAPDH protein expression was used to demonstrate uniform protein loading. (B) The regulation of HCK transcription by PAX5 was assessed by lentiviral knockdown of PAX5 with 2 distinct shRNAs in MYD88-mutated BCWM.1 and TMD-8 cells and compared with scrambled control vector. Quantitative RT-PCR was performed after day 5 of lentiviral transduction. HCK protein levels and knockdown efficiencies for PAX5 were analyzed by western blot at the same time as the sample collection for HCK mRNA quantification. GAPDH was used for loading control. (C) The regulation of PAX5 by mutated MYD88 was assessed by lentiviral-mediated knockdown of MYD88 in MYD88-mutated BCWM.1 and TMD-8 cells using 2 distinct shRNAs and compared with scrambled control vector. Protein levels of PAX5 are shown, and GADPH served as a protein loading control. (D) Transcriptome analysis depicting PAX5 transcript levels in CD19-selected bone marrow LPCs from MYD88-mutated WM patients, and MYD88 wild-type WM patients; peripheral CD19-selected B cells and CD19- and CD27-selected memory B cells from healthy donors; and CD138-selected bone marrow plasma cells from healthy donors. ***P < .001.
HCK transcription is PAX5 dependent in MYD88-mutated WM and ABC-DLBCL cells
Because PAX5 showed greatest reduction among TFs in promoter binding following addition of HCK promoter to nuclear extracts of unstimulated or LPS stimulated BCWM.1 cells, and highest number of consensus binding sites on the HCK promoter, we sought to clarify the role of PAX5 on the regulation of HCK transcription. We therefore performed knockdown of PAX5 in MYD88-mutated BCWM.1 WM and TMD-8 ABC-DLBCL cells. Knockdown of PAX5 resulted in a corresponding reduction of HCK transcription and protein expression in both BCWM.1 and TMD-8 cells, indicative of a PAX5-positive regulatory function on HCK gene expression (Figure 3B). Our next question is whether the PAX5 itself is transcriptionally regulated by mutated MYD88. We then detected PAX5 expression levels following MYD88 knockdown in MYD88-mutated cells. Knockdown of MYD88 did not modulate PAX5 transcription (data not shown) or protein expression in either MYD88-mutated BCWM.1 WM and TMD8 ABC-DLBCL cells (Figure 3C), consistent with our transcriptome findings in primary WM cells (Figure 3D). PAX5 is a TF that is essential for B-cell commitment and identity and is downregulated with B-cell to plasma cell differentiation. By transcriptome analysis, PAX5 is expressed in primary MYD88-mutated WM cells.7 However, relative to normal peripheral CD19+ B cells, as well as memory CD19+ CD27+ B cells from which WM is thought to be derived, PAX5 transcript levels are lower in MYD88-mutated WM cells. Similar levels of PAX5 transcription was observed in MYD88-mutated and wild-type cells. Conversely, PAX5 transcription was very low or absent in healthy donor CD138+ plasma cells (Figure 3D). These findings denote that PAX transcription shows intermediate levels in MYD88-mutated WM cells between that of healthy donor B cells and plasma cells and does not appear impacted by the presence of mutated MYD88 in primary WM cells.
ChIP studies show that the TFs STAT3, NF-kB-p65, and AP-1 bind directly to the HCK promoter
Because TF binding studies and PROMO-weighted analysis implicated STAT3, NF-κB, and AP-1 signaling in addition to PAX5 on HCK promoter activity, we next sought to confirm their regulatory involvement on HCK promoter activity by ChIP. All 3 TFs are known to be activated by mutated MYD88 and are expressed in WM and ABC-DLBCL cells that carry the MYD88 mutation. Based on the expression and activation patterns of these TFs (Figure 3A), ChIP grade antibodies to endogenous STAT3, NF-κB-p65, and the AP-1 components JunB and c-Jun were used. Following ChIP, an HCK promoter-specific quantitative PCR assay was used to detect HCK promoter sequences. The results demonstrated that STAT3, NF-κB-p65, and JunB had robust binding to the HCK promoter of MYD88-mutated cells in comparison with MYD88WT cells, whereas c-Jun bound to the HCK promoter in all DLBCL cell lines regardless of MYD88 mutation status. By comparison with DLBCL cells, c-Jun binding to the HCK promoter was low in MYD88-mutated WM cells (Figure 4).

ChIP studies assessing STAT3, NF-kB, and AP-1 TF binding to the HCK promoter. The fold enrichments of HCK promoter-specific sequence assessed by quantitative PCR following ChIP with ChIP grade antibodies to STAT3, NF-kB-p65, JunB, and c-Jun in MYD88-mutated WM (BCWM.1, MWCL-1) and ABC-DLBCL (TMD-8, HBL-1, OCI-Ly3) cells, and MYD88 wild-type lymphoma cells (OCI-Ly7, OCI-Ly19). Antibody to GAPDH was used as control antibody.
ChIP studies assessing STAT3, NF-kB, and AP-1 TF binding to the HCK promoter. The fold enrichments of HCK promoter-specific sequence assessed by quantitative PCR following ChIP with ChIP grade antibodies to STAT3, NF-kB-p65, JunB, and c-Jun in MYD88-mutated WM (BCWM.1, MWCL-1) and ABC-DLBCL (TMD-8, HBL-1, OCI-Ly3) cells, and MYD88 wild-type lymphoma cells (OCI-Ly7, OCI-Ly19). Antibody to GAPDH was used as control antibody.
JunB is activated by TLR/MYD88 signaling and responsible for HCK transcription regulation
Because the protein expression and activation patterns of JunB rather than c-Jun were shown to be more relevant to MYD88 mutation and HCK expression pattern in B-cell lymphoma cell lines we have tested (Figure 3A), we then evaluated the activation state of JunB and c-Jun following TLRs stimulation or overexpression of mutated MYD88. Compared with untreated cells, LPS-EB (TLR4) or ODN-2006 (TLR9) stimulation increased JunB phosphorylation, whereas c-Jun phosphorylation was not impacted (Figure 5A) in both MYD88-mutated BCWM.1 and MYD88WT Ramos cell lines. Similarly, overexpression of MYD88L265P but not MYD88WT enhanced JunB, but not c-Jun phosphorylation (Figure 5B). Further to this observation, the phosphorylation of JunB, but not c-Jun was reduced following MYD88 knockdown in MYD88-mutated BCWM.1 cells (Figure 5C). These data demonstrate JunB rather than c-Jun is activated by TLR/MYD88 signaling. Next, we investigated whether JunB could regulate HCK transcription directly. By lentiviral knockdown of JunB using 2 different shRNAs, the HCK expression levels were reduced in MYD88-mutated BCWM.1 WM and TMD8 ABC-DLBCL cell lines (Figure 5D). These data would therefore indicate that JunB activation is closely related to TLR/MYD88 signaling and HCK transcription.
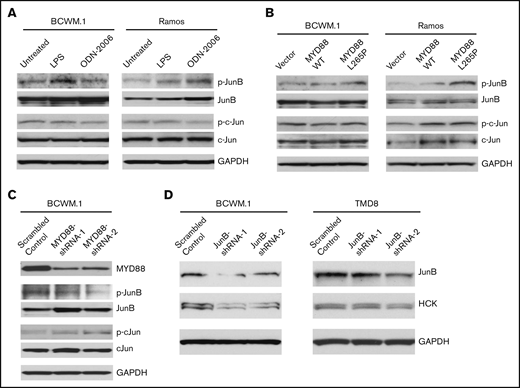
The regulation of JunB by TLR/MYD88 signaling and the impact of JunB on HCK transcription. The phosphorylation of JunB (Ser79) and c-Jun (Ser63) was assessed following TLR4 (by LPS-EB) and TLR9 (by ODN-2006) stimulation (A) as well as the lentiviral cells (C). HCK overexpression of MYD88 L265P mutant vs MYD88 WT (B) in both MYD88-mutated BCWM.1 cells and MYD88 wild-type Ramos cells. The phosphorylation of JunB (Ser79) and c-Jun (Ser63) was determined following MYD88 knockdown in MYD88-mutated BCWM.1 protein levels detected following lentiviral mediated knockdown of JunB in MYD88-mutated BCWM.1 and TMD-8 cells using 2 distinct shRNAs and compared with scrambled control vector (D). Protein levels of MYD88, JunB, c-Jun, and GADPH served as protein expression, knockdown efficiency, and loading controls.
The regulation of JunB by TLR/MYD88 signaling and the impact of JunB on HCK transcription. The phosphorylation of JunB (Ser79) and c-Jun (Ser63) was assessed following TLR4 (by LPS-EB) and TLR9 (by ODN-2006) stimulation (A) as well as the lentiviral cells (C). HCK overexpression of MYD88 L265P mutant vs MYD88 WT (B) in both MYD88-mutated BCWM.1 cells and MYD88 wild-type Ramos cells. The phosphorylation of JunB (Ser79) and c-Jun (Ser63) was determined following MYD88 knockdown in MYD88-mutated BCWM.1 protein levels detected following lentiviral mediated knockdown of JunB in MYD88-mutated BCWM.1 and TMD-8 cells using 2 distinct shRNAs and compared with scrambled control vector (D). Protein levels of MYD88, JunB, c-Jun, and GADPH served as protein expression, knockdown efficiency, and loading controls.
Deletion of STAT3, NF-kB, and AP-1 consensus binding sites on HCK promoter reduces its promoter activity
Because TNF-α can stimulate the STAT3, NF-κB, and AP-1 signaling pathways,13-16 we next examined the impact of STAT3, NF-κB, and AP-1 consensus binding site deletion on HCK promoter activity in HEK-293 cells transfected with an HCK promoter-driven luciferase reporter. Following TNF-α stimulation, the luminescence signal in HEK-293 cells expressing HCK wild-type promoter was significantly increased. Conversely, TNF-α–triggered HCK luminescence signaling was attenuated by the deletion of NF-κB, STAT3, and AP-1 consensus binding sites on the HCK promoter (Figure 6A). Compared with wild-type HCK promoter activity, NF-κB binding site mutants 1, 4, 5, a STAT3 binding site mutant, and the AP-1 binding site mutants 1, 4, 5, 6 (positions shown in Figure 2C) significantly reduced HCK promoter activity in TNF-α–stimulated HEK-293 cells.

Deletion of STAT3, AP-1, and NF-kB consensus DNA motifs reduces HCK promoter activity and their binding to HCK promoter in MYD88-mutated WM and ABC-DLBCL cells. HCK promoter activity was assessed following deletion of AP-1, STAT3, and NF-kB binding sites in TNF-α–stimulated HEK-293 cells (A), and MYD88-mutated BCWM.1 and TMD-8 cells (B). (C) The binding of biotin-labeled HCK promoter to the corresponding TFs NF-kB, STAT3, and AP-1 was also reduced by indicated mutants in pull-down assays. **P < .01; ***P < .001.
Deletion of STAT3, AP-1, and NF-kB consensus DNA motifs reduces HCK promoter activity and their binding to HCK promoter in MYD88-mutated WM and ABC-DLBCL cells. HCK promoter activity was assessed following deletion of AP-1, STAT3, and NF-kB binding sites in TNF-α–stimulated HEK-293 cells (A), and MYD88-mutated BCWM.1 and TMD-8 cells (B). (C) The binding of biotin-labeled HCK promoter to the corresponding TFs NF-kB, STAT3, and AP-1 was also reduced by indicated mutants in pull-down assays. **P < .01; ***P < .001.
To delineate whether TF binding site mutants could affect HCK promoter activities in MYD88-mutated WM and ABC-DLBCL cells, we next transduced wild-type and mutated HCK promoter vectors with luciferase reporters into MYD88-mutated BCWM.1 and TMD-8 cells. As was observed in HEK293 cells, NF-kB binding site mutants 1, 4, 5, a STAT3 binding site mutant, and the AP-1 binding site mutants 1, 4, 5, 6 showed significantly reduced HCK promoter activity in unstimulated BCWM.1 and TMD-8 cells (Figure 6B). Last, to validate if NF-κB, STAT3, or AP-1 binding site mutations could affect their corresponding HCK promoter binding, we performed pull-down experiments using either biotin-labeled wild-type or mutated HCK promoter sequences with a corresponding TF binding site deletion. The deletion of either NF-κB binding site 5, the STAT3 binding site, or AP-1 binding site 6 resulted in markedly reduced pull-down of NF-κB-p65, STAT3, and JunB levels, respectively, compared with the wild-type HCK promoter (Figure 6C).
Inhibitors to STAT3, NF-κB, and AP-1 attenuate HCK transcription in MYD88-mutated WM and ABC-DLBCL cells
To further examine the importance of STAT3, NF-kB, and AP-1 TFs in triggering HCK promoter activity in MYD88-mutated cells, we treated native or transduced BCWM.1 WM and TMD-8 ABC-DLBCL cells that expressed an HCK promoter-driven luciferase reporter with inhibitors to STAT3, NF-kB, and AP-1 at previously determined sublethal dosimetry (data not shown) and examined HCK promoter activity and HCK mRNA transcription. Treatment of BCWM.1 and TMD8 cells with either STAT3, NF-kB, or AP-1 inhibitors resulted in decreases in HCK promoter activity (Figure 7A) and transcript levels (Figure 7B). Combined use of inhibitors showed at least additive reductions of HCK transcription when compared with each inhibitor alone (Figure 7C).
![Inhibitors to STAT3, NF-κB, and AP-1 reduce HCK transcription in MYD88-mutated WM and ABC-DLBCL cells. (A) HCK promoter activities indicated by RLU% were determined by HCK promoter-driven luciferase reporter assay following treatment by inhibitors to STAT3 (STA-21), NF-kB (QNZ), and AP-1 (SR 11302) at one-fifth to one-twenty-fifth of their 50% effective concentration (EC50) levels for 6 hours in BCWM.1 WM and TMD-8 ABC-DLBCL cells. (B) HCK mRNA levels were determined by TaqMan Gene Expression Assay following treatment by inhibitors to STAT3 (Galiellalactone), NF-kB (ACHP), and AP-1 (SP100030) at one-fourth to one-twentieth of their EC50 levels for 24 hours in BCWM.1 WM and TMD-8 ABC-DLBCL cells. (C) HCK mRNA levels were determined by TaqMan Gene Expression Assay following treatment by the combination of STAT3 (Galiellalactone), NF-kB (Triptolide [PG490]), and AP-1 (SR 11302) inhibitors at less than one-tenth to one-fortieth of their EC50 levels for 24 hours in BCWM.1 WM and TMD-8 ABC-DLBCL cells. Inhibitors used for each TF are labeled, and Comb indicates the combination of inhibitors to all 3 TFs. *P < .05; **P < .01; ***P < .001. DMSO, dimethyl sulfoxide.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/1/10.1182_bloodadvances.2019000947/3/m_advancesadv2019000947f7.png?Expires=1764979699&Signature=I4qRNW9Kw7jRel2nnuNOO453yIIFfbrIJ5Ktlp~NkNqGIphouH6fk2COIphKkGmkMXJ8dhBJ73H7-uSxcE5kJ39JsHD7Ao12NhPsaoiA7YUo-26XxouChIw28gEEr1o-GdeGSRLmcsEBSYtn-~hT4ErbiFQYf9MKQpytzrJG5NeWJ2eHCauLrUnkUOZ4SZesBwO5fSMUlVKT5IRV1FHi~2KBUdu2nEHx3P~U7-Q3qqJ8xOThl1ytcnq6vF4jUt3RClb9TKB2b5kB9tlvSDbLXlxD2~ESK1vybj4RIXqpwIWVBTyOhkJcxUpr5jPRpgaIM0~aGYNJPPKY8IQSHTMC3w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Inhibitors to STAT3, NF-κB, and AP-1 reduce HCK transcription in MYD88-mutated WM and ABC-DLBCL cells. (A) HCK promoter activities indicated by RLU% were determined by HCK promoter-driven luciferase reporter assay following treatment by inhibitors to STAT3 (STA-21), NF-kB (QNZ), and AP-1 (SR 11302) at one-fifth to one-twenty-fifth of their 50% effective concentration (EC50) levels for 6 hours in BCWM.1 WM and TMD-8 ABC-DLBCL cells. (B) HCK mRNA levels were determined by TaqMan Gene Expression Assay following treatment by inhibitors to STAT3 (Galiellalactone), NF-kB (ACHP), and AP-1 (SP100030) at one-fourth to one-twentieth of their EC50 levels for 24 hours in BCWM.1 WM and TMD-8 ABC-DLBCL cells. (C) HCK mRNA levels were determined by TaqMan Gene Expression Assay following treatment by the combination of STAT3 (Galiellalactone), NF-kB (Triptolide [PG490]), and AP-1 (SR 11302) inhibitors at less than one-tenth to one-fortieth of their EC50 levels for 24 hours in BCWM.1 WM and TMD-8 ABC-DLBCL cells. Inhibitors used for each TF are labeled, and Comb indicates the combination of inhibitors to all 3 TFs. *P < .05; **P < .01; ***P < .001. DMSO, dimethyl sulfoxide.
Inhibitors to STAT3, NF-κB, and AP-1 reduce HCK transcription in MYD88-mutated WM and ABC-DLBCL cells. (A) HCK promoter activities indicated by RLU% were determined by HCK promoter-driven luciferase reporter assay following treatment by inhibitors to STAT3 (STA-21), NF-kB (QNZ), and AP-1 (SR 11302) at one-fifth to one-twenty-fifth of their 50% effective concentration (EC50) levels for 6 hours in BCWM.1 WM and TMD-8 ABC-DLBCL cells. (B) HCK mRNA levels were determined by TaqMan Gene Expression Assay following treatment by inhibitors to STAT3 (Galiellalactone), NF-kB (ACHP), and AP-1 (SP100030) at one-fourth to one-twentieth of their EC50 levels for 24 hours in BCWM.1 WM and TMD-8 ABC-DLBCL cells. (C) HCK mRNA levels were determined by TaqMan Gene Expression Assay following treatment by the combination of STAT3 (Galiellalactone), NF-kB (Triptolide [PG490]), and AP-1 (SR 11302) inhibitors at less than one-tenth to one-fortieth of their EC50 levels for 24 hours in BCWM.1 WM and TMD-8 ABC-DLBCL cells. Inhibitors used for each TF are labeled, and Comb indicates the combination of inhibitors to all 3 TFs. *P < .05; **P < .01; ***P < .001. DMSO, dimethyl sulfoxide.
Discussion
We and others previously have reported that activating mutations in MYD88 drive WM and ABC-DLBCL cell growth and survival through activation of multiple downstream signaling components, including NF-kB through transactivation of IRAK1/IRAK4 and BTK, STAT3, as well as AP-1.2,6,17-19 Mutated MYD88 also induces the expression, as well as activation of HCK, an SRC family member that is normally downregulated during late-stage B-cell ontogeny.1 HCK activates BTK, AKT, and ERK prosurvival signaling in MYD88-mutated cells. Both BTK and HCK are targets of ibrutinib, and mutations at the ibrutinib binding site of HCK lead to significant loss of ibrutinib activity in MYD88-mutated cells.2 Ibrutinib shows high levels of activity in MYD88-mutated patients, including those with WM, ABC-DLBCL, and primary CNS lymphoma.3-5
In this report, we sought to clarify the mechanisms that trigger HCK gene expression in MYD88-mutated B-cell lymphoma cells. We identified PAX5 as an essential regulator of HCK transcription in MYD88-mutated cells. PAX5 is normally expressed from the pro-B to mature B-cell stage and enforces B-cell commitment by repression of the BLIMP1.20-23 PAX5 showed the greatest reduction among any TFs following HCK promoter addition to both unstimulated and LPS stimulated BCWM.1 cells nuclear extracts in a promoter binding TF profiling assay. Importantly, knockdown of PAX5 significantly reduced HCK expression in MYD88-mutated WM and ABC-DLBCL cells. However, transcription and protein expression of PAX5 was not impacted by knockdown of MYD88 in MYD88-mutated BCWM.1 and TMD8 cells, nor did PAX5 expression vary among MYD88-mutated and wild-type lymphoma cell lines. Moreover, our transcriptome studies showed normal B-cell stage-specific expression that was not impacted by MYD88 mutation status in WM cells. Taken together, these findings affirm that PAX5 expression is not impacted by mutated MYD88 signaling per se, but its presence is required to facilitate or permit mutated MYD88 signaling.
PAX5 functions include recruitment of chromatin-remodeling, histone-modifying, and basal TF complexes to its target genes.24 This may reflect its functional role with regard to HCK transcriptional regulation wherein PAX5 induces active chromatin at the HCK promoter region, thereby permitting recruitment of TFs activated by mutated MYD88 signaling. Indeed, high levels of PAX5 are observed in most MYD88-mutated diseases, including WM, ABC-DLBCL, and primary CNS lymphoma.25-27 Although PAX5 gene expression does not appear to be regulated by MYD88 per se, it remains possible that activation of PAX5 may be regulated by mutated MYD88. Indeed, differences in phosphorylation patterns for BCR vs TLR/MYD88 pathway signaling following use of receptor-specific agonists in MYD88 wild-type cells have been observed28-31 and may contribute to upregulation of HCK transcription following TLR/MYD88 signaling. These findings also support that HCK induction is likely a normal, on TLR/MYD88 path event, that becomes constitutively activated by mutated MYD88 signaling. The facilitators for such ongoing signaling are likely to involve NF-κB, STAT3, and AP-1 TFs that are triggered by mutated MYD88 signaling, but are dependent on the presence of PAX5.
Among AP-1 TFs, our studies showed that JunB is an important mediator of mutated MYD88 signaling in WM and ABC-DLBCL cells. Overexpression of mutated but not wild-type MYD88 triggered activation of JunB but not c-Jun, and knockdown of MYD88 in MYD88-mutated BCWM.1 cells reduced JunB but had no impact on c-Jun phosphorylation. Importantly, selective deletion of consensus binding sites for NF-κB, STAT3, or AP-1, or use of inhibitors that target these TFs, resulted in modest decreases in HCK transcription, and more robust reductions when the latter was combined, signifying the necessity for multiple TFs in mediating the full effects of mutated MYD88 signaling. Interestingly, HCK was one of the target genes in the STAT3 ChIP gene list in MYD88-mutated TMD8 and OCI-Ly10 cell lines,32 and also among the downregulated genes by AP1 components JunB and c-Jun double knockdown in OCI-Ly3 and OCI-Ly10 MYD88-mutated ABC-DLBCL cells.33 The binding of STAT3 to the promoter of the regulated gene could be enhanced by NF-κB,34 and synergistic regulatory effects among these 3 TFs have also been reported in B-cell malignancies.35-37 As such, efforts to individually target these TFs from a therapeutics point of view are unlikely to silence HCK signaling. Conversely, targeting mutated MYD88 directly or HCK itself is more likely to produce sustained inhibition of HCK signaling.
In summary, we identified PAX5, and the MYD88 downstream signaling mediators STAT3, NF-κB, and AP-1, as important drivers of HCK transcription. Knockdown of PAX5, a crucial regulatory factor required for B-cell commitment and identity, abrogated HCK transcription in MYD88-mutated lymphoma cells. Among AP-1 complex components, JunB showed greatest relevance to TLR/MYD88 signaling and HCK transcription regulation. The findings therefore provide new insights into the transcriptional regulation of HCK prosurvival signaling by mutated MYD88, and the importance of JunB as a downstream mediator of the MYD88-directed signaling apparatus.
Acknowledgments
The authors gratefully acknowledge the generous support of Peter Bing, the David and Janet Bigham Research Fund of the International Waldenstrom's Macroglobulinemia Foundation, the Leukemia and Lymphoma Society TRP Grant R6507-18, the National Institutes of Health, National Cancer Institute Spore Grant 2P50 CA100707-16A1, the Edward and Linda Nelson Fund for WM Research, the Kerry Robertson Fund for WM Research, the Bauman Family Trust, and the WM patients who provided samples for these studies.
Authorship
Contribution: G.Y. and S.P.T. conceived and designed the experiments and wrote the manuscript; G.Y. performed the data analysis; X.L. and J.G.C. performed cell culture, transduction experiments, drug treatments, PCR-based studies, and immunoblotting; X.L. performed TF profiling and ChIP studies; Z.R.H. and G.G.C. performed the transcriptome and bioinformatics analysis; L.X., M.M., N.T., M.G.D., M.L.G., A. Kofides, and C.J. prepared samples; and J.J.C., K.M., A. Keezer, C.J.P., and S.P.T. provided patient care, obtained consent, and were responsible for sample collection.
Conflict-of-interest disclosure: S.P.T., J.J.C., Z.R.H., and G.Y. have received consulting fees and/or research support from Pharmacyclics Inc and Janssen Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Steven P. Treon, Bing Center for Waldenström's Macroglobulinemia, Dana-Farber Cancer Institute, M548, 450 Brookline Ave, Boston, MA 02115; e-mail: steven_treon@dfci.harvard.edu.

